

Abstract
The University of California Santa Cruz (UCSC) Genome Browser (genome.ucsc.edu) is a popular Web-based tool for quickly displaying a requested portion of a genome at any scale, accompanied by a series of aligned annotation “tracks”. The annotations—generated by the UCSC Genome Bioinformatics Group and external collaborators—display gene predictions, mRNA and expressed sequence tag alignments, simple nucleotide polymorphisms, expression and regulatory data, phenotype and variation data, and pairwise and multiple-species comparative genomics data. All information relevant to a region is presented in one window, facilitating biological analysis and interpretation. The database tables underlying the Genome Browser tracks can be viewed, downloaded, and manipulated using another Web-based application, the UCSC Table Browser. Users can upload data as custom annotation tracks in both browsers for research or educational use. This unit describes how to use the Genome Browser and Table Browser for genome analysis, download the underlying database tables, and create and display custom annotation tracks.
Keywords: Genome Browser, Table Browser, UCSC, human genome, genome analysis, comparative genomics, human variation, Bioinformatics, Bioinformatics Fundamentals, Biological Databases
INTRODUCTION
The rapid progress of public sequencing and analysis efforts on vertebrate genomes has increased the demand for tools that offer quick and easy access to the data and annotations at many levels and facilitate comparative data analysis. The University of California Santa Cruz (UCSC) Genome Bioinformatics Web site at http://genome.ucsc.edu provides links to a variety of genome analysis tools, most notably the UCSC Genome Browser (Kent et al., 2002; Kuhn et al., 2009), a graphical tool for viewing a specified region of a genome and a collection of aligned annotation “tracks.” Another tool on the Web site—the UCSC Table Browser—supplies convenient access to the MySql database tables (Karolchik et al., 2003) underlying the Genome Browser annotations. Both browsers support a custom annotation tracks feature that enables users to upload their own data for display and comparison.
The main protocol of this unit (see Basic Protocol) describes how to display and navigate through a specific section of a genome and its associated annotation tracks in the Genome Browser, configure the view to focus on annotations of interest and optimize comparative analysis, link to external information, and download sequence or annotation data. Support Protocol 1 explains the process for creating and displaying a custom annotation track based on the user’s own data. Support Protocol 2 provides a basic overview of the UCSC Table Browser, describing the most commonly used functions, how to set up a simple query, and an introduction to some of the advanced features. The Genome Browser annotations and software continually evolve as new data and techniques become available; therefore, it is recommended that the user consult the UCSC Web site (http://genome.ucsc.edu) and the current version of the User’s Guide (http://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html) for the latest information on new releases and features.
BASIC PROTOCOL: USING THE UCSC GENOME BROWSER
The Genome Browser software and data may be accessed on the Internet from the UCSC Genome Bioinformatics Group Web site at http://genome.ucsc.edu.
Necessary Resources
Hardware
Unix, Windows, or Macintosh workstation with an Internet connection and a minimum display resolution of 800 × 600 dpi.
Software
An up-to-date Internet browser that supports JavaScript, such as Firefox 3.0 and higher (http://www.mozilla.com/firefox); Internet Explorer 6.0 and higher (http://www.microsoft.com/ie); or Safari 3.0 and higher (http://www.apple.com/safari). The browser must have cookies enabled.
Files
None
Navigate to the Genome Browser window to a specific genomic position
-
1.
Open the UCSC Genome Bioinformatics Group home page, at http://genome.ucsc.edu, in a Web browser.
The UCSC Genome Bioinformatics home page provides links to the Genome Browser application and a variety of other useful tools: BLAT (Kent et al., 2002), for quickly mapping sequences to a genome assembly; the Table Browser (Karolchik et al., 2004; Kuhn et al., 2009), for viewing and manipulating the data underlying the Genome Browser; the Gene Sorter (Kent et al., 2005), for exploring relationships (expression, homology, etc.) among groups of genes; VisiGene, for browsing through a large collection of in situ mouse and frog images to examine expression patterns; the Proteome Browser (Hsu et al., 2005), for viewing information about a selected protein; an in silico PCR tool for rapidly searching a sequence database with a pair of PCR primers; and Genome Graphs, a tool for viewing quantities plotted along chromosomes. General information about the Genome Browser tool suite can be found in the User’s Guide—accessed via the Help link—and the FAQ. From the home page, the user can also download the genomic sequence and annotation data, display contributed custom tracks and older archived data, review a log of released data, and access helpful utilities, training materials, credits for contributors and collaborators, mirror information, and related publications.
-
2.
Click the Genome Browser link in the left-hand sidebar menu to open the Genome Browser Gateway page.
On the Gateway page (Fig. 1.4.1), the user can set the parameters that determine which portion of a genome the Genome Browser will initially display. The bottom portion of the page provides information about the currently selected genome assembly and a list of sample position queries that can be used to open the browser.
Alternatively, the Genome Browser can be accessed by clicking on the BLAT link on the home page and then searching a DNA or protein sequence for regions of homology (step 16).
-
3.
Select the clade, genome, and assembly of interest, then type one or more search terms or a set of genomic coordinates into the “position or search term” text box to specify the genome region to display. Click the “submit” button.
The position search supports direct positional queries such as chromosome bands or chromosome coordinate ranges, as well as queries related to genomic features such as gene symbols, mRNA or EST accession numbers, author names, or other descriptive terms likely to occur in GenBank (Benson et al., 2009). The Gateway page shows examples of valid position requests applicable to the selected genome assembly.
If the position query is resolved to a single location, the Genome Browser will display a page containing an annotation track image specific to the position query, accompanied by navigation controls and display controls (Fig. 1.4.2). Frequently, the position search returns a list of several matches in response to a query rather than immediately displaying the Genome Browser page. When this occurs, click on the item of interest and the Genome Browser will open to that location. Invalid position queries (e.g., withdrawn gene names, abandoned synonyms, misspelled identifiers, and data added after the last Genome Browser database update) will result in a warning message and the previous or default position will be retained.
A custom annotation track can be uploaded into the Genome Browser by clicking the “add custom tracks” button on the Gateway page. For more information on creating and uploading custom annotation tracks, see Support Protocol 1.
To access an older genome assembly that is no longer available from the assembly menu, look in the Genome Browser archives at http://genome-archive.cse.ucsc.edu
. Several aspects of the Genome Browser display can be customized by clicking the “configure tracks and display” button (see step 8).
Figure 1.4.1.
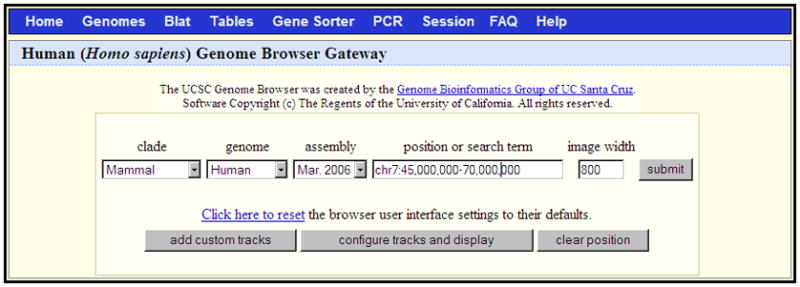
The Genome Browser Gateway page, set up to span the central portion of chromosome 7 (chr7:45,000,000-70,000,000) in the March 2006 human assembly (NCBI36). Custom annotation tracks (Support Protocol 1) can be uploaded by clicking the “add custom tracks” button. The initial Genome Browser display may be configured by clicking the “configure tracks and display” button. The lower portion of this page (not shown) displays a description of the selected assembly, relevant links, and examples of queries that may be entered in the “position or search term” box.
Figure 1.4.2.
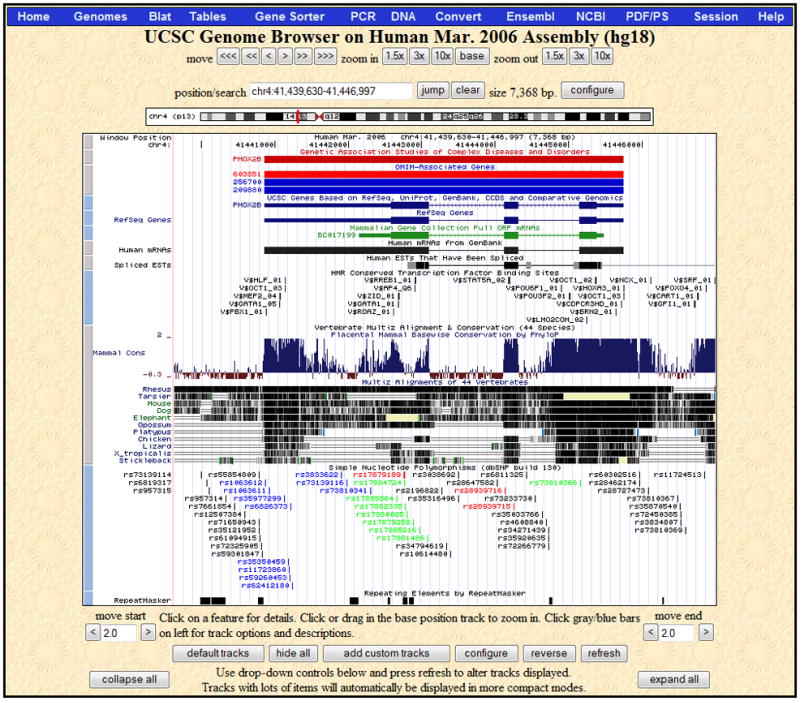
The Genome Browser annotation track page zoomed in to display the PHOX2B gene on human chromosome 4, March 2006 assembly (NCBI36). The navigation and configuration buttons are visible above and below the image. The red rectangle in the ideogram directly above the annotation tracks image indicates the location of the currently displayed region of the chromosome. The SNPs (130) track visibility has been changed from dense to pack to show individual SNPs, some of which are colored according to gene region (e.g. UTR, coding-synonymous or coding-nonsynonymous). Three additional tracks have been added to the display by changing their visibilities from hide to pack: GAD View and OMIM Genes in the Phenotype and Disease Associations group, and TFBS Conserved in the Regulation group. PHOX2B is a developmental gene that has also been associated with cancer; move the mouse over the PHOX2B item in the GAD View track in order to see a list of diseases associated with the gene. In the Vertebrate Multiz Alignment & Conservation track, note the areas of high conservation peaking in the upstream region (to the right because PHOX2B is on the antisense strand), UTRs, most exons as well as part of the first intron.
