

Abstract
Telomeres of most somatic cells progressively shorten, compromising the regenerative capacity of human tissues during aging and chronic diseases and after acute injury. Whether telomere shortening reduces renal regeneration after acute injury is unknown. Here, renal ischemia-reperfusion injury led to greater impairment of renal function and increased acute and chronic histopathologic damage in fourth-generation telomerase-deficient mice compared with both wild-type and first-generation telomerase-deficient mice. Critically short telomeres, increased expression of the cell-cycle inhibitor p21, and more apoptotic renal cells accompanied the pronounced damage in fourth-generation telomerase-deficient mice. These mice also demonstrated significantly reduced proliferative capacity in tubular, glomerular, and interstitial cells. These data suggest that critical telomere shortening in the kidney leads to increased senescence and apoptosis, thereby limiting regenerative capacity in response to injury.
As the population ages, the number of elderly patients with chronic kidney disease is expected to grow, making kidney aging an issue of great clinical importance. Old kidneys show a decline in function as reflected by increased renal vascular resistance, reduced renal plasma flow, and increased filtration fraction.1 Morphologic changes include cortical thinning2 and a histology indicating deterioration, such as tubular atrophy, interstitial fibrosis, and glomerulosclerosis.3 In the aging population, acute kidney injury is significantly more common, the clinical course is more severe, and kidney function is less likely to recover.4 The elderly are more likely to develop end-stage renal failure than the young.5 Donor age is one of the most important predictors of long-term graft survival, and older donor kidneys are more likely to fail when they experience rejection, even when the rejection is relatively mild.6 Taken together, the phenotype of renal aging is described not only by the loss of function and mass but also by the loss of an appropriate response toward injury.
Telomere shortening and dysfunction are crucial determinants for human lifespan and the regenerative capacity of organs exposed to extrinsic stresses.7 Telomeres consist of repetitive DNA elements at the end of linear chromosomes and protect the DNA ends from degradation and recombination. Because of the “end-replication problem,” telomeres shorten progressively in humans with every cell division.7 Eventually, telomeres reach a critically short length, behaving as double-stranded DNA breaks that activate p53 and result in telomere-initiated replicative senescence or apoptosis. Replicative senescence is mediated via the p53/p21 pathway and specifically mediates the proaging effect of short telomeres.8
Telomere length is maintained by telomerase, an enzyme that counteracts telomere shortening by addition of tandem repeats of the TTAGGG sequence.9 The telomerase reverse transcriptase (TERT in human, Tert in mouse) generates telomere repeats by using an associated RNA molecule (telomerase RNA component, TERC for human, Terc for mouse) as a template. TERC is indispensable for telomerase to maintain telomere length.10 In humans, robust telomerase activity is predominantly restricted to some embryonic and adult stem/progenitor cell compartments, to germ cells, and to proliferating lymphocytes.11 Thus, telomere attrition is observed with increasing age in all human tissues in which it has been tested, including the kidney.7,12 Various human diseases associated with aging, such as cardiovascular disease, ulcerative colitis, liver cirrhosis, and infections, show accelerated telomere shortening.7 A correlation between telomere length and risk for death from heart disease or infections has been reported.13 Laboratory rodents have long telomeres relative to humans.7,14 Murine telomere length can be reduced to a “human length” by serial intercrossing of Terc−/− mice.10 Late-generation Terc−/− mice with critically short telomeres show defects in homeostasis of proliferative organs,15 but these are not the only organ systems that are affected by premature aging phenotypes.16 Liver regeneration after acute and chronic stresses is diminished in late-generation Terc−/− mice and was associated with an increased number of senescent liver cells.17
We had hypothesized that the increased susceptibility of older kidneys toward acute injury and the inability of an appropriate repair is the result of more cells being senescent.18,19 In this study, we demonstrate that ischemia-reperfusion injury (IRI) of the kidney induces stronger acute and chronic damage in late-generation Terc−/− mice with short telomeres (G4) compared with G1 Terc−/− and wild-type Terc+/+ mice. Greater damage in G4 Terc−/− is accompanied by an increase in critically shortened telomeres, higher expression of the downstream mediator p21, increased apoptosis, and reduced cellular regeneration measured by proliferation marker Ki-67.
