

Abstract
Two different mutations of the active-site Lys-296 in rhodopsin, K296E and K296M, have been found to cause autosomal dominant retinitis pigmentosa (ADRP). In vitro studies have shown that both mutations result in constitutive activation of the protein, suggesting that the activated state of the receptor may be responsible for retinal degeneration in patients with these mutations. Previous work has highlighted the potential of retinylamine analogs as active-site directed inactivators of constitutively active mutants of rhodopsin with the idea that these or related compounds might be used therapeutically for cases of ADRP involving mutations of the active-site Lys. Unfortunately, however, amine derivatives of 11-cis-retinal, although highly effective against a K296G mutant of rhodopsin, were without affect on the two naturally occurring ADRP mutants, presumably because of the greater steric bulk of Glu and Met side chains in comparison to Gly. For this reason we synthesized a retinylamine analog one carbon shorter than the parent 11-cis-retinal and show that this compound is indeed an effective inhibitor of both the K296E and K296M mutants. The 11-cis C19 retinylamine analog 1 inhibits constitutive activation of transducin by these mutants and their constitutive phosphorylation by rhodopsin kinase, and it does so in the presence of continuous illumination from room lights.
Retinitis pigmentosa is an inherited degenerative disease of the retina that is found in X-linked, autosomal recessive, and autosomal dominant inheritance patterns (1, 2). Within a given inheritance group the clinical presentation is markedly heterogeneous, and mutations in many different genes are found to cause the disease. For example, autosomal dominant retinitis pigmentosa (ADRP) is known to be caused by mutations in at least eight different genes, including the visual pigment rhodopsin.
More than 70 different mutations of rhodopsin have been identified as the causative lesion in patients with ADRP. The mutations are found to be spread relatively evenly over the primary structure of rhodopsin and, in general, are thought to disrupt proper folding and/or transport of the protein within the rod photoreceptor cell. Although most of the mutants can be fit cleanly into one of these two categories (folding or transport defects; refs. 3 and 4), there are a few for which the mechanism of cellular dysfunction is less clear. For example, mutation of Arg-135, located at the cytoplasmic surface of the third transmembrane segment, to Leu or Trp has been claimed to prevent activation of the G protein transducin, but is otherwise without apparent effect on folding or stability of rhodopsin (5). Given that the disease is expressed in heterozygotes, it is puzzling to consider how cell death and retinal degeneration follow as a consequence of a mutation that displays a signal transduction defect in vitro.
A similar conundrum is encountered for two other ADRP mutations that involve the active site Lys-296 in rhodopsin. Lys-296 is the residue to which the 11-cis-retinal chromophore is bound covalently in the protein. Mutation of Lys-296 to either Glu or Met results in a particularly severe form of retinitis pigmentosa with early onset and the development of cataracts by the third or fourth decade of life (6, 7). The mutant proteins have been studied extensively in vitro, and although they cannot bind 11-cis-retinal, they do appear to be folded properly because they display one glaring phenotypic anomaly: they are constitutively activated (8–11). That is, the mutant proteins activate transducin in the absence of chromophore and in the absence of light. Under identical conditions wild-type opsin is inactive. Here again the connection between a defect in signal transduction and retinal cell death is not obvious although in this case at least the constitutive activity is consistent with a dominant inheritance pattern.
Although it is difficult to establish a firm causative connection between constitutively activated opsin and retinal cell death, it is likely that the activated state of rhodopsin in these mutants is responsible for the disease. If so, the therapeutic response is obvious: synthesize an active-site directed reagent that binds to the mutant proteins and irreversibly locks them in an inactive conformation. A preliminary study to test the feasibility of this approach has been carried out with a related rhodopsin mutant containing a change of Lys-296 to Gly (12). K296G opsin, like the Glu and Met mutants, does not bind 11-cis-retinal and is constitutively active. The protein does bind, however, a Schiff base derivative of 11-cis-retinal to form a pigment with near wild-type spectral properties (13). When bound to the Schiff base complex, the mutant protein is inactive in the dark but can be activated by light to a specific activity that is essentially identical to that of the wild-type protein (13). If instead of the Schiff base derivative the mutant protein is given a retinylamine analog [Fig. 1, 11-cis-retinylpropylamine (2); derived from the Schiff base by reduction with sodium cyanoborohydride], the protein is then inactive in the dark and remains inactive even after exposure to light (12). Thus, the 11-cis-retinylpropylamine functions as an active-site directed irreversible inactivator for the constitutively active K296G mutant.
Figure 1.
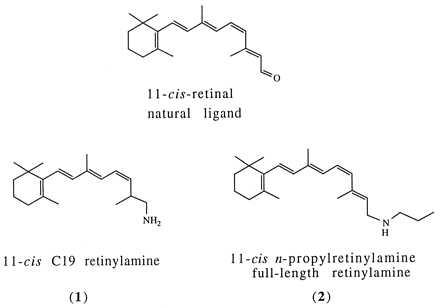
Structure of retinal and retinylamine analogs synthesized and used in this work.