Browse and configure the annotation tracks display
-
4.
Examine the Genome Browser annotation tracks page (Fig. 1.4.2).
This image displays a set of annotation tracks aligned beneath a Base Position track (the “ruler”) indicating genomic coordinate positions. Tracks are organized into groups reflecting the nature of their data. The first time the Genome Browser is opened, the application’s default values are used to configure this display. Any preferences and configurations set during the session will be retained for use in subsequent sessions on the same Web browser. To reset the display to the set of default tracks for the selected assembly, click the “default tracks” button.
The annotation tracks image is accompanied by controls to configure the display and navigate through the sequence. For selected assemblies, a chromosome band ideogram directly above the image graphically indicates the location of the currently displayed region on the overall chromosome. Custom annotation tracks can be uploaded to the current assembly by clicking the “custom tracks” button below the image (see Support Protocol 1 for more information).
Figure 1.4.2 shows the annotation track image opened to the position of the gene PHOX2B on chromosome 4. To reach this position, enter “PHOX2B” in the position/search box, select the first matching item (the UCSC Genes PHOX2B), and then click the zoom out 1.5x button. Note that the Genome Browser automatically changes the text in the Position box to show the chromosomal position of the resulting display. In most annotation tracks, the aligned regions are represented by vertical bars or blocks. In the Spliced ESTs track shown in this example, the degree of darkness of the block shading corresponds to the number of features aligning to the region. In the mRNA and gene prediction tracks, the thicker regions (usually coding exons) are connected by thin horizontal lines representing gaps (usually spliced-out introns). Thinner blocks on the leading and trailing ends of the aligning regions in gene tracks represent the 5′ and 3′ untranslated regions (UTRs). In full or pack display mode, arrowheads on the connecting lines indicate the direction of transcription.
Note the comparative genomics annotations displayed in Figure 1.4.2. The Conservation track shows a measure of evolutionary conservation among multiple species, which tends to indicate functional regions of the genome. The lower section of the track shows pairwise alignments of each species to the reference sequence; the top section displays the evolutionary conservation scores assigned by the phyloP program in the PHAST package (Siepel et al., 2005). At this level of detail, the phyloP scores highlight exons, untranslated regions (UTRs) and other regions that show signs of conservation across species.
To generate a high-quality image of this annotation tracks image in PostScript or PDF format, click the PDF/PS link in the top menu bar.
-
5.
Change the display mode of an annotation track by locating the track’s name in the Track Controls section below the image, selecting a display mode from the track’s pull-down menu, and then clicking the “refresh” button.
Depending on individual display modes, annotation tracks may be hidden from view (hide mode), displayed with all features collapsed into a single line (dense mode), or fully expanded with each feature on a separate line (full mode). Many tracks feature two additional display modes: pack mode, in which each feature is displayed and labeled, but not necessarily on a separate line, and squish mode, which is similar to pack mode, but displays unlabeled features at half-height. To quickly toggle between dense and full (or pack) modes in the annotation track image, click on the track’s label. To hide all the tracks in the display, click the “hide all” button beneath the annotation tracks image.
By adjusting the display modes of the tracks in the annotation track graphic the user can restrict the display to data of interest, reduce clutter, and improve speed. Dense display mode is useful to get an overview of an annotation or to reduce the space used by a track when the individual feature details of an annotation track are not required. Squished and packed displays show individual feature details of densely populated tracks while conserving space. Use full mode sparingly: in some tracks, the number of features that may potentially align at a selected position can be quite large. When the feature count is excessive in full display mode, the browser displays the track in pack mode if possible; if the track does not support pack mode, it displays the first 250 items individually, then groups the remaining items into a single line in dense mode at the bottom of the track.
-
6.
To change the image to a new genomic position, type a different set of search terms into the position/search box, then click the “jump” button.
Figure 1.4.3 shows the larger region obtained by entering the query 22q13.32;22q13.33 on the March 2006 (NCBI36) human genome assembly. Several tracks that display best in large regions due to the sparseness of their annotations have been added to the display, and several tracks whose many items would saturate the display have been hidden. At this large scale, the completeness of the assembly is indicated by the sparse gaps, and it is easy to see regions of relative gene density or scarcity Coarse measures such as population genetic statistics have more of a perceivable signal, while fine-scale measures such as the per-base Conservation scores have almost no signal due to averaging over large numbers of bases.
-
7.
Use the mouse drag-and-zoom feature or the “zoom” and “move” buttons to increase or decrease the breadth of the displayed coordinate range, or to shift one or both ends of the coordinate range to the left or right.
To quickly zoom in to an exact coordinate range, click on the desired leftmost coordinate in the Base Position track and drag the mouse to the right to highlight the region of interest. The navigation buttons are useful for generally focusing the display on a position. “Zoom” buttons increase or decrease the displayed coordinate range by 1.5-, 3-, or 10-fold. To zoom in by 3-fold on a particular coordinate, click the Base Position track at that location. To rapidly zoom in to the base composition of the sequence underlying the current annotation track image, click the zoom-in “base” button. “Move” buttons shift the displayed coordinates in the indicated direction by approximately 10%, 50%, or 95% of the displayed size. To scroll the coordinate position of one side of the track display while holding the position of the opposite end static, click the corresponding “move start” or “move end” arrow button. For example, to preserve the left-hand display coordinate but increase the right-hand coordinate, click the “move end” forward arrow. To increase or decrease the scroll interval, edit the number in the “move start” or “move end” text box.
-
8.
Click the “configure” button above or below the annotation tracks image to access a Web page for changing display characteristics (such as the image width and text size), hiding, showing, or reordering track groups, and displaying the chromosome ideogram, the track controls section, and image labels. Click the “submit” button on this page to apply the changes and return to the annotation tracks page.
The default display width of the annotation tracks graphic is optimized for smaller monitors with lower resolutions. Most displays are no longer subject to these limitations; in these situations the visible portion of the genome can be increased and the need for screen redraws can be reduced by setting the image width to a larger number.
Exercise caution when using the “show all” option in the track configuration section: if the group or assembly has a large amount of annotation data, the Web browser session may freeze or terminate before the data sets are loaded.
-
9.
Click the vertical button to the left of a displayed track to view additional information about the annotation and (in many cases) to filter or configure the features displayed in the track.
The description page can also be displayed by clicking the track’s name in the “track controls” section.
Click the button adjacent to the UCSC Genes track to view an example of a typical description page. This page contains a configuration/filter section (when applicable) followed by a description of the annotation track, information about interpreting and configuring the track display, a discussion of the methods used to collect and compute the data, credits for authors and contributors, associated references, and in this case, restrictions on the use of the data. Additional credits can be found by clicking the Credits link on the home page.
Most of the tracks in the Genome Browser have filter or configuration options that modify the graphical characteristics or restrict the display to features that match filtering criteria. Filters are useful for focusing attention on relevant features when a track contains large amounts of data. Some of the more complex graphical annotations, such as the continuous value graph (“wiggle”) display featured in the Conservation track, offer an extensive set of configuration options. In most cases, detailed configuration information can be found in the “Display Conventions and Configuration” section on the description page.
Filter and configuration settings are persistent from session to session on the same Web browser. To revert to the original default settings for a track, manually restore the settings on the description page; to undo all changes that have been made to default settings for any track or tool, click the “Click here to reset” link on the Gateway page.
-
10.
Click on the label of a feature in a track shown in pack or full display mode to view detailed information about the feature and access links to additional information.
The types of information available vary by track. Enter HOXA1 into the position/search box, click the jump button, and select the first matching item under UCSC Genes. In the track display image, click on the HOXA1 gene label in the UCSC Genes track in Figure 1.4.3 to view an extensive collection of information about the gene, including the associated UniProt (The UniProt Consortium, 2009) and RefSeq (Pruitt et al., 2009) descriptions, microarray expression data, links to associated information about this gene in several UCSC tools (such as the Gene Sorter, Proteome Browser, and Table Browser) as well as links to related records in external databases, including Online Mendelian Inheritance in Man (OMIM; Amberger et al., 2009; UNIT 1.2), Entrez Gene (Sayers et al., 2009), GeneLynx (Lenhard et al., 2001), GeneCards (Safran et al., 2003), AceView, PubMed (Sayers et al., 2009; UNIT 1.3), the HUGO Gene Nomenclature Committee Database (HGNC; Bruford et al., 2008), the Cancer Genome Anatomy Project (CGAP; Strausberg et al., 2001), PDB (Deshpande et al., 2005), ModBase (Pieper et al., 2006), InterPro (Hunter et al., 2009), Pfam (Finn et al., 2008), the Stanford SOURCE, Jackson Lab, and the Allen Brain Atlas. The page also includes hyperlinks that will display the corresponding protein, mRNA, and genomic sequences for HOXA1. These sequences are a useful source of input into the BLAT tool, which will be discussed in step 16.
The Genome Browser also provides direct links to the Ensembl Browser (Hubbard et al., 2009; UNIT 1.15) and NCBI’s Map Viewer (Sayers et al., 2009; UNIT 1.5), when available. To view the complementary annotation in one of these browsers, return to the annotation tracks page and click the Ensembl or NCBI link in the top menu bar.