Results
Histopathology of the Kidney after IRI
Acute tubular damage assessed for the cortical and corticomedullary region was highest on days 1 and 3 in Terc+/+, G1 Terc−/−, and G4 Terc−/− (Figure 1, A and B). On day 3, G4 Terc−/− showed significantly more damage when compared with Terc+/+ and G1 Terc−/−. On days 7 and 30, acute tubular damage decreased, ranging approximately 10%, without significant differences between the groups.
Figure 1.

Histopathology after renal IRI is shown. (A) Acute tubular damage, shown as percentage of damage to total tubular area, was quantified 1, 3, 7, and 30 d after 30 min of IRI of the left kidney in Terc+/+, G1 Terc−/−, and G4 Terc−/− kidneys. (B) Representative periodic acid-Schiff stainings 3 d after IRI showing more acute tubular damage in G4 Terc−/− compared with G1 Terc−/− and Terc+/+. * areas of acute tubular damage. (C) Chronic tubular deterioration and (D) interstitial fibrosis—both reflecting chronic kidney damage—were quantified 1, 3, 7, and 30 d after IRI in Terc+/+, G1 Terc−/−, and G4 Terc−/−. (E) Representative Masson Trichrome stainings showing increased interstitial fibrosis (blue staining, indicated by black arrowhead) in G4 Terc−/− on day 30 after IRI. □, Terc+/+;  , G1 Terc−/−; ■, G4 Terc−/−. Data are means ± SEM; significances are indicated. Magnification, ×200.
, G1 Terc−/−; ■, G4 Terc−/−. Data are means ± SEM; significances are indicated. Magnification, ×200.
Histopathologic alterations classified as chronic tubular damage were observed starting from day 7 and significantly increased by day 30 in Terc+/+, G1 Terc−/−, and G4 Terc−/− (Figure 1C). There was a tendency toward a higher incidence in G4 Terc−/−, but this was significant only when the corticomedullary junction was assessed separately (P = 0.01 for the comparison of G4 Terc−/− with Terc+/+ on day 30; data not shown).
Interstitial fibrosis was seen only on day 30 in Terc+/+, G1 Terc−/−, and G4 Terc−/− with significantly higher levels in G4 Terc−/− kidneys (Figure 1, D and E). Sham-operated and nonoperated controls showed neither acute or chronic tubular damage nor any interstitial fibrosis (data not shown).
Renal Expression of Connective Tissue Growth Factor after IRI
Connective tissue growth factor (Ctgf) is recognized as an important player in fibrogenic pathways. Because of the significant increase in interstitial fibrosis in G4 Terc−/− kidneys, we performed Western analysis for Ctgf in a subgroup of animals on days 7 and 30 after IRI (Figure 2). For day 7, we saw no significant differences among the three groups (Figure 2, A and B). Highest values for Ctgf were seen for G1 Terc−/− and G4 Terc−/− kidneys on day 30, but the differences between groups were NS (Figure 2, A and C).
Figure 2.