Unfortunately, the same analog was ineffective against the two naturally occurring ADRP mutants, K296E and K296M, as was also the smaller primary amine 11-cis-retinylamine, presumably because of the larger steric bulk of the Glu and Met side chains relative to that of Gly (12). For this reason, we have designed and synthesized 1 (C19 retinylamine§), a retinylamine analog that is one carbon shorter than the naturally occurring 11-cis-retinal. We show here that this analog binds and irreversibly inactivates both of the naturally occurring ADRP mutant opsins.
EXPERIMENTAL PROCEDURES
Materials.
11-cis-Retinal was the generous gift of Peter Sorter (Hoffman-La Roche, Nutley, NJ). The plasmid pCMV5-RK used to express rhodopsin kinase in COS cells was generously provided by James Inglese and Robert Lefkowitz (Duke University). The Microsorb C18 reverse-phase HPLC column (10.0 × 250.0 mm) was from Rainin Instruments.
Methods.
1H NMR spectra were recorded at 300 MHz on a Varian XL-300 spectrometer; chemical shifts are reported in δ (parts per million downfield from tetramethylsilane). Mass spectra were obtained on a JEOL JMS-AX505H spectrometer at the Mass Spectra Lab at the Chemistry Department of Harvard University.
β-Ionylidenethyltriphenylphosphonium Bromide (Figs. 2 and 7).
Figure 2.
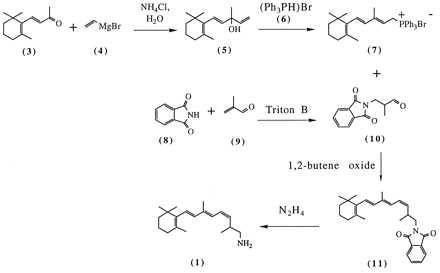
Synthetic scheme for 11-cis C19 retinylamine 1. The numbering of the compounds is as described in Experimental Procedures. The trans isomers of 1 and 11 are not shown.
β-vinylionol (5) was prepared by the literature procedure (14, 15). Triphenylphosphonium bromide (6) was prepared by bubbling HBr through a solution of triphenylphosphine (26.2 g, 0.1 mol) in 250 ml of anhydrous ether at room temperature (16). The reaction was stopped when no more HBr was absorbed. The solid was collected by filtration, washed with ether, and dried in vacuo over P2O5 to give 15.0 g (44%) of 6 as a white powder.
β-vinylionol (5, 5.5 g, 25 mmol) and 6 (8.55 g, 25 mmol) in 100 ml of methanol were allowed to react at room temperature for 48 h by the literature procedure (14, 17). During this time, the triphenylphosphonium bromide dissolved, and the solution turned yellow. The solvent was evaporated in vacuo, and the yellow residue was dissolved in a minimum of acetone. After addition of ether, 12.0 g (44%) of β-ionylidenethyltriphenylphosphonium bromide (7) crystallized as pale yellow prisms: 1H NMR (CDCl3) δ 7.80 (m, 15 H, arom. H), 6.00 (s, 2 H, CH=CH), 5.5–5.1 (m, 1 H, CH2-CH=), 4.75 (dd, JHP = 15 Hz, J = 8 Hz, P-CH2), 2.2-1.8 (m, 2 H, allyl-CH2), 1.63 (s, 3 H, =C-CH3), 1.6–1.1 (m, 4 H, CH2-CH2), 1.47 (s, 3 H, =C-CH3), 0.97 (s, 6 H, C(CH3)2).
(3Z,5E,7E)-2,6-Dimethyl-8-(2,6,6-Trimethylcyclohex-1-Enyl)-Octa-3,5,7-Trienyl-1-Amine (1, C19 Retinylamine).
A solution of phosphonium salt 7 (12.5 g, 23 mmol) in 50 ml of anhydrous dimethylformamide under N2 was cooled to 0°C. 6.0 g (28 mmol) of aldehyde 10 (18) was added, and 5 ml of 1,2-butene oxide was added dropwise (14, 17, 19). The mixture was stirred for 16 h at room temperature and then for 4 h at 60°C. Petroleum ether (100 ml, 30–60°C fraction) was added, and the solution was poured into 150 ml of 20% aqueous H2SO4. The organic phase was separated, and the aqueous phase was extracted twice with 100 ml of petroleum ether. The organic phases were combined, dried over Na2SO4, and evaporated in vacuo. The resulting yellow oil was purified by flash chromatography (silica gel, 90:10 hexane/ether), giving 1 g (11%) of an approximately 1:1 mixture of 11 and the corresponding all-trans isomer: 1H NMR (CDCl3) δ (11) 5.26 (dd, J = 10.5, 10.5 Hz, 1 H, H12); all-trans isomer 5.60 (dd, J = 14.5, 9.5 Hz, 1 H, H12).