Figure 1.4.3.

The Genome Browser annotation track page displaying chromosome bands 22q13.32 and 22q13.33 in the March 2006 human assembly (NCBI36). Several tracks useful for display of large regions have been made visible: from the Mapping and Sequencing Tracks group, Chromosome Bands, Recombination Rate and Gap; from the Phenotype and Disease Associations, GAD View, OMIM Genes and RGD Human QTLs; and from the Variation group, HGDP Smoothed FST (fixation index), HGDP XP-EHH (estimated likelihood of positive selection), Tajima’s D (measure of nucleotide diversity) and DGV Structural Variation. “Squish” display mode (Basic Protocol 1, step 5) has been set for UCSC Genes and DGV structural variation, in order to show the density of items in those tracks along the genome. Several tracks have been hidden because they have so many items in this large region that they would display as solid bars in dense mode, or take up large amounts of vertical space if displayed in pack or squish mode.
Examine the underlying data and download the sequence and annotation data tables
-
11.
Click the DNA link on the annotation tracks page menu bar to view the DNA sequence underlying the features in the image. This option allows the user to change the formatting and coloring of the text that represents the sequence to highlight features of interest.
The initial window that displays provides options for marking or masking repeats, changing the case of the letters that represent the DNA, showing the reverse complement of the sequence, and displaying additional sequence upstream or downstream of the selected sequence. Click the “extended case/color options” button to display additional font and color configuration options.
The Extended DNA Case/Color Options page is useful for highlighting features within a genomic sequence, pointing out overlaps between two types of features, or masking out unwanted features. In Figure 1.4.4, the configuration has been set to display exons from the UCSC Genes track in uppercase letters. The Spliced EST track is configured to reflect the level of coverage by setting its color to RGB value (0,64,0). When the Submit button is clicked, the Extended DNA Output window displays exons in uppercase. Nucleotides covered by a single EST appear as dark green, while regions with more EST alignments appear progressively brighter, saturating at four ESTs.
Be careful when requesting complex formatting for a large chromosomal region: when all the HTML tags have been added to the output page, the file size may exceed the limits that the Web browser, clipboard, and other software can display.
-
12.
Click on the Tables link on the annotation tracks page menu bar to examine the database tables underlying the Genome Browser annotation tracks.
The Table Browser tool provides a graphical interface for viewing and manipulating Genome Browser data. Support Protocol 2 gives a brief introduction to using the Table Browser. Additional information can be found in the Table Browser User’s Guide accessible from the Help link in the Table Browser top menu bar.
-
13.
Click the Home link on the top menu bar to return to the UCSC Genome Bioinformatics home page, then click the Downloads link on the side bar to display a listing of sequence files and database tables available for downloading.
The Downloads page contains links to all the Genome Browser assemblies, annotations, and source code available on the Genome Browser downloads server. To access older assembly versions, it may be necessary to look in the archives (http://genome-archive.cse.ucsc.edu). Data is also downloadable at the Genome Browser FTP site (ftp://hgdownload.cse.ucsc.edu/goldenPath/). FTP or rsync is recommended for large data downloads. All data in the Genome Browser are freely available, except where noted in the README.txt file specific to a particular downloads directory. The Genome Browser and BLAT source are freely available for academic, noncommercial, and personal use; commercial licensing information can be found via the Licenses link on the home page.
Figure 1.4.4.
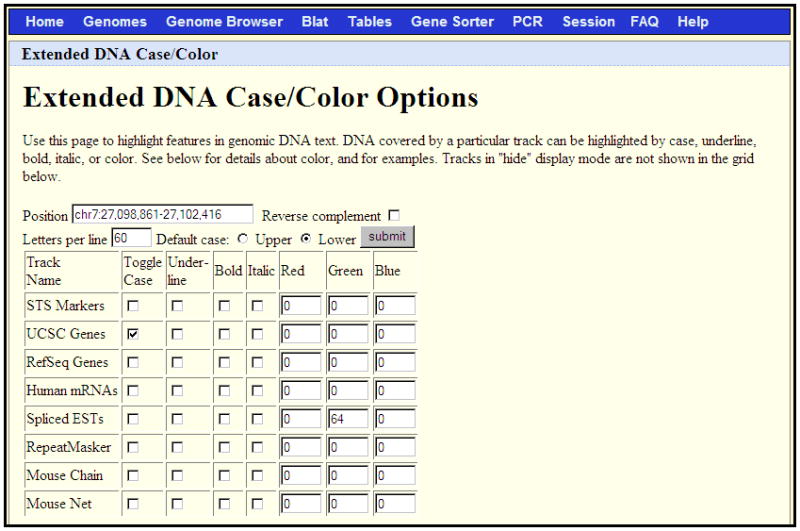
An extended DNA Case/Color Options request to display the DNA for the chr7:27,098,861-27,102,416 region of the March 2006 human assembly. This configuration will show UCSC Genes in uppercase, all other regions in lowercase, and Spliced ESTs in varying intensities of green, depending on the level of coverage.
Convert coordinates in the displayed range to a different assembly using the Convert, LiftOver, or BLAT tools
-
14.
Return to the annotation tracks page. Click the Convert link in the menu bar to convert the coordinates in the displayed range to those of a different assembly.
The coordinate conversion tool is useful for locating the position of a feature of interest in a different genome assembly. Coordinates of features frequently change from one assembly to the next as gaps are closed, strand orientations are corrected, and duplications are reduced. For example, to map the location of a sequence in the May 2004 human assembly to the March 2006 human assembly, open the May 2004 Genome Browser to the desired position, click the Convert link, select the Mar. 2006 option in the New Assembly pull-down menu, then click the “Submit” button. If successful, the Convert tool displays one or more coordinate ranges in the March 2006 assembly to which the May 2004 sequence maps.
-
15.
To convert multiple sets of sequence coordinates between assemblies or to exert control over the parameters used in the conversion, use the LiftOver batch coordinate conversion tool.
The LiftOver tool can be accessed from the Utilities link on the Genome Bioinformatics home page. Enter the list of coordinate ranges in the large text box, one per line, or upload the list from a file. Detailed information about parameter settings can be found at the bottom of the page, as well as information about a Linux command-line version of the tool.
-
16.
Alternatively, use BLAT to map a sequence to a different assembly:
To determine the sequence underlying the region currently displayed on the annotation tracks image, click the DNA link in the top menu bar on the annotation tracks page, then click the “get DNA” button. For more information on the Get DNA utility, see step 11. To find the sequence of a specific feature within the annotation track image, click on the feature label to display its details page. In most cases, the sequence is available as a link from this page. Note that BLAT limits input to 25,000 bases.
Using the Web browser’s copy function, copy the entire sequence onto the clipboard. Return to the previous page and click the BLAT link in the top menu bar.
On the BLAT Web page, paste the sequence into the large text box (Fig. 1.4.5). Select the genome and assembly to which to map the sequence, then click the “submit” button. If successful, BLAT will display a list of search results sorted by score (Fig. 1.4.6).
To view the details of the matching alignments, click the “details” link; to display the sequence in the Genome Browser, click the “browser” link.
This procedure demonstrates one use of the BLAT search tool. This tool, which can be accessed from the BLAT link on the top menu bar of most Genome Browser pages, is a very fast sequence alignment tool similar to BLAST (UNITS 3.3 & 3.4), but optimized for inputs with high similarity, e.g. sequences from the same species. For more information on BLAT, refer to the Genome Browser User’s Guide.
Figure 1.4.5.
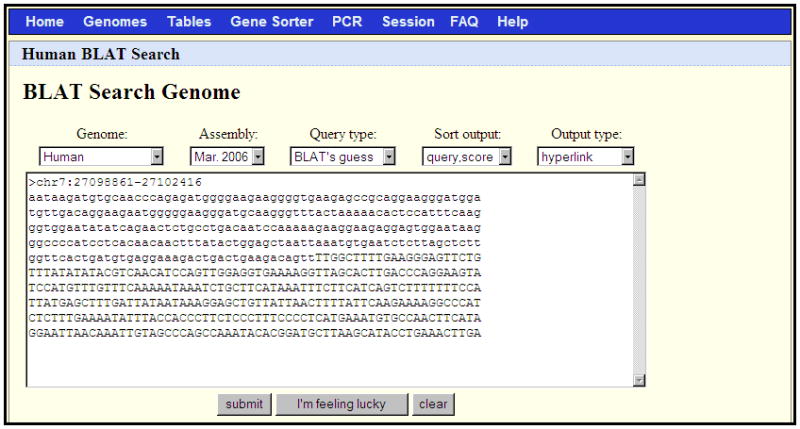
A BLAT search set up to align the FASTA sequence in the text box against the March 2006 human genome assembly. This sequence was obtained by copying and pasting the first 600 bases of output from the Get DNA search illustrated in Figure 1.4.4.
Figure 1.4.6.
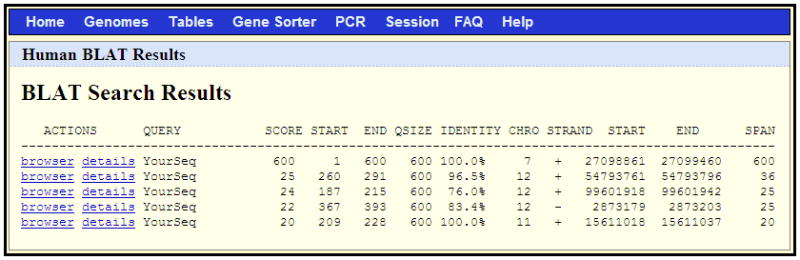
The results returned by the BLAT search shown in Figure 1.4.5. Clicking on the “browser” link for a given line will display the data in the Genome Browser; the “details” link will display a page showing a base-by-base of the alignment to the genome.