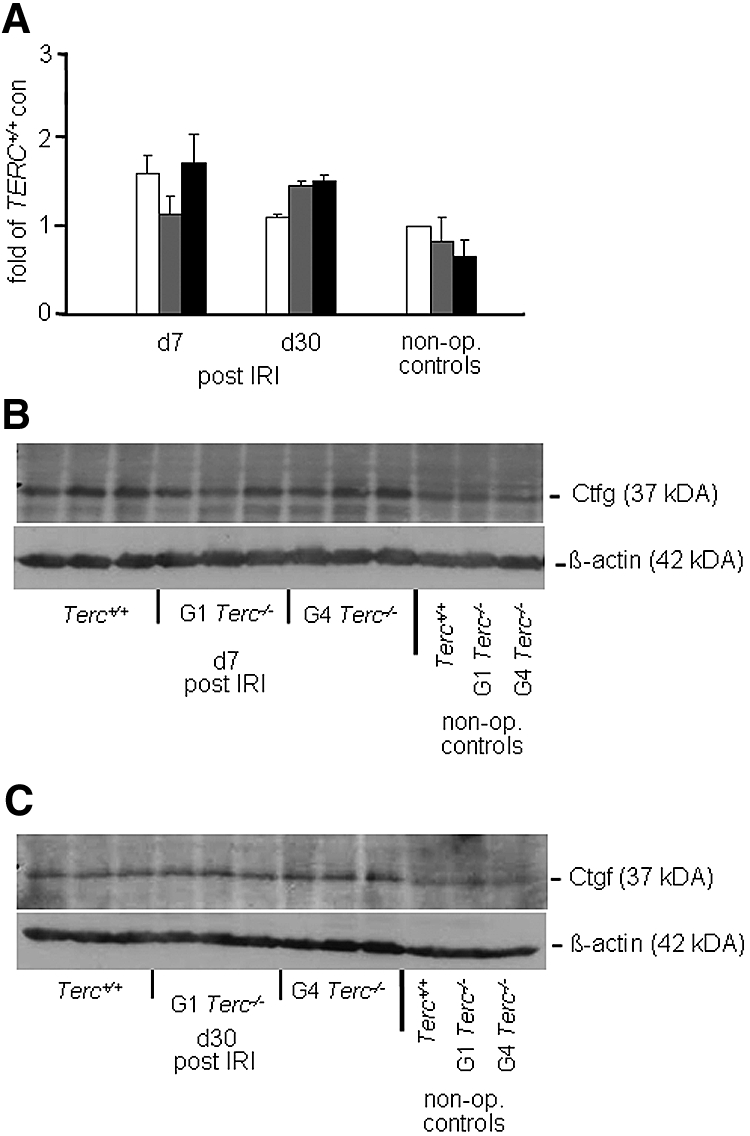
Renal Ctgf expression after IRI is shown. (A) Analysis of Ctgf protein expression given as fold expression on the basis of the expression of Terc+/+ nonoperated controls. (B and C) Western blots showing Ctgf at 37 kD and β-actin as a loading control at 42 kD for day 7 (B) and day 30 (C). □, Terc+/+; , G1 Terc−/−; ■, G4 Terc−/−. Data are means ± SEM; significances are indicated.
Telomere Quantitative Fluorescence In Situ Hybridization Analysis
We analyzed telomere length in a subgroup of animals by quantitative fluorescence in situ hybridization (Q-FISH) analysis (Figure 3, A and B). In nonoperated controls, mean telomere fluorescence intensity (TFI) in tubular cells from Terc+/+ kidneys was significantly higher when compared with G4 Terc−/− kidneys (99.7 versus 73.8; P < 0.05). Analysis of kidney sections from day 30 after IRI revealed that in both groups, the injury led to significant telomere shortening (TFI at d30 53.8 [in Terc+/+] versus 44.5 [in G4 Terc−/−]). Even though the difference in mean TFI was no longer significant, the percentage of critically shortened telomeres, defined as a TFI ≤40, was significantly higher in G4 Terc−/− kidneys (48.6 versus 14.0% in Terc+/+; P < 0.001).
Figure 3.

Telomere Q-FISH and renal gene expression analysis are shown. (A) Representative pictures for Q-FISH analysis showing tubular cross-sections from nonoperated Terc+/+ and G4 Terc−/− controls and from Terc+/+ and G4 Terc−/− kidneys 30 d after IRI. (B) Distribution of TFI in kidneys showing that critically shortened telomeres, defined as TFI ≤40, are rarely found in nonoperated Terc+/+ and G4 Terc−/− controls, even though there is a significant difference in mean TFI between the two groups; however, kidneys from G4 Terc−/− mice 30 d after IRI show a significantly higher number of critically shortened telomeres when compared with Terc+/+ 30 d after IRI. (C) p21 expression was significantly higher in G4 Terc−/− compared with Terc+/+ and G1 Terc−/− for days 3, 7, and 30 after IRI as well as for sham-operated controls. A similar tendency was seen for nonoperated controls. *Sham-operated controls (P < 0.005 for Terc+/+, P < 0.001 for G1 Terc−/−, and P < 0.05 for G4 Terc−/−) as well as **nonoperated controls (P < 0.005 for Terc+/+, P < 0.001 for G1 Terc−/−, and P < 0.05 for G4 Terc−/−) showed significantly lower p21 expression levels compared with days 1, 3, 7, and 30 after IRI. (D) Cell-cycle inhibitor p16INK4a showed a continuous increase after IRI in Terc+/+, G1 Terc−/−, and G4 Terc−/− kidneys with significantly higher levels on day 30 compared with sham-operated (***P < 0.05 for Terc+/+, P < 0.001 for G1 Terc−/−, and P < 0.001 for G4 Terc−/−) and nonoperated controls (****P < 0.01 for Terc+/+, P < 0.001 for G1 Terc−/−, and P < 0.001 for G4 Terc−/−). Apart from day 30, significantly higher values of p16INK4a were seen in G4 Terc−/− kidneys when compared with Terc+/+ (day 3), G1 Terc−/− (day 7), or both (day 1). □, Terc+/+; , G1 Terc−/−; ■, G4 Terc−/−. Data are means ± SEM; significances are indicated. Figure 3 is also provided in color as Supplemental Figure 3.