The entire sample was dissolved in 20 ml of ethanol, 2 ml of hydrazine hydrate was added, and the mixture was kept at room temperature under N2 for 4 days as a white precipitant formed gradually. The mixture was filtered to remove the phthalylhydrazide precipitate. The filtrate then was dried over Na2SO4 and the solvent evaporated in vacuo giving 0.5 g (76%) of a mixture of 1 and the all-trans isomer: MS (CI, ammonia) m/z 274 (MH+).
An ethanolic solution of the cis- and trans-isomers was separated by purification on semi-preparative reverse-phase C18 HPLC (Microsorb C18/Rainin, particle size 5 μm, pore size 100 Å, 10.0 × 250.0 mm; gradient of 60:40 to 90:10 acetonitrile/0.5% Et3N and 0.3% trifluoroacetic acid in H2O, 4.7 ml/min). The HPLC chromatogram contained two major peaks with a 1-min separation and widths of about 1 min. The faster eluting peak was identified as the cis isomer 1 (J11,12 = 10.5 Hz), and the slower peak as the corresponding trans-isomer (J11,12 = 14.5 Hz), on the basis of 1H NMR spectroscopy, which showed all the expected absorptions. The concentrations of solutions of 1 (and the trans isomer) were estimated from UV/visible spectra by using the absorption coefficient of 24,800 M−1 cm−1 for 14-hydroxyl-13-methoxyretinylacetate (20) as representative of both compounds.
The cis isomer 1: 1H NMR (CDCl3) δ 6.56 (dd, J = 11 Hz, 10.5 Hz, 1 H, H11), 6.25 (d, J = 11 Hz, 1 H, H10), 6.23 (d, J = 16.5 Hz, 1 H, H7), 6.13 (d, J = 16.5 Hz, 1 H, H8), 5.12 (dd, J = 10.5, 10.5 Hz, 1 H, H12), 2.7–2.5 (m, 2 H, H14), 2.3 (m, 1 H, H13), 2.0 (m, 2 H, H4), 1.9 (s, 3 H, 9-Me), 1.6–1.4 (m, 4 H, H2, H3), 1.7 (s, 3 H, 5-Me), 1.1 (d, J = 4 Hz, 3 H, 13-Me), 1.0 (s, 6 H, 1-Me); HPLC (10.6 min); UV, 290.2 nm.
The trans isomer: 1H NMR (CDCl3) δ 6.59 (dd, J = 14.5, 11 Hz, 1 H, H11), 6.17 (d, J = 16 Hz, 1 H, H7), 6.04 (d, J = 16 Hz, 1 H, H8), 5.98 (d, J = 11 Hz, 1 H, H10), 5.41 (dd, J = 14.5, 9.5 Hz, 1 H, H12), 2.7–2.5 (m, 2 H, H14), 2.3 (m, 1 H, H13), 2.0 (m, 2 H, H4), 1.9 (s, 3 H, 9-Me), 1.6–1.4 (m, 4 H, H2, H3), 1.7 (s, 3 H, 5-Me), 1.1 (d, J = 4 Hz, 3 H, 13-Me), 1.0 (s, 6 H, 1-Me); HPLC (11.6 min); UV, 290.2 nm.
11-Cis n-Propylretinylamine (Figs. 1 and 2, Full-Length Retinylamine).
Compound 2 was prepared from 11-cis-retinal and n-propylamine in the dark, as has been described previously (12). Ethanolic n-propylamine (6.7 μl of a 1.2 M solution) and 100 μl of 7.5 mM ethanolic 11-cis-retinal were mixed together and allowed to react for 1 hr at room temperature. The resulting Schiff base then was reduced by adding 20 mg of NaCNBH3. After a 10-min incubation, the solid was removed by centrifugation, and the n-propylretinylamine product (2) was isolated by evaporation of the solvent. It then was dissolved in ether, washed sequentially with H2O and a saturated solution of NaCl, and finally dried over Na2SO4. The ether was evaporated with a stream of N2, and the retinylamine analog was dissolved in ethanol. The concentration of the amine analog was measured spectrophotometrically by using an absorption coefficient of 34,300 M−1 cm−1 at 320 nm (12).
Purification and Reconstitution of Wild-Type and Mutant Opsin.