SUPPORT PROTOCOL 1: CREATING A CUSTOM ANNOTATION TRACK
Custom annotation tracks enable users to upload personal data for temporary use in the Genome Browser and Table Browser. Custom tracks are viewable only on the machine from which they are uploaded, and the data may be accessed only by the users on that machine. Tracks are kept for 48 hr after the last time accessed or until the user switches to a different genome assembly; no permanent archives are created. Optionally, users can make custom annotations viewable by others as well.
Typically, custom annotation tracks are displayed under the corresponding genomic positions on the Base Position track. Each custom track has its own track control and persists even when not displayed in the Genome Browser window (e.g., if the position changes to a range that no longer includes the track).
Since space is limited in the annotation track graphic, many excellent genome-wide tracks must be excluded from the set provided with the browser. A Web page with links to several user-contributed custom tracks can be found by clicking the Custom Tracks link on the home page. The information in this section provides an overview of custom annotation tracks. For a more detailed discussion of formats and syntax, refer to the Genome Browser custom annotation track documentation Web page at http://genome.ucsc.edu/goldenPath/help/customTrack.html.
Necessary Resources
Hardware
Unix, Windows, or Macintosh workstation with an Internet connection and a minimum display resolution of 800 × 600 dpi.
Software
Text editor (APPENDIX 1C)
Files
None
-
Format the data set to be analyzed as a tab-separated file using general feature format (GFF) or one of the formats designed specifically for the Human Genome Project or UCSC Genome Browser. These include gene transfer format (GTF), pattern space layout (PSL), browser extensible data (BED), wiggle format (WIG), multiple alignment format (MAF), or binary indexed formats (bigBed, bigWig) (see http://genome.ucsc.edu/FAQ/FAQformat).
Each line of data in the file provides display and positional information for a feature line within the displayed annotation track. The browser ignores empty lines and lines starting with a pound sign (#).
For detailed information on data formats, refer to the Genome Browser’s custom annotation track documentation. Data in PSL, GFF, and GTF files must be tab-delimited rather than space-delimited in order to display correctly. More than one data set may be included in an annotation file, but all lines within a single annotation track must be in the same format.
Figure 1.4.7 shows examples of data in BED, PSL, and GFF format.
An easy way to create correctly formatted data for an annotation file is by collecting PSL output from BLAT or downloading data from the Table Browser.
-
Add one or more optional browser lines to the beginning of the formatted data file to specify the configuration of the Genome Browser window in which the custom annotation track will be displayed.
Browser lines define the genome position to which the browser will initially open, the width of the display, and the configuration of the other annotation tracks that are shown (or hidden) in the initial display. The Genome Browser custom annotation track documentation describes the browser line syntax and options.
In the sample BED annotation track shown in Figure 1.4.7, the initial display position is set to chr22:10000000-10007500, and all tracks are hidden except the custom annotation track. If the browser position is not explicitly set in the annotation file, the initial display will default to the position setting most recently used by the user, which may not be an appropriate position for viewing the annotation track.
-
Add a track line immediately above the formatted data in the file to define the display attributes for the annotation track.
The track line defines the track’s name, description, colors, initial display mode, use score, and associated URL. The Genome Browser custom annotation track documentation contains a complete description of the track line syntax and options. If more than one data set is included in the annotation file, insert a track line at the beginning of each new set of data. In Figure 1.4.7 the left-hand label of the BED annotation track is ‘BED track’; the center label is ‘BED track example’. The track labels will be displayed in green and the features will be fully displayed. Because the useScore attribute is set to 1, the level of shading of each feature will reflect its score value.
-
Upload the annotation file into the Genome Browser by clicking the “add custom tracks” button on the Gateway page (Fig. 1.4.1) or the “add custom tracks” button on the annotation tracks page (Fig. 1.4.2).
If the file is located on a local machine, enter the file name in the “upload” text box in the “URLs or data” section. To open an annotation through a URL or to manually enter the track data, type or paste the information into the large text box in this section. Multiple tracks may be uploaded simultaneously by including all the track data or URLs (on separate lines) in the text box or grouping the tracks into one uploaded file. Figure 1.4.8 shows the custom track that displays when the BED sample track in Figure 1.4.7 is uploaded into the Genome Browser. Optionally, associated track descriptive text may be uploaded or inserted in the “optional track documentation” section.
To make the annotation file viewable on a different machine or at a different site, put a copy of the file on a Web server and create a custom annotation track URL that allows the file to be uploaded over the Internet. The URL must contain two bits of information specific to the annotation data file: the UCSC genome assembly on which the annotation is based and the URL of the annotation file on the Web site. The Genome Browser FAQ (http://genome.ucsc.edu/FAQ/FAQreleases#release1) lists the UCSC genome assembly codes. The URL can also include the position within the genome to which the Genome Browser should initially open.
For example, placing the BED track in Figure 1.4.7 in a file named test.bed on the genome-test.cse.ucsc.edu Web site enables it to be uploaded using the following custom annotation track URL: http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg18&position=chr22&hgt.customText=http://genome-test.cse.ucsc.edu/goldenPath/help/test.bed.
This URL sets the assembly database to the hg18 (March 2006) assembly of the human genome, initializes the display position to chromosome 22, and loads the annotation track file http://genome-test.cse.ucsc.edu/test.bed. In this case, the position initialization in the URL is extraneous; it will be overwritten by the position defined in the custom track file.
Figure 1.4.7.

Sample custom annotation tracks containing BED, PSL, and GFF data formats. To load correctly, the track line data in the PSL and GFF examples must be tab-separated. Some of the line breaks shown in the BED and PSL examples are artificial (to make the text fit on the page); in each of these formats, each item must appear on a single line.
Figure 1.4.8.

The annotation track display that results when the BED track example in Figure 1.4.7 is uploaded into the Genome Browser. Note that the lower score value in the ItemB data results in lighter shading of this feature.
SUPPORT PROTOCOL 2: USING THE UCSC TABLE BROWSER
The UCSC Table Browser provides a powerful and flexible graphical interface for querying and manipulating the data in the Genome Browser annotation database.
The Table Browser can be used to: (1) retrieve the annotation data or DNA sequence underlying Genome Browser tracks for the entire genome, a specific coordinate range, or a set of accessions; (2) view a list of the tables affiliated with a particular Genome Browser track; (3) view the schema of an annotation table; (4) organize table data into formats that can be used in other applications, spreadsheets or databases; (5) combine data from multiple tables or custom tracks into a single set of output data; (6) filter out certain records in a table based on certain field values; (7) display basic statistics calculated over a selected range of table data; and (8) conduct structured or free-form SQL queries on the annotation data.
The information in this section provides an overview of the Table Browser, which can be accessed on the Internet from the UCSC Genome Bioinformatics home page at http://genome.ucsc.edu. For a more detailed discussion of Table Browser options, advanced queries, and several practical examples, refer to the Table Browser User’s Guide at http://genome.ucsc.edu/goldenPath/help/hgTablesHelp.html. For complex queries, you may want to use the Galaxy interactive genome analysis tool (http://main.g2.bx.psu.edu/).
Necessary Resources
Hardware
Unix, Windows, or Macintosh workstation with an Internet connection and a minimum display resolution of 800 × 600 dpi.
Software
An up-to-date Internet browser that supports JavaScript, such as Firefox 3.0 and higher (http://www.mozilla.com/firefox); Internet Explorer 6.0 and higher (http://www.microsoft.com/ie); or Safari 3.0 and higher (http://www.apple.com/safari). The browser must have cookies enabled.
Files
None
Set up a simple Table Browser query
-
1.
On the UCSC Genome Bioinformatics home page (Basic Protocol, step 1), click the Table Browser link in the left-hand sidebar menu to display the Table Browser Web page.
The Table Browser is also accessible from the Tables link in the top menu bar of most Genome Browser pages.
The top section of the Table Browser Web page (Fig. 1.4.9) contains options for setting up a data query, many of which are optional when conducting simple queries. Each of the options is briefly described at the bottom of the Web page. To view the complete Table Browser User’s Guide, click the Help link in the top menu bar.
-
2.
Select the clade, genome, and assembly.
The clade, genome, and assembly pull-down menus correspond to those found on the Genome Browser Gateway page. The current Genome Browser settings are used when the Table Browser is started from the menu bar on a Genome Browser page.
For this example, set the clade to “Mammal,” the genome to “Human,” and the assembly to “Mar. 2006.”
-
3.
Select the group, track, and table of interest.
The options in the group and track menus directly correspond to the annotation groups and tracks available in the Genome Browser for the currently selected genome assembly. The track list—which shows all tracks contained in the selected group—automatically updates when a different group is selected.
The table menu lists all the tables in the annotation database that are affiliated with the selected track. Many annotation tracks are based on data from multiple tables joined by common fields. By default, the primary table underlying the track’s display in the Genome Browser is listed first.
Click the “describe table schema” button to view the SQL schema for the selected table. The schema page also lists other tables in the annotation database that are joined to the selected table by a particular field, as well as a description of the Genome Browser annotation track associated with the table (when applicable).
The All Tracks and All Tables options in the group menu provide convenient shortcuts if the name of the desired track or table is already known.
For this example, a subset of data in the UCSC Genes track will be examined. Select the “Genes and Gene Prediction Tracks” group, the “UCSC Genes” track, and the “knownGene” (default) table.
-
4.
Specify the query region.
Click the “genome” region setting to view annotation data for the entire genome. To limit the data output to a specific query region, click the “position” region setting and type a query into the adjacent text box. The Table Browser accepts the same types of queries that are valid for the Genome Browser (see Basic Protocol,, step 3). Click the “lookup” button to convert a nonpositional query (e.g., an accession or keyword) to a coordinate range.