p21, p16INK4a, and p53 mRNA Expression in the Kidney after IRI
There was a significant increase in cell-cycle inhibitor p21 mRNA expression in Terc+/+, G1 Terc−/−, and G4 Terc−/− kidneys for all time points after IRI when compared with sham-operated and nonoperated controls (Figure 3C). Apart from day 1, G4 Terc−/− showed significantly higher levels of p21 when compared with either Terc+/+ or G1 Terc−/−. This was also true for G4 Terc−/− controls that were sham-operated, and the same tendency was seen in nonoperated animals.
Cell-cycle inhibitor p16INK4a showed a continuous increase after IRI in Terc+/+, G1 Terc−/−, and G4 Terc−/− kidneys with significantly higher levels on day 30 compared with sham-operated and nonoperated controls (Figure 3D). Apart from day 30, significantly higher values of p16INK4a were seen in G4 Terc−/− kidneys when compared with either Terc+/+ (day 3), G1 Terc−/− (day 7) or both (day 1). On day 30, ANOVA revealed statistically significant differences among the three groups for p16INK4a levels (P < 0.05), but the higher values seen now for G4 Terc−/− and G1 Terc−/− kidneys compared with Terc+/+ animals were no longer significant after post hoc testing with Bonferroni correction (P = 0.07 and P = 0.09, respectively).
p53 mRNA levels were significantly upregulated after IRI on days 1 and 3 in Terc+/+, G1 Terc−/−, and G4 Terc−/− mice compared with sham-operated controls (Supplemental Figure 1) and continuously decreased with time. Apart from day 3, for which G4 Terc−/− kidneys showed higher values when compared with Terc+/+ and G1 Terc−/−, there were no statistically significant difference among the three groups.
Proliferation and Apoptosis after IRI
Proliferation was measured using Ki-67 immunostaining. Quantification of positively stained nuclei revealed significantly lower proliferation rates in tubular and interstitial cells of G4 Terc−/− kidneys on day 3 (Figure 4A) and in tubular, glomerular, and interstitial cells of G4 Terc−/− kidneys on day 30 (Figure 4B). As previously shown by our group20 and other investigators,21 we confirmed very low proliferation rates in renal cells of sham-operated (Figure 4C) and nonoperated controls (data not shown), without significant differences among the three groups. Representative stainings are shown in Figure 4D.
Figure 4.