The K296G (8, 13), K296E (8), and K296M (11) mutants used in this study have been described. The wild-type and mutant opsins were prepared from COS cells that had been transfected with a synthetic gene encoding the bovine opsin protein according to procedures that have been described previously (8, 11, 13, 21–24). Two different preparations of protein were used in these studies: (i) COS cell membranes (8), and (ii) purified opsin reconstituted into asolectin vesicles (11). The COS cell membrane preparation was convenient for routine isolation of the protein and could be used in transducin assays. However, it was found not to be adequate for assays with rhodopsin kinase (11). Consequently, we show here only the results from assays that used reconstituted asolectin vesicles and note that similar results with the inhibitors were obtained in transducin assays that used COS cell membrane preparations. For reasons that are at present unknown, inactivation of the mutant opsins with 1 required much longer incubation with the inhibitor when using COS cell membranes than when using asolectin vesicles. Typically, COS cell membranes required incubation with inhibitor for 4–5 hr before assay, whereas inhibition was nearly complete in asolectin vesicles after 30 min.
Opsin was purified for reconstitution into asolectin vesicles from transfected COS cells after solubilization of the cell membrane with asolectin/3-[(3-chlolamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) mixtures essentially as described (11) except that the concentrations of asolectin and CHAPS were as specified in the figure legends. Vesicles were formed by removal of CHAPS on a Sephadex G-50 column exactly as previously published (11).
Assay for Activation of Transducin.
The transducin assay was done essentially as previously described (8, 11, 13). The catalytic activities of wild-type rhodopsin as well as the mutant opsins were assessed by following the binding of [35S]GTPγS with time. The reaction mixture contained 2.5–6.5 nM opsin in 10 mM Tris buffer, pH 7.4 (or Mes buffer, pH 6.4)/100 mM NaCl/1 mM MgCl2/1 mM CaCl2/0.1 mM EDTA/1 mM DTT/2.5 μM transducin/3 μM [35S]GTPγS (3 Ci/mmol) in a final volume of 100 μl. The reaction was initiated by addition of [35S]GTPγS. For all of the following assays, a time point was taken every 30 s after initiation of the reaction. More data points were taken at every 30-s interval beginning from the first data point. The mutants K296E and K296M were preincubated either without or with 100 μM of 1 (120 μM of 2 with the K296G mutant) for 30 min at room temperature (50 min at 4°C for 2 and K296G) before being diluted 10-fold into the reaction mixture and assayed for activity. For control experiments, wild-type opsin was preincubated without or with 100 μM of 1 for 30 min at room temperature followed by incubation with 231 μM 11-cis-retinal for 30 min at room temperature before the transducin assay. Alternatively, wild-type opsin was preincubated with 231 μM of 11-cis-retinal and 100 μM of 1 simultaneously for 30 min at room temperature before the transducin assay.
Preparation of Rhodopsin Kinase from Transfected COS Cells.
Rhodopsin kinase was partially purified from COS cells transfected with the vector pCMV5-RK (25) essentially as described (11, 26) with the following exceptions. Transfected cells were lysed by forcing through a 25-gauge needle six times, and the resulting suspension was centrifuged in a Beckman TLA 120.2 rotor for 20 min at 95,000 rpm and 4°C. The supernatant fraction was chromatographed on heparin-Sepharose (1 × 10 cm column) as reported previously (11), but Tween-80 was omitted from all of the buffers used in the protocol.
Assay for Phosphorylation by Rhodopsin Kinase.
Phosphorylation of wild-type and mutant opsins purified and reconstituted into asolectin vesicles was essentially as described (11). The reaction mixture was in a total volume of 30 μl and contained 75 mM Tris buffer, pH 7.4 (or Mes buffer, pH 6.4), 10 mM MgCl2, 5 mM DTT, 100 μM [γ-32P]ATP (2,000 cpm/pmol), a 6-μl aliquot of the partially purified and concentrated rhodopsin kinase (a final concentration of roughly 1 nM as estimated from a silver staining gel), and about 0.7 pmol of opsin in asolectin vesicles. The mutants K296E and K296M were preincubated either without or with 100 μM of 1 (120 μM 2 with the K296G mutant) for 30 min at room temperature (50 min at 4°C for 2 and K296G). The reactions were initiated by the addition of rhodopsin kinase, allowed to continue for 40 min, and then stopped by the addition of 8 μl of 5× SDS/PAGE load buffer (300 mM Tris buffer, pH 6.8/10% SDS/30% sucrose/0.026% bromophenol blue) and 2 μl of β-mercaptoethanol. For control experiments, wild-type opsin was preincubated without or with 100 μM 1 for 30 min at room temperature followed by incubation with 231 μM 11-cis-retinal for 30 min before being assayed for light-dependent phosphorylation by rhodopsin kinase. Alternatively, wild-type protein was preincubated with 100 μM of 1 and 231 μM of 11-cis-retinal simultaneously, as indicated in the figure legends. After the incubation with the kinase and termination of the reaction with load buffer, all samples were applied to a 10% SDS/PAGE gel. After electrophoretic separation of the proteins, the gel was dried on filter paper by using a Bio-Rad Model 583 gel dryer (1 h at 60°C), and the phosphorylated proteins detected by autoradiography on Kodak X-Omat AR film by using an intensifying screen at −70°C.