On the hg17 and hg18 genome assemblies, which have ENCODE pilot project annotations (The ENCODE Project Consortium, 2004; Thomas et al., 2007), an additional “ENCODE” region setting is available that restricts output to data located in the 41 ENCODE Pilot regions. There is no need to use this setting for the genome-wide production phase ENCODE data found on hg18 and later assemblies.
For many tables, the query region can be further defined by restricting the output to a set of specific identifiers, such as UCSC Gene IDs or mRNA accession numbers. Upload the identifiers as a space- or line-separated list by clicking the “paste list” or “upload list” button. For this type of query to return successfully, the identifiers in the list must conform to the format specified for identifiers in the selected table.
For this example, several UCSC Gene identifiers from chromosome 7 are included in the query. Select the “position” region setting, then type chr7 in the text box. Click the “paste list” button, then type the following items in the large text box, one per line: NM_014390, NM_022143, D49487, NM_018077.
-
5.
Select an output format.
The help text at the bottom of the Table Browser page describes the output formats. Not all options may be available for a given query. The “all fields… “ format displays the entire set of fields for each record in the output. The “selected fields… ” format is useful when the user wishes to create output that contains only a subset of fields that will be used as input for further data processing or if the user desires to link in fields from an associated table (see step 7). The “sequence” option returns the sequence underlying the annotation in FASTA format. The GTF, BED, and custom track options are useful for saving the output into a format that can be displayed as a custom track in the Genome Browser. The “data points” format, which is available only for “wiggle” and Conservation tracks, is useful for displaying the conservation scores associated with individual base locations; in contrast, the Conservation track’s “MAF” format displays the multiple species alignments underlying the conservation scores. To display a set of search results in the Galaxy genome analysis tool, check the “Send output to Galaxy” box.
For this example, choose the “selected fields… “ output format.
-
6.
Click the “get output” button to submit the query and display the results.
By default the Table Browser displays the query output in the user’s Web browser. To save the data to a file on the local computer, type a file name in the “output file” text box and select the plain or compressed file type option before clicking the “get output” button. Many output formats—including the “selected fields… ” format used in the example—require an additional setup step before the output is displayed. On the setup page associated with our example, check the “name,” “chrom”, txStart”, and “txEnd” boxes, then click the “get output” button. The Table Browser will display the following output:#name chrom txStart txEnd uc003vmi.1 chr7 127079437 127519895 uc003vmk.1 chr7 127454359 127458238 uc003vmm.1 chr7 127679279 127682185 uc003vmo.2 chr7 127737671 127762969 uc003vmp.2 chr7 127737671 127771198 uc010lle.1 chr7 127145885 127519895 uc010llf.1 chr7 127455967 127457929 -
7.
Link in additional data from tables associated with the table being queried.
The linked tables feature included on the “selected fields… “ output format setup page provides a convenient way to pull in data from additional tables without having to conduct multiple queries.
In the previous query, it is easy to display additional data associated with the selected genes by linking in the associated tables. The kgXref table, linked by default when the UCSC Genes track is selected, provides a convenient cross-reference among gene IDs and information from several different sources such as RefSeq, Swiss-Prot, HGNC, etc. At the top of the selected fields setup page, check the “name,” “chrom,” txStart”, and “txEnd” boxes in the hg18.knownGene section, and the “geneSymbol” and “refseq” fields in the hg18.kgXref section. Scroll down to the Linked Tables section, check the box in front of the “hg18/kgAlias” table/field, then click the “Allow selection from checked tables” button at the bottom of the page. Check the “alias” field in the hg18.kgAlias section. Click the “get output” button. The Table Browser will display a comma-separated list of aliases, followed by the HGNC gene symbol and the RefSeq accession associated with each UCSC Genes record in the output shown in step 6.
-
8.
Click the “summary/statistics” button to display a table of basic statistics about the current query.
The Summary Statistics page profiles data and query characteristics. This information can be useful in determining such information as the percent of bases in a query region that is covered by items returned from the query (or by their exons, if applicable).
Figure 1.4.9.
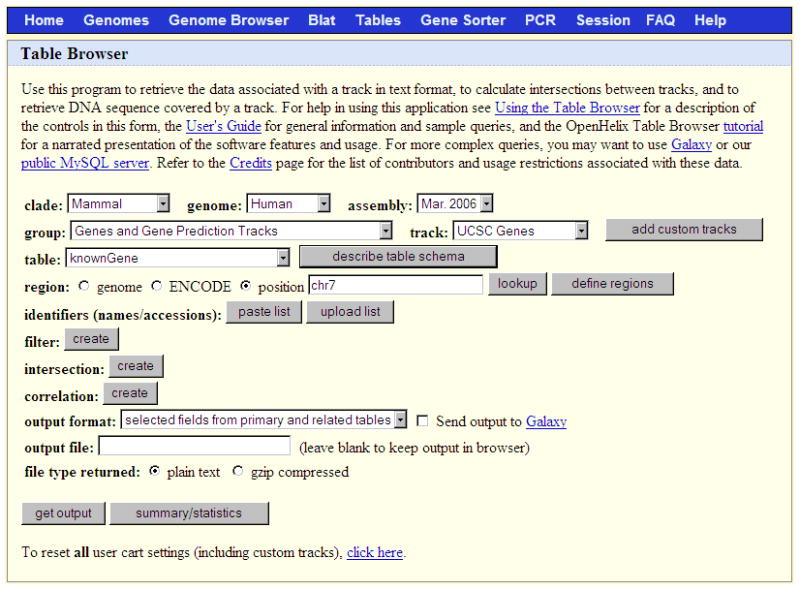
The Table Browser tool provides access to the database tables underlying the Genome Browser annotations; in this case, the chromosome 7 data in the knownGene table on the March 2006 human assembly.
Explore advanced query options
-
9.
Create a custom track from a subset of table data using the “custom track” output format option.
The custom track output format allows the user to save query results into a custom annotation file that can be loaded into the Table Browser for further data manipulation or uploaded for display in the Genome Browser.
For this example, repeat steps 1 to 3. Select the “genome” region setting. If you have not reset your session since trying the previous examples, click the “clear list” button. Select the “custom track” output format option, then click “get output”. On the custom track setup page, configure the header of the custom track (optional). Select the “Coding exons” option, then click the “get custom track in table browser” button. The track is now loaded into the Table Browser. The data in the track can now be viewed and manipulated by selecting the “Custom Tracks” group option and setting the track list to the name of the user’s custom track.
-
10.
Click the filter “create” button to set up a filter on one or more fields in a data table.
The filter utility allows the user to fine-tune a query to produce a restricted data set that meets a certain set of criteria, such as a minimum threshold or a specific set of IDs or keywords.
For this example, set the clade, genome, and assembly as described in the example in step 2. Select the “Comparative Genomics” group, the “Conservation” track, and the “phyloPNwayGroup” table (where N represents the number of species present in the multiple alignment and Group is a subset of species with a name like “Primate” or “Mammal”, such as phyloP44WayPrimate). Set the position to chr7. On the filter page, set dataValue >0.98, then click the “submit” button. Select the “data points” format, then click “get output”. This query will return the first 100,000 bases in the Conservation track that are associated with the peaks where the multiple species conservation score exceeds 0.98 (i.e., regions with a high amount of evolutionary conservation). By default, the number of output data points from wiggle data is limited to 100,000. You can increase this limit on the filter page. To find out how many data points would be returned from the query without any limit, click the summary/statistics button.
-
11.
Click the intersection “create” button to combine the data from two different tables into a single output file using an intersection or union.
The intersection feature lets the user compare the positions of features in different annotations to identify points of overlap or nonoverlap, establish thresholds for the amount of overlap, and conduct feature-by-feature or base-wise comparisons.
In this example, select the “Variation and Repeats” option in the group menu and “Simple Repeats” from the track menu. Select the “genome” region setting. Click the intersection “create” button. On the intersection setup page, select the “Custom Tracks” group option,, then set the track menu to the track created in step 9. Select “All Simple Repeats records that have at least [80%] overlap with tb_knownGene”, change “80%” to “100%”, and click “submit.” Back on the main Table Browser page, select the “hyperlinks” output format, then click “get output.” The Table Browser will return a list of links to view simple repeats completely overlapped by coding exons of UCSC Genes in the Genome Browser.
Note that the “all fields… “ and “selected fields… “ output format options are not available when an intersection is active in the current query. Although the intersection utility restricts combinations to two tables, additional tables can be included in an intersection by saving the initial intersection to a custom track, then performing subsequent intersections using the custom track.
GUIDELINES FOR UNDERSTANDING RESULTS
The Genome Browser can be used for genome analysis and interpretation at many different levels. With the annotation track image zoomed out to display several million bases or an entire chromosome, the tool provides a good overview of the coverage and completeness of the region. At a reduced display scale, the Genome Browser is useful for viewing splicing patterns or searching for evidence of previously unidentified genes. By presenting a large collection of annotation tracks in a single view, the browser facilitates interpretations based on a visual correlation of features. However, care must be taken when drawing conclusions. Information presented in the Genome Browser is only as accurate as the underlying data. It is essential to gather supporting evidence when making an analysis, rather than basing judgments on a single track that may contain erroneous or misleading data.
It is important to consider the methods and criteria used to compute an annotation track. Consult the track’s description page (Basic Protocol, Step 9) for a discussion of the sources and methods used to generate the track. In many cases, the page will provide links to additional information about the annotation (such as a seminal publication or related Web site), estimates of accuracy, and caveats for use.
The track details pages (Basic Protocol 1, step 10) are another good source for supporting documentation. Many pages contain links to feature-specific information in external public databases. The OMIM database (UNIT 1.2), for example, contains hand-curated experimental literature summaries. Entrez, GeneLynx, GeneCards, AceView, and PubMed (UNIT 1.3) are other good sources for supplementary information.