Proliferation and apoptosis after renal IRI are shown. (A through C) Quantification of Ki-67–positive tubular, glomerular, and interstitial nuclei in Terc+/+, G1 Terc−/−, and G4 Terc−/− animals 3 (A) and 30 d (B) after IRI and in sham-operated controls (C). Lower proliferation rates were seen in tubular and interstitial cells of G4 Terc−/− kidneys on day 3 and in tubular, glomerular, and interstitial cells of G4 Terc−/− kidneys on day 30 compared with Terc+/+ and G1 Terc−/−. Very low proliferation rates were detected in renal cells from sham-operated controls with no significant differences among the three groups. (D) Representative Ki-67 immunostainings on day 30 after IRI showing reduced numbers of positively stained nuclei in G4 Terc−/−. (E) Representative TUNEL stainings on day 30 after IRI showing increased numbers of apoptotic cells in G4 Terc−/−. TUNEL-positive cells are marked by arrowheads. (F through H) Quantification of TUNEL-positive tubular, glomerular, and interstitial nuclei in Terc+/+, G1 Terc−/−, and G4 Terc−/− animals 3 (F) and 30 d (G) after IRI and in sham-operated controls (H). For both time points after IRI, G4 Terc−/− kidneys showed a significantly higher number of TUNEL-positive tubular and interstitial cells compared with Terc+/+. The same was true for the comparison of G4 Terc−/− with G1 Terc−/− kidneys, with the exception of interstitial cells 3 d after IRI. Very low numbers of apoptotic tubular and interstitial cells and no apoptotic glomerular cells were detected in sham-operated controls, with no statistical differences between groups. □, Terc+/+; , G1 Terc−/−; ■, G4 Terc−/−. Data are means ± SEM; significances are indicated. Magnification, ×200.
Apoptotic cells were detected by terminal deoxynucleotidyl transferase–mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) staining (Figure 4E). On days 3 and 30 after IRI, G4 Terc−/− kidneys showed a significantly higher number of TUNEL-positive tubular and interstitial cells compared with Terc+/+. The same was true for the comparison of G4 Terc−/− with G1 Terc−/− kidneys, with the exception of interstitial cells 3 d after IRI (Figure 4, F and G). As expected, sham-operated controls showed very low numbers of apoptotic tubular and interstitial cells, and no apoptotic cells were seen in glomeruli (Figure 4H). No statistical differences were found between controls.
Inflammatory Response after IRI
We characterized the infiltrating cell populations using CD3 (T lymphocytes) and CD68 (macrophages) immunostaining at days 3, 7, and 30 (Table 1). For CD3+ cells, we found no differences among the three groups at days 3 and 7; however, on day 30, the amount of CD3+ cells was significantly lower in G4 Terc−/− when compared with G1 Terc−/− kidneys. The increases in CD3+ cells over time, which were seen for all three groups, were significant only for Terc+/+ (ANOVA P = 0.009) and G1 Terc−/− (ANOVA P < 0.001) animals. The number of CD68+ cells was higher when compared with CD3+ cells. Comparing the three groups, we found differences only at day 7, with less CD68+ cells in G1 Terc−/− when compared with G4 Terc−/−. This difference was no longer seen at day 30. There seemed to be no tendency for CD68 infiltrating cells over time, even though ANOVA became significant for the group of G1 Terc−/− (P = 0.009) mice because of the low number of cells at day 7.
Table 1.
Characterization of infiltrating cells after IRI in Terc mice
| Parameter | Terc+/+ | G1 Terc−/− | G4 Terc−/− | P, ANOVA |
|---|---|---|---|---|
| Day 3 | ||||
| CD3 | 9.1 ± 1.6 | 8.1 ± 0.9 | 6.8 ± 1.9 | NS |
| CD68 | 76.7 ± 18.9 | 129.5 ± 12.4 | 106.7 ± 8.1 | NS |
| Day 7 | ||||
| CD3 | 34.2 ± 9.4 | 25.6 ± 5.4 | 11.8 ± 3.3 | NS |
| CD68 | 99.4 ± 5.5 | 70.6 ± 2.4 | 120.0 ± 11.3a | 0.002 |
| Day 30 | ||||
| CD3 | 46.8 ± 8.0 | 62.1 ± 5.1 | 20.0 ± 7.6b | <0.004 |
| CD68 | 107.2 ± 14.8 | 121.4 ± 16.1 | 103.1 ± 17.2 | NS |
aP = 0.001 for the comparison with G1 Terc−/−.
bP = 0.003 for the comparison with G1 Terc−/− and P = 0.06 for the comparison with Terc+/+.