RESULTS
Synthesis of the C19 Retinylamine 1.
The strategy used for synthesis of the C19 retinylamine 1 was to assemble the final product from two smaller fragments, a 15-carbon fragment 7 (14–17) and a 4-carbon fragment 10 (18), by using a Wittig reaction to form the double bond between carbon atoms 11 and 12 (common numbering system used for 11-cis-retinal). The Wittig reaction product (11%) was purified by flash chromatography to a 1:1 mixture of 11 and the trans isomer at the newly formed double bond. In 11, H12 absorbs at δ 5.26 as a dd, with J11,12 = 10.5 Hz, whereas H12 in the trans isomer absorbs at δ 5.60 as a dd, with J11,12 = 14.5 Hz. The low yield of the Wittig reaction was not a concern because HPLC separation of the isomers was the limiting factor.
After hydrazinolysis, 1 and the corresponding all-trans isomer were separated and purified by reverse-phase HPLC, and the identity of each isomer was verified by 1H NMR spectroscopic analysis. The faster eluting peak with a retention time of 10.6 min displayed the H12 splitting pattern of the cis isomer 1, whereas the slower eluting peak, with a retention time of 11.6 min, displayed the H12 splitting pattern of the trans isomer.
The UV/visible absorption spectrum of 1 has a maximum at 290 nm, which does not change on exposure to room light for over 24 h (not shown). Furthermore, exposure to room light does not alter the ratio of mixtures of the two isomers as judged by 1H NMR spectroscopy, nor does it affect the NMR spectrum of purified 1 (not shown). These data show that the 11,12-double bond of 1 is stable to photoisomerization under conditions of normal room lighting.
Inhibition of Phosphorylation of K296G with 11-cis n-Propylretinylamine 2.
Previous work from this laboratory has shown that 11-cis n-propylretinylamine 2 irreversibly inhibits constitutive activation of transducin by the mutant opsin K296G (Fig. 3A) whereas 2 inhibits neither the binding of retinal to nor the subsequent light-dependent activation of transducin by wild-type opsin (12). As is shown in Fig. 3B, constitutive phosphorylation of K296G by rhodopsin kinase (11) also is inhibited by 2.
Figure 3.
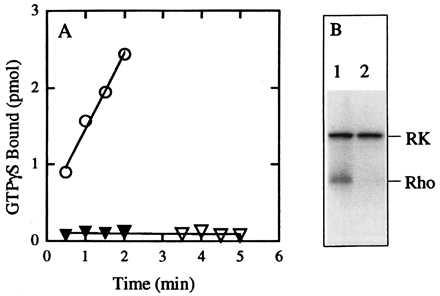
Effect of 11-cis n-propylretinylamine 2 on constitutive activity of the K296G mutant. The K296G mutant was purified by using 1 mg/ml asolectin and 1% CHAPS and then reconstituted into vesicles by removal of CHAPS on a Sephadex G-50 column. Assays were at pH 7.4. (A) Transducin activation by K296G in the absence (circles) or presence (triangles) of 11-cis n-propylretinylamine 2. Closed symbols, assay conducted in the dark; open symbols, assay conducted after exposure to light. (B) Phosphorylation of K296G by rhodopsin kinase in the absence (lane 1) or presence (lane 2) of 2. Phosphorylation reactions were performed in the presence of continuous illumination from room lights. RK indicates location of autophosphorylated rhodopsin kinase. Rho indicates location of rhodopsin.
Effect of the C19 Retinylamine 1 on the K296E and K296M Mutants.
As is shown in Fig. 4, preincubation of asolectin vesicles containing either K296E (Fig. 4A) or K296M (Fig. 4C) opsin with 100 μM of 1 for 30 min at room temperature resulted in essentially complete inhibition of the ability of these mutants to activate transducin (even in the illuminated environment of normal room lighting). Furthermore, the constitutive phosphorylation of both mutants by rhodopsin kinase (11) was similarly inhibited by preincubation with 1 under the same conditions (Fig. 4 B and D). Thus, although the larger analog 2 had no effect (12), the retinylamine analog 1 binds to and irreversibly inhibits the ability of the two naturally occurring retinitis pigmentosa mutants K296E and K296M to activate transducin and to be phosphorylated by rhodopsin kinase.
Figure 4.