Many regions–particularly in unfinished areas of a genome–may exhibit discrepancies among the various gene prediction tracks, EST evidence, and cross-species orthology tracks. Tracks generated by gene prediction methods vary considerably in their degrees of sensitivity and specificity. Kent (2002) illustrated some of these differences in a comparison of the correlation of EST, cross-species homology, and ab initio gene prediction tracks with the RefSeq Genes track across the entire genome, along with a similar comparison to the Sanger Centre chromosome 22 gene annotations in the Sanger22 Genes track. It is better to use correlations between EST, cross-species homologies, and ab initio gene predictions to look for evidence of unidentified genes, rather than relying on the information in a single annotation track. There is no gene prediction tool that integrates all the annotation evidence into a single track as yet.
ESTs often exhibit sequencing errors due to the nature of the techniques used. EST databases contain contamination from mRNA and genomic sequence. Because of this, a single unspliced EST should be viewed with considerable skepticism, and alternate splicing predictions should be evaluated by examining the quality of the EST/genomic alignment. Cross-species BLAT alignments that match too perfectly may also be suspect. Those with greater than 97% identity may simply reflect the contamination of one genome by the other.
In several of the annotation tracks generated at UCSC, attempts have been made to filter out data that might provide misleading results. For example, the mRNA and EST alignments on which several of the browser tracks are based are filtered to reduce the presence of pseudogenes, paralogs, and assembly errors. Filtering removes a significant number of alignments in the tracks, particularly very short ones. The Spliced EST track applies additional splicing criteria that greatly reduce the level of contamination from EST databases, although at the expense of eliminating genuine ESTs. Since the maximum intron length allowed by BLAT is 500,000 bases, some ESTs with very long introns are eliminated that otherwise might align.
In summary, good judgment should be used when using any genome-browsing tool. To work effectively in a bioinformatics area subject to errors, it is a good idea to seek supporting data for any unusual findings. Often, the ultimate supporting evidence for a conclusion must be generated in the lab.
For a general discussion of the advantages and potential pitfalls of genomic data analysis using genome browsers, see Cline and Kent (2009).
COMMENTARY
Background Information
History and development of the UCSC browser
The need for interactive software to search and display a genome at a variety of levels predates the inception of the UCSC Genome Browser. Research on the nematode C. elegans in the mid-1990s prompted the creation of A Caenorhabditis elegans Database (ACeDB; Eeckman and Durbin, 1995; UNIT 9.1) to track strains and genetic crosses. As ACeDB grew in functionality, the software was adopted by the C. elegans community, and over the years has been enhanced and extended to support a large number of organisms.
The UCSC Genome Browser was originally developed as an alternative to ACeDB to examine RNA splicing for gene predictions in C. elegans (Kent and Zahler, 2000a). This set of Web-based tools—initially called the Intronerator—displayed EST and full-length cDNA tracks from GenBank aligned to the C. elegans genomic sequence. The Intronerator was subsequently expanded to include tracks showing homology with C. briggsae (Kent and Zahler, 2000b). With the completion of the assembled human genome working draft on the horizon, the software underwent major revisions to accommodate the human genome assembly, which was 30 times larger than that of C. elegans. The resulting UCSC Genome Browser retains the speed and performance of its predecessor while displaying the vastly larger data sets of vertebrate genomes. The initial mouse (Mus musculus) draft assembly (Waterston et al., 2002) was added to the Genome Browser in 2002, and the browser has subsequently grown to include a large array of genomes: In mid 2009, this totaled 47 organisms, including 14 mammals, 10 non-mammalian vertebrates, 3 deuterostomes, 13 insects, 6 nematodes, and a yeast. Older assemblies are archived as newer versions are released; the UCSC Web site maintains complete assembly archives of the more popular genomes.
In the years since its public debut, the Genome Browser has gained broad popularity among research scientists worldwide for its speed, stability, extensibility, annotation tracks, and the consistency of its user interface. Of the alternative existing tools that provide a somewhat similar functionality to the Genome Browser, the Ensembl Genome Browser (http://www.ensembl.org/; UNIT 1.15) and the National Center for Biotechnology Information (NCBI) Entrez Map Viewer (http://www.ncbi.nlm.nih.gov/mapview/; UNIT 1.5) are perhaps the most widely known. The UCSC browser provides links to both of these tools from the menu bar at the top of the annotation tracks page. To support the growing demand for tools that can handle complex analysis of sequence data, the Genome Browser annotation set is continually evolving, including the introduction of several new graphical display types, such as wiggle displays and composite tracks comprised of several related subtracks. Additionally, the Genome Browser application set has grown to include several tools that analyze different aspects of the data: the BLAT alignment tool, Table Browser, Gene Sorter, Proteome Browser, VisiGene, in silico PCR tool, Genome Graphs, and Sessions.
How are various annotation tracks determined?
The Genome Browser annotation tracks are grouped by functionality into the following basic categories: mapping and sequencing, phenotype and disease associations, genes and gene predictions, mRNA and EST data, expression, regulation, comparative genomics, variations and repeats, and annotations specific to ENCODE Pilot regions. More groupings will undoubtedly be added as scientific technologies improve and analytical efforts branch into new areas. The browser offers a large selection of annotations in each of these categories for the more highly studied genomes, such as the human and mouse; other assemblies may feature only a subset of these annotations. The following discussion highlights some of the tracks featured in the browser; for an in-depth description of the tracks, see Kent et al. (2002), the Genome Browser updates in the annual Nucleic Acid Research database issue (e.g. Kuhn et al. (2009)), and the individual track description pages.
The Phenotype and Disease Associations group includes annotations from the Genetic Association Database (GAD, Becker et al., 2004) and Online Mendelian Inheritance in Man (OMIM, UNIT 1.2). Several Quantitative Trait Loci (QTLs) tracks are available in the Human May 2006 browser: Human QTLs collected by the Rat Genome Database (RGD, Dwinell et al., 2009), as well as Rat QTLs from RGD and Mouse QTLs from Mouse Genome Informatics (Blake et al.,2009) that are mapped to the human assembly using whole-genome alignments. The cross-species mappings of QTLs are extremely coarse and should be critically evaluated using the cross-species Net tracks and any other relevant data.
Gene prediction tracks within the UCSC Genome Browser vary in the evidence used for genes they report, their coverage of bases in known coding regions, and their specificity. The UCSC Genes track is generated by an automated process that combines evidence from RefSeq, GenBank, Consensus CDS (CCDS) and UniProt. This is a moderately conservative set of predictions, requiring the support of one GenBank RNA sequence plus at least one additional line of evidence, with the exception of the RefSeq RNAs, which require no additional evidence. The track includes both protein-coding and putative non-coding transcripts. The UCSC Genes annotation is based on the earlier Known Genes track (Hsu et al., 2006), which was updated in 2005 to increase the quality and coverage through more stringent filtering and the inclusion of more supporting evidence (refer to the UCSC Genes description page for more details). Other UCSC-generated gene prediction tracks of note include the RefSeq Genes track, based on human RefSeq mRNAs in GenBank that have been aligned against the genome with BLAT and stringently filtered; the Mammalian Gene Collection (MGC) Genes track, showing genes for which high-quality clones are available from the MGC Project (Gerhard et al., 2004), and the CCDS Genes track, which shows a high-quality, consistently annotated core set of human protein-coding genes obtained from the CCDS project (Pruitt et al., 2009) and identified by consensus among the Ensembl, Vega (Ashurst et al., 2005), and RefSeq gene annotation sets.
The browser displays several tracks based on mRNA alignments. The mRNA and EST sequences are extracted from databases in GenBank (UNIT 1.3; APPENDIX 1B), and are aligned against the genome using the BLAT search tool (see Basic Protocol). The set of alignments undergoes several filtering steps (detailed on the individual track description pages) prior to its presentation in the Genome Browser. As mentioned in the Guidelines for Understanding Results section, these filtering methods reduce the occurrence of misleading and erroneous data in the tracks at the expense of eliminating some genuine data. The mRNA data in the Genome Browser are incrementally updated from GenBank nightly; EST data are updated weekly.
The Genome Browser provides a wealth of comparative genomics annotations. In addition to the cross-species homology mRNA and EST tracks found in the mRNA and EST group, the Comparative Genomics group contains a wide variety of pairwise chain and net alignment tracks (Kent et al., 2003; Schwartz et al., 2003) that can be used to look for orthologous regions between organisms, large-scale rearrangements, duplications and deletions, and processed pseudogenes. The chain tracks can also be used to identify paralogs. The Conservation track is based on multi-species alignments generated by multiz (Blanchette et al., 2004) from a set of pairwise net alignments. Pairwise net alignments from a subset of the species are displayed in a condensed form. Above the alignments is a graph of estimated basewise probability of evolutionary conservation computed on the alignments by the program phyloP (Siepel et al., 2006) using a phylogenetic hidden Markov model. This track is highly customizable, allowing the user to adjust the display to the species of interest and vary several of the graph characteristics. The Most Conserved subtrack provides an alternative simplified view of the Conservation track that highlights the parts of the genome that are most likely conserved by purifying selection.
The Expression and Regulation groups feature microarray and expression data from several sources, conserved transcription factor binding sites (TFBS), regulatory potential scores, DNaseI-hypersensitive sites, microRNA regulatory targets, and more. Several high-level map tracks are included in the Mapping and Sequencing section: FISH clones, which shows the locations of FISH-mapped BAC clones from the BAC Resource Consortium (Cheung et al., 2001) along the draft assembly sequence; the Chromosome Bands, which uses the locations of FISH-mapped clones on the cytogenetic map and the assembly to approximate the Giemsa-stained chromosome bands at an 800-band resolution; the Sequence-Tagged Site (STS) Markers track, which displays the positions of markers used in constructing several genetic, radiation hybridization (RH), and yeast artificial chromosome (YAC) maps, as well as markers from the UniSTS database; and the BAC End and Fosmid End Pairs tracks, which show mappings of paired BAC and fosmid end reads.