Kidney Function after 15 Min of IRI to the Left Kidney and Right Nephrectomy
We performed additional IRI experiments using 15 min of ischemia to the left kidney followed by nephrectomy of the right kidney. Longer ischemia times had led to death in G4 Terc−/− animals. On day 30, creatinine clearances in G4 Terc−/− mice were significantly lower compared with Terc+/+ (Table 2). A similar, almost significant tendency had been seen for the comparison at day 7. No significant differences were seen for nonoperated or sham-operated control animals.
Table 2.
Kidney function in Terc+/+, G1 Terc−/−, and G4 Terc−/− mice
| Parameter | Creatinine Clearance (ml/min) |
P, ANOVA | ||
|---|---|---|---|---|
| Terc+/+ | G1 Terc−/− | G4 Terc−/− | ||
| Controls | ||||
| nonoperated | 0.145 ± 0.051 (n = 6) | 0.220 ± 0.024 (n = 5) | 0.110 ± 0.019 (n = 5) | NS |
| sham-operated | 0.197 ± 0.024 (n = 3) | 0.240 ± 0.031 (n = 3) | 0.147 ± 0.009 (n = 3) | NS |
| Mice after IRI (15 min of ischemia to left kidney, right nephrectomy) | ||||
| day 7 | 0.130 ± 0.012 (n = 6) | 0.110 ± 0.023 (n = 7) | 0.065 ± 0.011 (n = 6)a | 0.06 |
| day 30 | 0.157 ± 0.017 (n = 6) | 0.151 ± 0.017 (n = 7) | 0.098 ± 0.006 (n = 6)b | <0.05 |
aP = 0.06 for the comparison G4 Terc−/− vs. Terc+/+.
bP < 0.05 for the comparison G4 Terc−/− vs. Terc+/+.
Discussion
Critically short telomeres in kidneys of late-generation Terc−/− mice increase the susceptibility to acute cell death and reduce long-term regenerative capacity of the kidney after IRI. Telomere attrition is observed with increasing age in human tissues, including the kidney,11,12 and is accelerated in various age-associated human diseases (for review, see reference7). Our study provides a mechanistic approach by investigating the effect of critically short telomeres on kidney regeneration after acute replicative stress. Reduced long-term regeneration in G4 Terc−/− mice was reflected by more chronic deterioration and reduced organ function. Significantly lower creatinine clearances in G4 Terc−/− compared with Terc+/+ were seen 30 days after IRI, and G4 Terc−/− mice did not show compensatory hypertrophy of the right kidney after unilateral clamping without contralateral nephrectomy. The excess in acute damage after IRI in late-generation Terc−/− reflects the reduced ability of cells with critically short telomeres to respond adequately toward stresses that under normal circumstances result in increased proliferation. The pronounced chronic deterioration found in G4 Terc−/− with increased interstitial fibrosis on day 30 supports this and argues for a loss of parenchyma as a result of absent tubular regeneration during early days, resulting in replacement of lost cells by fibrotic tissue. Our results are in line with studies in liver from late-generation Terc−/− mice showing reduced regenerative capacity after acute stress through genetic, surgical, and chemical ablation of liver cells.22
DNA damage signaling in response to telomere dysfunction induces transient cell-cycle arrest, senescence, or apoptosis, depending on cell type and severity of stress.23 In late-generation Terc−/− mice, apoptosis is the dominant phenotype in thymic lymphocytes and male germline,15,24 whereas cell-cycle arrest/senescence is the major checkpoint limiting liver regeneration.17 In our model of renal IRI, G4 Terc−/− kidneys show significantly higher numbers of TUNEL-positive cells, but with a maximum of only 1 to 3% apoptotic cells, it seems that senescence pathways are driving the failure of kidney regeneration seen in G4 Terc−/−. Most senescent cells remain present and are not simply lost. The phenotype of senescent fibroblasts in culture is associated with a typical expression pattern: Increases in cell-cycle inhibitors (e.g., p16INK4a, p21) and extracellular matrix proteins (e.g., collagen, fibronectin) as well as decreases in genes that are involved in cell-cycle progression. Senescent cells secrete degradative proteins, inflammatory cytokines, and growth factors (e.g., Ctgf25) that may promote tissue aging and probably possess a complex role in chronic disease processes.26 We previously showed that senescence features correlate with the presence of interstitial fibrosis in aged human kidneys,3 and it is conceivable that interstitial fibrosis simply reflects the accumulation of fibroblasts that have turned senescent.