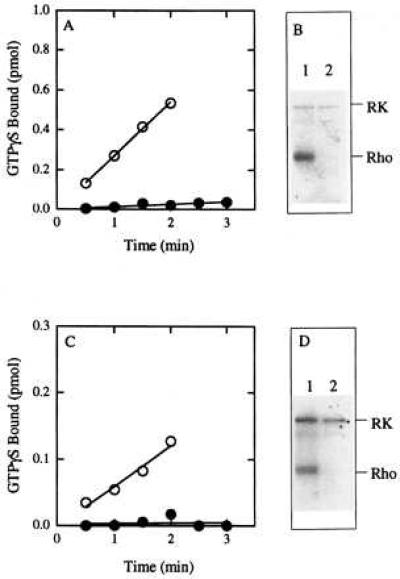
Effect of 11-cis C19 retinylamine 1 on constitutive activity of the K296E and K296M mutants. The K296E and K296M mutants were purified by using 0.375 mg/ml asolectin and 0.25% CHAPS and then reconstituted into vesicles by removal of CHAPS on a Sephadex G-50 column. (A) Transducin activation by K296E in the absence (○) or presence (•) of 1 (pH 7.4). (B) Phosphorylation of K296E by rhodopsin kinase (pH 7.4): Lane 1, in the absence of 1; lane 2, in the presence of 1. The film was exposed for 2 days. (C) Transducin activation by K296M in the absence (○) or presence (•) of 1 (pH 6.4). (D) Phosphorylation of K296M by rhodopsin kinase (pH 6.4): Lane 1, in the absence of 1; lane 2, in the presence of 1. The film was exposed for 4 days. All reactions were performed in the presence of continuous illumination from room lights.
Effect of Retinylamine Analog 1 on Wild-Type Opsin.
To determine if the C19 retinylamine analog inhibited activity of the wild-type protein, 100 μM of 1 was incubated with opsin in the dark for 30 min at room temperature either (i) in the presence of 231 μM 11-cis-retinal or (ii) alone and then 231 μM 11-cis-retinal added and the incubation continued for an additional 30 min at room temperature. The samples then were assayed for light-dependent activation of transducin and phosphorylation by rhodopsin kinase. As shown in Fig. 5, preincubation of wild-type opsin with 1 results in a significant reduction in the ability of the protein to bind 11-cis-retinal as evidenced by a greater than 50% reduction in the light-dependent activation of transducin (Fig. 5A). Similarly, preincubation with 1 inhibits light-dependent phosphorylation of the protein by rhodopsin kinase (Fig. 5B). Thus, it appears that retinylamine analog 1 can bind to and inactivate wild-type opsin in addition to the K296E and K296M mutants. Although this is an undesirable side effect, we note that simultaneous incubation with 11-cis-retinal completely protects the protein from inactivation as monitored by either the transducin (Fig. 5A) or rhodopsin kinase assays (Fig. 5B).
Figure 5.
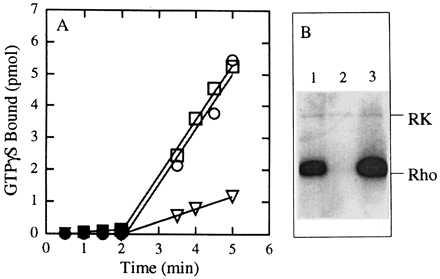
Effect of 11-cis C19 retinylamine 1 on wild-type opsin. Wild-type opsin was purified by using 0.375 mg/ml asolectin and 0.25% CHAPS and then reconstituted into vesicles by removal of CHAPS on a Sephadex G-50 column. (A) Activation of transducin. Closed symbols, in the dark; open symbols, after exposure of the reaction mixture to light for 30 sec. Circles, opsin incubated in the absence of the analog for 30 min at room temperature then with 11-cis-retinal (231 μM) for 30 min before the assay. Squares, opsin incubated with 1 (100 μM) and 11-cis-retinal (231 μM) simultaneously for 30 min at room temperature before the assay. Triangles, opsin incubated with 1 (100 μM) for 30 min at room temperature then with 11-cis-retinal (231 μM) for 30 min at room temperature before the assay. All the assays were at pH 7.4. (B) Phosphorylation of wild-type opsin. Lane 1, wild-type opsin that was incubated in the absence of the analog for 30 min at room temperature then with 231 μM of 11-cis-retinal for 30 min; lane 2, incubation with 100 μM of 1 for 30 min at room temperature then with 231 μM of 11-cis-retinal for 30 min; and lane 3, incubation with 100 μM of 1 and 231 μM of 11-cis-retinal simultaneously for 30 min at room temperature.