The Variation and Repeats section features annotations of polymorphisms, measures of selection and population variance, probe locations of common assay platforms and repetitive sequences. dbSNP (Sayers et al., 2009) provides a comprehensive set of simple nucleotide polymorphisms (SNPs) observed in humans. The International HapMap Project (HapMap; The International HapMap Consortium, 2003; The International HapMap Consortium, 2005) assayed 4 million SNPs in individuals from four populations and 1 million of those SNPs in individuals from 7 additional populations. The Human Genome Diversity Project (HGDP, http://www.stanford.edu/group/morrinst/hgdp.html) assayed 660,000 polymorphisms in individuals from 53 populations. The Genome Variants track contains single nucleotide differences from several published personal genome sequences. The HapMap LD Phased track shows linkage disequilibrium (LD) scores computed from HapMap genotypes that have been phased. Other measures of population variance include Tajima’s D and several per-continent measures from HGDP: FST, Heterozygosity, iHS and XP-EHH. Probe mappings from several commonly used SNP assaying platforms are shown in the SNP Arrays track. The Database of Genomic Variants (DGV) is a curated collection of published structural variations. Discordant clone end mappings from the Human Genome Structural Variation Project (HGSV) provide a more detailed view of possible structural variation in eight individuals. The Segmental Dups track shows reference genome regions of at least 1,000 bases that have at least a 90% similarity to other regions. Repetitive sequences are annotated in the RepeatMasker (Smit, 1999), Interrupted Repeats, Simple Repeats (Benson, 1999), Microsatellite and Self Chain tracks.
Critical Parameters and Troubleshooting
Use caution when interpreting the information displayed in the UCSC Genome Browser, particularly if the chromosomal region under scrutiny is incompletely assembled. The Genome Browser annotation tracks are generated from publicly available data, and therefore are only as accurate as the data on which they are based. Assembly errors and sequence gaps may occur well into the genome sequencing process due to regions that are intrinsically difficult to sequence, and incorrect data may be propagated into the public databases. The browser cannot fill in sequencing gaps or correctly assign strand information in the absence of good coverage data. Artifactual duplications arise as unavoidable compromises during a genome assembly build, causing misleading matches in genome coordinates found by alignment. Conclusions about the data should never be made based on the information available in a single track. Instead, gather supporting evidence and correlation from other tracks aligned to the same region to identify problematic areas. Cross-check information in the public databases such as Entrez Gene and OMIM (UNIT 1.2).
A common source of confusion among users is the positional differences that result when genome assembly versions are interchanged. New genome versions are added to the UCSC Genome Browser on a regular basis. Unless a feature lies on a completely sequenced and unrevised chromosome, its coordinates are likely to change between one assembly and the next. Often the position of a genomic feature cited in the literature will not coincide with the location displayed in the browser. When faced with such a discrepancy, compare the assembly date of the genome in the reference with that of the genome displayed in the browser. In most cases, the newer assembly will have the most accurate information. When feasible, it is usually best to work with the most current assembly even if it lacks a complete set of annotation tracks. Two procedures are described (see Basic Protocol, steps 14, 15, and 16) that can be used to map the position of genomic sequence in one assembly version to that of a newer version.
UCSC makes a concerted effort to provide uninterrupted Browser and BLAT service to the research community. In the event of the occasional power or equipment failure, there are multiple mirror sites that replicate the UCSC Genome Browser environment. To view a list of actively maintained mirror sites, click the Mirrors link on the UCSC Genome Bioinformatics home page.
Troubleshooting custom annotation track problems
Custom annotation track display problems usually stem from syntax or formatting errors in the annotation track file. A spurious line break in one of the browser, track, or data lines is a frequent source of errors. Another common cause of problems is GFF or GTF data that are separated by spaces rather than tabs. Refer to the troubleshooting section in the Custom Annotation Track section of the User’s Guide (http://genome.ucsc.edu/goldenPath/help/customTrack.html) for more information.
Suggestions for Further Analysis
The UCSC Genome Bioinformatics home page offers links to several tools that facilitate analysis of the genomic and annotation data underlying the browser’s graphical presentation. The Table Browser and BLAT tools were introduced in the main part of the unit (see Basic Protocol). The BLAT tool can be used for a large number of functions, such as finding the genomic coordinates of an mRNA or protein in an assembly, determining the exon structure of a gene, displaying a coding region within a full-length gene, searching for gene family members, or finding homologs of a query from another species. The output of a BLAT or Table Browser search can be saved in a custom track format (PSL or BED, respectively) for direct upload into the browser, or can be downloaded into a spreadsheet or text editor (APPENDIX 1C) for further manipulation.
The Gene Sorter
The Gene Sorter is accessible from the top menu bar on most of the browser Web pages. It provides a simple interface for studying the relationship among a group of genes based on protein-level homology, the similarity of gene expression profiles, genomic proximity, or other parameters, which in turn facilitates the study of the evolution of genes and their functions. This tool can be used to gather a collection of genes that share similar properties for statistical analysis or to filter a large group of genes into a small subset of interesting features, based on specific properties.
The VisiGene image browser
The VisiGene image browser is available from a link on the Genome Bioinformatics Group home page. It can be used to browse images from in situ RNA hybridization, reporter genes, and other techniques that show where a gene, enhancer, or promoter is active in an organism. In 2009 the VisiGene image database contained nearly 100,000 images from several high-throughput gene projects, as well as images from literature curated by the model organism databases.
The Proteome Browser
The Proteome Browser can be accessed from the home page or Genome Browser UCSC Genes track details pages for selected assemblies. It provides a large variety of information about individual protein characteristics such as polarity, hydrophobicity, amino acid anomalies, domains, exons, and much more, displayed as a series of tracks and histograms. This browser is tightly integrated into the Genome Browser UCSC Genes annotation and in turn links to several external databases and Web sites containing related information.
The in silico PCR utility
The in silico PCR utility is available from the menu bar on most of the Genome Browser Web pages. It provides a means to quickly search genomic sequences or (on human and mouse assemblies) transcribed sequences with a pair of PCR primers, returning a FASTA output file that contains all sequence in the database that lie between and include the primer pair.
Genome Graphs
The Genome Graphs utility, which is available from a link on the left sidebar of the home page, displays data plotted along all chromosomes in a single image. Users can upload their own data (e.g. Whole Genome Analysis Study results) using a very simple text format or import Genome Browser tracks that will be condensed into density plots. The display is configurable. Clicking on a region in the image leads to a Genome Browser view of that region. Other functions include finding the correlation (Pearson’s R) coefficient of two tracks, browsing regions that have scores above a given threshold, and jumping to the Gene Sorter with a list of genes in regions scoring above the threshold.
Genome Browser Sessions
The Sessions utility is available from the menu bar on most of the Genome Browser web pages. It enables the saving, loading and sharing of user session information (i.e. all configuration choices, track visibility changes, filter settings, etc.) that have been set by the user since the session was last reset or loaded. Through the use of Genome Browser sessions, the user can save or load highly tailored views of specific genomic regions with selected tracks enabled, which can be shared as text files, URLs, or emailed to colleagues. Use of many of the session management features requires a valid login at genomewiki.ucsc.edu (see below). UCSC makes its best attempt to preserve sessions stored on the UCSC server, but users are advised to back up their sessions locally, especially any custom track data that may be deleted if they have not been accessed in 48 hours.
genomewiki.ucsc.edu
The website genomewiki.ucsc.edu is a user-editable forum for sharing information about the Genome Browser and associated tools and data. Both the Genome Browser staff and users have contributed technical articles and how-to examples. Registration is not required to search and view the contents, but users are encouraged to register so that they can edit and add content, and use the UCSC storage feature of the Sessions utility described above.
Additional resources for Genome Browser information
In addition to the analytical tools available through the Genome Browser, the track description and details pages provide links to many external resources that present a wealth of related information. For a demonstration of the use of the Genome Browser in comparative genomics analysis, see Bejerano et al. (2005). For a general primer on using genome browsers for data analysis, see Cline and Kent (2009).
Three active mailing lists provide sources for Genome Browser information. The genome@soe.ucsc.edu mailing list provides a moderated discussion forum about the Genome Browser software, databases, genome assemblies, and related tools. The genome-announce@soe.ucsc.edu mailing list provides announcements of data and software releases, system maintenance. Finally, the genome-mirror@soe.ucsc.edu mailing list offers a moderated discussion forum for Genome Browser mirror sites.
Online training materials and tutorials on the Genome Browser are available via the “Training” link on the home page.
In upcoming years, users can expect the Genome Browser to provide more support for the visualization and analysis of medical sequence data, including more genotype/phenotype data sets and new types of whole-genome data, as well as better controlled access for confidential data sets. The authors also plan to expand and enhance the array of comparative genomics analytical tools and data, provide high-performance solutions for mapping, visualizing, and analyzing next-generation sequencing data, and offer more variation data. The browser will continue to add annotated assemblies of vertebrate and model invertebrate genomes as they become available.
Key References
Kent et al., 2002. See above.
A description of the UCSC Genome Browser tool and the underlying conceptual and technical framework.
Kuhn et al., 2009. See above.
The 2009 update of Kent et al. (2002) that includes software enhancements and additions, new genome assemblies, and new annotations.
Internet Resources
The UCSC Genome Bioinformatics and Genome Browser home page.
http://hgdownload.cse.ucsc.edu/downloads.html
The UCSC Genome Browser downloads server.
http://genome-mysql.cse.ucsc.edu
The Genome Browser public MySql server.
http://genome.ucsc.edu/goldenPath/help/hgTracksHelp.html
The UCSC Genome Browser User’s Guide.
http://genome.ucsc.edu/goldenPath/help/hgTablesHelp.html
The UCSC Table Browser User’s Guide.
http://genome.ucsc.edu/goldenPath/help/customTrack.html
Information for constructing and uploading a custom annotation track.