Significantly higher levels of p21 in G4 Terc−/− controls are in line with the concept of an increased induction of senescence as a result of dysfunctional telomeres even before any injury.27,28 Telomere dysfunction leads to formation of DNA-damage foci that activate ATM/ATR signaling,29 and a downstream cascade stabilizes p53 and, beside others, upregulates p21.8 The cyclin-dependent kinase inhibitor p21 mediates p53-dependent and -independent induction of senescence.30 p21 deletion can prolong the lifespan of human cells31 and of mice with dysfunctional telomeres.8 Our finding of increased p21 expression after IRI reconfirms observations showing p21 induction after IRI.30 The upregulation of p21 is more pronounced in G4 Terc−/− mice probably as a result of more telomeres becoming critically shortened. The concept of an increase in senescent cells in G4 Terc−/− mice is further confirmed by reduced proliferation 3 and 30 days after IRI. p53 has been shown to be upregulated in some cell types from Terc−/− mice as telomeres reach a critically short length.32 In our study, p53 mRNA expression in total kidney specimens was induced in Terc+/+, G1 Terc−/−, and G4 Terc−/− after IRI without significant differences among the groups. The discrepancy to other reports might be explained by the fact that we did not perform a cell type–specific evaluation for p53 expression or investigate posttranscriptional modifications of p53, as suggested by some.33 Conversely, high levels of p21 mRNA were induced in kidneys of p53 “null” mice, demonstrating that p21 gene activation can also occur through a p53-independent pathway.30
Another major regulator of senescence, the cell-cycle inhibitor p16INK4a, is progressively induced after IRI. p16INK4a expression was higher in G4 Terc−/− kidneys for the early days after IRI, but 30 days after IRI, there was no longer a difference between expression levels in kidneys from both generations of Terc−/− mice. p16INK4a is the key mediator for STASIS (stress or aberrant signaling induced senescence), a pathway that can induce senescence independent of telomere dysfunction.34 The interplay between replicative senescence and STASIS is not completely unraveled yet, but there are data suggesting connections under certain circumstances. As primary cells divide and telomeres shorten, p16INK4a levels progressively increase.35 p16INK4a can enforce a G1/S arrest in response to DNA damage,36 and telomere damage can also elicit a G1/S arrest through p16INK4a, especially in cells lacking p53 function.37 Our results may be explained by the observation that even the first generation of telomerase-deficient mice show a shortened lifespan,38 and one could speculate that telomere shortening per se, even in the absence of critically short telomeres, has an effect on regulation of p16INK4a. A reason for this could be the missing protection of telomeres through telomerase.7
Impaired regeneration in late-generation telomerase-deficient mice is also seen with regard to the immune system. Immunosenescence mainly affects the specific immune responses with a reduction of germinal center reactivity upon immunization and a reduced proliferative capacity of T and B cells.7,15 The increase in CD3+ cells that was significantly less pronounced in G4 Terc−/− animals confirms these observations; however, the unspecific immune response, as reflected by CD68-infiltrating cells, seemed to be conserved in G4 Terc−/− mice.
Late-generation Terc−/− mice mimic the situation seen in aged human kidneys with their inadequate ability to respond to injury. We provide evidence that a reason for this restriction of old kidneys could be telomere shortening and dysfunction, leading to impaired replication and regeneration. The changes induced through the activation of certain senescence proteins contribute to the collapse of tissue integrity, leading to a disturbed organ homeostasis. On the basis of this study, a pathogenetic role of telomere dysfunction for the development of renal insufficiency through structural and functional impairment is likely.
Concise Methods
Animals
All procedures performed on animals were done in accordance with institutional guidelines for animal research and were approved by the local government authorities.
Generation and Genotyping of G4 Terc−/− Mice
Terc+/− mice on C57BL/6J background were purchased from Jackson Laboratories.10 G1 Terc−/− and Terc+/+ control animals were derived from heterozygous intercrosses according to previously published breeding strategies.16 Mating of G1 Terc−/− animals to each other generated G2 Terc−/− animals. Following this mating scheme, Terc−/− animals up to the fourth generation were bred. G4 Terc−/− mice displayed the typical phenotype that has been described before,16 including decreased survival, reduced fertility, premature hair graying, alopecia, and ulcerative skin lesions.