DISCUSSION
The goal of this work was to develop an effective inhibitor of the two constitutively active, ADRP-related rhodopsin mutants, K296E and K296M. Although activating mutations in other G protein-coupled receptors are known to cause human disease (27–31), and other mutations in rhodopsin found associated with congenital stationary night blindness present a plausible explanation for pathophysiology of rod cell dysfunction in these patients (32), a clear mechanism connecting constitutive activity of the K296E and M mutants with the clinical symptoms of retinitis pigmentosa has not been established. Indeed, the results from a transgenic mouse model suggest that the K296E mutation does not constitutively activate transducin in vivo (33). However, it is likely nonetheless that the constitutively active state of the receptor is responsible for the cellular dysfunction, perhaps as a result of the persistent phosphorylation of the protein by rhodopsin kinase and the consequent large-scale accumulation in the cell of inactive complexes of phosphorylated opsin and arrestin (11, 33). An alternate possibility is that the K296E and M mutants are inherently more unstable and prone to denaturation than the wild-type protein because they cannot bind 11-cis-retinal and, therefore, exist exclusively in the apoprotein form (cf. ref. 34). We have taken into consideration both possibilities in our approach to this work. We began with the goal of designing an active-site directed reagent that would irreversibly inactivate the constitutively active mutants, but reasoned that if we had such a reagent it also would stabilize the protein to denaturation.
Previous work suggested that a retinylamine analog of retinal would make an ideal inhibitor (12). However, it was also clear that an inhibitor derived from 11-cis-retinal would be too large to be accommodated in the ligand binding pocket of the K296E or K296M mutants (12, 13). Therefore, we set out to synthesize a retinylamine analog that was one carbon shorter than the parent 11-cis-retinal.
We presented here the synthesis and characterization of the 11-cis C19 retinylamine analog 1. We also showed that analog 1 was indeed an effective inactivator of the K296E and K296M ADRP mutants, inhibiting the ability of both proteins to constitutively activate transducin and to be constitutively phosphorylated by rhodopsin kinase. Most importantly, inhibition was complete even in the presence of constant illumination from room lights. Thus, the smaller size of this analog appears to permit access of the ligand to the active site of the mutants.
This analog does not have the same degree of specificity for mutant opsin over the wild type that was observed with the full-length 11-cis-retinylpropylamine and K296G mutant (12). As was shown in Fig. 5, the C19 retinylamine 1 has a significant inhibitory effect on the wild-type protein. It is not clear at this time why the C19 analog is less specific than the full-length amine. Perhaps the shorter carbon chain places the positively charged ammonium ion closer to the Glu-113 counterion and further from the Lys-296 side chain (35). Alternatively, loss of sp2 hybridization and planarity about carbons 13 and 14 may result in significant distortion within the ligand occupied binding pocket such that there is less distinction between mutant and wild-type proteins.
We have not extensively characterized the affinity of the C19 analog for the mutant opsins because of complications presented by the presence of lipid vesicles, but it is clear from the limited analysis we have done that the apparent affinity is not high and an analog concentration in the tens of μM range is needed for inhibition.
In conclusion, we have shown that the 11-cis C19 retinylamine 1 is an effective inhibitor of the naturally occurring constitutively active mutant rhodopsins, K296E and K296M, found in certain patients with ADRP. Future work will focus on improvements to the affinity and specificity of this reagent.
Acknowledgments
We thank Eric Gerber from Rainin for advice on purification of the analog with HPLC, Fadila Derguini for advice on the NMR analysis of the different isomers, and Jeanne Rim for help in setting up the kinase assays. This work was supported by National Institutes of Health Grant EY07965. We also acknowledge support for the Volen Center for Complex Systems by the W. M. Keck Foundation.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: ADRP, autosomal dominant retinitis pigmentosa; CHAPS, 3-[(3-chlolamidopropyl)dimethylammonio]-1-propanesulfonate.
This compound is a nor dihydro retinylamine that is properly named 15-nor-13,14-dihydroretinyl-14-amine or (Z,E,E)-2,6-dimethyl-8-(2,6,6-trimethyl-1-cyclohexen-1-yl)-3,5,7-octatrien-1-amine.