User-editable website for sharing information related to the browser.
genome@soe.ucsc.edu
Mailing list for questions and discussions about the browser software, database, and genome assemblies.
genome-announce@soe.ucsc.edu
Mailing list for announcements about releases of browser software and data, server maintenance, etc.
genome-mirror@soe.ucsc.edu
Mailing list for questions and discussion about mirroring the UCSC Genome Browser.
Acknowledgments
The Genome Browser project is funded by grants from the National Human Genome Research Institute (NHGRI), the Howard Hughes Medical Institute (HHMI) and the National Cancer Institute (NCI). The authors would like to acknowledge the faculty, staff, students, and systems administrators listed at http://genome.ucsc.edu/staff.html who have contributed to the UCSC Genome Browser project, as well as the collaborators listed at http://genome.ucsc.edu/goldenPath/credits.html. The authors would also like to thank their many users for their feedback and support.
Footnotes
Contributed by Donna Karolchik, Angie S. Hinrichs, and W. James Kent, University of California Santa Cruz, Santa Cruz, California
Contributor Information
Donna Karolchik, Email: donnak@soe.ucsc.edu, Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, Phone: (831) 459-1571, Fax: (831) 459-1809.
Angie S. Hinrichs, Email: angie@soe.ucsc.edu, Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, Phone: (831) 459-1544, Fax: (831) 459-1809
W. James Kent, Email: kent@soe.ucsc.edu, Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, Phone: (831) 459-1401, Fax: (831) 459-1809.
Literature Cited
- Amberger JS, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man. Nucleic Acids Res. 2009;37:D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashurst JL, Chen CK, Gilbert JG, Jekosch K, Keenan S, Meidl P, Searle SM, Stalker J, Storey R, Trevanion S, Wilming L, Hubbard T. The Vertebrate Genome Annotation (Vega) database. Nucleic Acids Res. 2005;33:D459–D465. doi: 10.1093/nar/gki135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KG, Barnes KC, Bright TJ, Wang SA. The Genetic Association Database. Nat Genetics. 2004;36:431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- Bejerano G, Siepel AC, Kent WJ, Haussler D. Computational screening of conserved genomic DNA in search of functional noncoding elements. Nat Methods. 2005;2:535–545. doi: 10.1038/nmeth0705-535. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2009;37:D26–D31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:12–17. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE the Mouse Genome Database Group. The Mouse Genome Database genotypes::phenotypes. Nucleic Acids Res. 2009;37:D712–D719. doi: 10.1093/nar/gkn886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, Haussler D, Miller W. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruford EA, Lush MJ, Wright MW, Sneddon TP, Povey S, Birney E. The HGNC Database in 2008: a resource for the human genome. Nucleic Acids Res. 2008;36:D445–D448. doi: 10.1093/nar/gkm881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, Furey TS, Kim UJ, Kuo WL, Livier M. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409:953–958. doi: 10.1038/35057192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Kent WJ. Understanding genome browsing. Nat Biotechnol. 2009;27:153–5. doi: 10.1038/nbt0209-153. [DOI] [PubMed] [Google Scholar]
- Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, Green RK, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE. The RCSB Protein Data Bank: A redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwinell MR, Worthey EA, Shimoyama M, Bakir-Gungor B, DePons J, Laulederkind S, Lowry T, Nigram R, Petri V, Smith J, Stoddard A, Twigger SN, Jacob HJ the RGD Team. The Rat Genome Database 2009: variation, ontologies and pathways. Nucleic Acids Res. 2009;37:D744–D749. doi: 10.1093/nar/gkn842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckman FH, Durbin R. ACeDB and Macace. Methods Cell Biol. 1995;48:583–605. [PubMed] [Google Scholar]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz H, Ceric G, Forslund C, Eddy SR, Sonnhammer ELL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard DS, Wagner L, Feingold EA, Shenmen CM, Grouse LH, Schuler G, Klein SL, Old S, Rasooly R, Good P, Guyer M, Peck AM, Derge JG, Lipman D, Collins FS, Jang W, Sherry S, Feolo M, Misquitta L, Lee E, Rotmistrovsky K, Greenhut SF, Schaefer CF, Buetow K, Bonner TI, Haussler D, Kent J, Kiekhaus M, Furey T, Brent M, Prange C, Schreiber K, Shapiro N, Bhat NK, Hopkins RF, Hsie F, Driscoll T, Soares MB, Casavant TL, Scheetz TE, Brown-stein MJ, Usdin TB, Toshiyuki S, Carninci P, Piao Y, Dudekula DB, Ko MS, Kawakami K, Suzuki Y, Sugano S, Gruber CE, Smith MR, Simmons B, Moore T, Waterman R, Johnson SL, Ruan Y, Wei CL, Mathavan S, Gunaratne PH, Wu J, Garcia AM, Hulyk SW, Fuh E, Yuan Y, Sneed A, Kowis C, Hodgson A, Muzny DM, McPherson J, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Madari A, Young AC, Wetherby KD, Granite SJ, Kwong PN, Brinkley CP, Pearson RL, Bouffard GG, Blakesly RW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Griffith M, Griffith OL, Krzywinski MI, Liao N, Morin R, Palmquist D, Petrescu AS, Skalska U, Smailus DE, Stott JM, Schnerch A, Schein JE, Jones SJ, Holt RA, Baross A, Marra MA, Clifton S, Makowski KA, Bosak S, Malek J and MGC Project Team. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection. Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Pringle TH, Kuhn RM, Karolchik D, Diekhans M, Haussler D, Kent WJ. The UCSC Proteome Browser. Nucleic Acids Res. 2005;33:D454–D458. doi: 10.1093/nar/gki100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Hubbard TJ, Aken BL, Ayling S, Ballester B, Beal K, Bragin E, Brent S, Chen Y, Clapham P, Clarke L, Coates G, Fairley S, Fitzgerald S, Fernandez-Banet J, Gordon L, Graf S, Haider S, Hammond M, Holland R, Howe K, Jenkinson A, Johnson N, Kahari A, Keefe D, Keenan S, Kinsella R, Kokocinski F, Kulesha E, Lawson D, Longden I, Megy K, Meidl P, Overduin B, Parker A, Pritchard B, Rios D, Schuster M, Slater G, Smedley D, Spooner W, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wilder S, Zadissa A, Birney E, Cunningham F, Curwen V, Durbin R, Fernandez-Suarez XM, Herrero J, Kasprzyk A, Proctor G, Smith J, Searle S, Flicek P. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, Finn RD, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Laugraud A, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Mulder N, Natale D, Orengo C, Quinn AF, Selengut JD, Sigrist CJ, Thimma M, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. InterPro: the integrative protein signature database (2009) Nucleic Acids Res. 2009;37:D224–228. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. The UCSC Genome Browser database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. The intronerator: Exploring introns and alternative splicing in Celegans. Nucleic Acids Res. 2000a;28:91–93. doi: 10.1093/nar/28.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zahler AM. Conservation, regulation, synteny, and introns in a large-scale C. briggsae-C. elegans genomic alignment. Genome Res. 2000b;10:1115–1125. doi: 10.1101/gr.10.8.1115. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci USA. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Hsu F, Karolchik D, Kuhn RM, Clawson H, Trumbower H, Haussler D. Exploring relationships and mining data with the UCSC Gene Sorter. Genome Res. 2005;15:737–741. doi: 10.1101/gr.3694705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, Rosenbloom KR, Rhead B, Raney BJ, Pohl A, Pheasant M, Meyer L, Hsu F, Hinrichs AS, Harte RA, Giardine B, Fujita P, Diekhans M, Dreszer T, Clawson H, Barber GP, Haussler D, Kent WJ. The UCSC Genome Browser Database: update 2009. Nucleic Acids Res. 2009;37:D755–D761. doi: 10.1093/nar/gkn875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhard B, Hayes WS, Wasserman WW. GeneLynx: A gene-centric portal to the human genome. Genome Res. 2001;11:2151–2157. doi: 10.1101/gr.199801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper U, Eswar N, Davis FP, Braberg H, Madhusudhan MS, Rossi A, Marti-Renom M, Karchin R, Webb BM, Eramian D, Shen MY, Kelly L, Melo F, Sali A. MODBASE: A database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 2006;34:D291–D295. doi: 10.1093/nar/gkj059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S, Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J, Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, Hart J, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD, Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D, Hubbard T, Ostell J, Durbin R, Lipman D. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;37:D32–D36. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Chalifa-Caspi V, Shmueli O, Olender T, Lapidot M, Rosen N, Shmoish M, Peter Y, Glusman G, Feldmesser E, Adato A, Peter I, Khen M, Atarot T, Groner Y, Lancet D. Human gene-centric databases at the Weizmann Institute of Science: GeneCards, UDB, CroW 21 and HORDE. Nucleic Acids Res. 2003;31:142–146. doi: 10.1093/nar/gkg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Mizrachi I, Ostell J, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Yaschenko E, Ye J. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2009;37:D5–D15. doi: 10.1093/nar/gkn741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison R, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs A, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Gen Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Greenhut SF, Grouse LH, Schaefer CF, Buetow KH. In silico analysis of cancer through the Cancer Genome Anatomy Project. Trends Cell Biol. 2001;11:S66–S71. doi: 10.1016/s0962-8924(01)02104-3. [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Rosenbloom KR, Clawson H, Hinrichs AS, Trumbower H, Raney BJ, Karolchik D, Barber GP, Harte RA, Hillman-Jackson J, Kuhn RM, Rhead BL, Smith KE, Thakkapallayil A, Zweig AS, Kent WJ The ENCODE Project Consortium Haussler D. The ENCODE project at UC Santa Cruz. Nucleic Acids Res. 2007;35:D663–667. doi: 10.1093/nar/gkl1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009;37:D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O’Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]