References
- 1.Humphries P, Kenna P, Farrar J G. Science. 1992;256:804–808. doi: 10.1126/science.1589761. [DOI] [PubMed] [Google Scholar]
- 2.Berson E L. Proc Natl Acad Sci USA. 1996;93:4526–4528. doi: 10.1073/pnas.93.10.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nathans J. Biochemistry. 1992;31:4923–4931. doi: 10.1021/bi00136a001. [DOI] [PubMed] [Google Scholar]
- 4.Sung C-H, Makino C, Baylor D, Nathans J. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Min K C, Zvyaga T A, Cypess A M, Sakmar T P. J Biol Chem. 1993;268:9400–9404. [PubMed] [Google Scholar]
- 6.Keen T J, Inglehearn C F, Lester D H, Bashir R, Jay M, Bird A C, Jay B, Bhattacharya S S. Genomics. 1991;11:199–205. doi: 10.1016/0888-7543(91)90119-y. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan J M, Scott K M, Falls H F, Richards J H E, Sieving P A. Invest Ophthalmol Visual Sci. 1993;34:1149. (abstr.). [Google Scholar]
- 8.Robinson P R, Cohen G B, Zhukovsky E A, Oprian D D. Neuron. 1992;9:719–725. doi: 10.1016/0896-6273(92)90034-b. [DOI] [PubMed] [Google Scholar]
- 9.Cohen G B, Oprian D D, Robinson P R. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- 10.Cohen G B, Yang T, Robinson P R, Oprian D D. Biochemistry. 1993;32:6111–6115. doi: 10.1021/bi00074a024. [DOI] [PubMed] [Google Scholar]
- 11.Rim J, Oprian D D. Biochemistry. 1995;34:11938–11945. doi: 10.1021/bi00037a035. [DOI] [PubMed] [Google Scholar]
- 12.Govardhan C P, Oprian D D. J Biol Chem. 1994;269:6524–6527. [PubMed] [Google Scholar]
- 13.Zhukovsky E A, Robinson P R, Oprian D D. Science. 1991;251:558–560. doi: 10.1126/science.1990431. [DOI] [PubMed] [Google Scholar]
- 14.Tietze L F, Eicher T. Reactions and Syntheses In the Organic Chemistry Laboratory. Mill Valley, CA: Univ. Sci. Books; 1989. pp. 464–468. [Google Scholar]
- 15.Ishikawa Y. Bull Soc Chem Jpn. 1964;37:207–209. [Google Scholar]
- 16.Seyferth D, Grim S O, Read T O. J Am Chem Soc. 1961;83:1617–1620. [Google Scholar]
- 17.Pfander H, Lachenmeier A, Hadorn M. Helv Chim Acta. 1980;63:1377–1382. [Google Scholar]
- 18.Bergmann E D, Migron Y. Org Prep Proc Int. 1976;8:75–80. [Google Scholar]
- 19.Buddrus J. Chem Ber. 1974;107:2050–2061. [Google Scholar]
- 20.Yamauchi R, Miyake N, Kato K, Ueno Y. Biosci Biotech Biochem. 1992;56:1529–1532. [Google Scholar]
- 21.Ferretti L, Karnik S S, Khorana H G, Nassal M, Oprian D D. Proc Natl Acad Sci USA. 1986;83:599–603. doi: 10.1073/pnas.83.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oprian D D, Molday R S, Kaufman R J, Khorana H G. Proc Natl Acad Sci USA. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhukovsky E A, Oprian D D. Science. 1989;246:928–930. doi: 10.1126/science.2573154. [DOI] [PubMed] [Google Scholar]
- 24.Oprian D D. In: Methods in Neurosciences. Hargrave P A, editor. Vol. 15. New York: Academic; 1993. pp. 301–306. [Google Scholar]
- 25.Lorenz W, Inglese J, Palczewski K, Onorato J J, Caron M G, Lefkowitz R J. Proc Natl Acad Sci USA. 1991;88:8715–8719. doi: 10.1073/pnas.88.19.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palczewski K. In: Methods in Neurosciences. Hargrave P A, editor. Vol. 15. New York: Academic; 1993. pp. 217–225. [Google Scholar]
- 27.Lefkowitz R J. Nature (London) 1993;365:603–604. doi: 10.1038/365603a0. [DOI] [PubMed] [Google Scholar]
- 28.Parma J, Duprez L, Van Sande J, Cochaux P, Gervy C, Mockel J, Dumont J, Vassart G. Nature (London) 1993;365:649–651. doi: 10.1038/365649a0. [DOI] [PubMed] [Google Scholar]
- 29.Shenker A, Laue L, Kosugi S, Merendino J J, Jr, Minegishi T, Cutler G B., Jr Nature (London) 1993;365:652–654. doi: 10.1038/365652a0. [DOI] [PubMed] [Google Scholar]
- 30.Laue L, Chan W-Y, Hsueh A J, Kudo M, Hsu S Y, Wu S-M, Blomberg L, Cutler G B., Jr Proc Natl Acad Sci USA. 1995;92:1906–1910. doi: 10.1073/pnas.92.6.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schipani E, Kruse K, Juppner H. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 32.Rao V R, Oprian D D. Annu Rev Biophys Biomol Struct. 1996;25:287–314. doi: 10.1146/annurev.bb.25.060196.001443. [DOI] [PubMed] [Google Scholar]
- 33.Li T, Franson W K, Gordon J W, Berson E L, Dryja T P. Proc Natl Acad Sci USA. 1995;92:3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liri T, Herzmark P, Nakamoto J M, Van Dop C, Bourne H R. Nature (London) 1994;371:164–168. doi: 10.1038/371164a0. [DOI] [PubMed] [Google Scholar]
- 35.Han M, Smith S O. Biochemistry. 1995;34:1425–1432. doi: 10.1021/bi00004a037. [DOI] [PubMed] [Google Scholar]