Ischemia-Reperfusion Injury
Ischemia-reperfusion surgery was performed in 3- to 4-mo-old age-matched male Terc+/+, G1 Terc−/−, and G4 Terc−/− mice according to current standard protocols. Briefly, mice were anesthetized with inhalational isoflurane and were kept on a heating pad to maintain body temperature during surgery. A midline laparotomy was made, and the bowel was gently placed aside by retractors. The renal pedicles were exposed, and adjacent fat tissue was removed carefully. The left renal pedicle (artery and vein) was clamped for 30 min, using nontraumatic microsurgical vascular clips (Aesculap, Tuttlingen, Germany). Occlusion of blood flow was confirmed by visual inspection of the kidneys. The right kidney was left in situ to reduce otherwise high mortality rates. After removal of the clip, the kidneys were observed for approximately 5 min to ensure blood reflow, and then fascia and skin were sutured in two layers with 6-0 silk and polyethylene. All animals received the same volume of warm saline instilled in the peritoneal cavity during the surgical procedure and were allowed to recover with ad libitum access to food and water. Mice for the group with 30 min of renal ischemia were killed after 1 (Terc+/+, n = 7; G1 Terc−/−, n = 8; G4 Terc−/−, n = 8), 3 (Terc+/+, n = 8; G1 Terc−/−, n = 8; G4 Terc−/−, n = 6), 7 (Terc+/+, n = 6; G1 Terc−/−, n = 8; G4 Terc−/−, n = 8), or 30 (Terc+/+, n = 8; G1 Terc−/−, n = 8; G4 Terc−/−, n = 7) days.
In addition, we performed ischemia-reperfusion surgery in mice of the three groups but used only 15 min ischemia to the left renal pedicle followed by nephrectomy of the right kidney to generate functional data (i.e., creatinine clearances). Mice in this group were killed after 7 (Terc+/+, n = 6; G1 Terc−/−, n = 7; G4 Terc−/−, n = 6) or 30 (Terc+/+, n = 6; G1 Terc−/−, n = 7; G4 Terc−/−, n = 6) days.
Controls consisted of nonoperated (Terc+/+, n = 8; G1 Terc−/−, n = 7; G4 Terc−/−, n = 8) and sham-operated animals (Terc+/+, n = 3; G1 Terc−/−, n = 3; G4 Terc−/−, n = 3). Sham-operated animals underwent anesthesia, laparotomy, and renal pedicle dissection only. They were killed 7 days after surgery.
After the mice were killed, the kidneys were excised, adjacent tissue was carefully removed, kidneys were decapsulated, and weight was determined. Part of each kidney was immediately snap-frozen in liquid nitrogen for later RNA extraction; a second part was fixed in 4% formaldehyde and paraffin-embedded.
Creatinine Clearance Determination
Except for day 1, all mice were kept in metabolic cages for the last 24 h before being killed. Twenty-four-hour urine was collected, urine volume was recorded, and aliquots were stored at −80°C for subsequent analysis. Blood was obtained by cardiac puncture under deep anesthesia right before the mice were killed. Plasma and urinary creatinine were determined using an enzymatic method that has been validated in rodents.39 Creatinine clearance (ml/min) was derived from the following formula: Urinary creatinine × urine volume × 1440 min−1 × plasma creatinine−1.
Body and Kidney Weight in Terc+/+ and Terc−/− Mice
G4 Terc−/− mice showed reduced body weights compared with Terc+/+ and G1 Terc−/−, as has been described before.16 The mean weight differences between Terc+/+ and G4 Terc−/− per group for each day ranged between 18 and 26%. The body weights of G1 Terc−/− mice were comparable to those of Terc+/+ mice (data not shown). Weight of the clamped left kidney after IRI related to body weight did not show differences among Terc+/+, G1 Terc−/−, and G4 Terc−/−; however, the weight of the right kidney increased significantly as a result of compensatory hypertrophy only in Terc+/+ and G1 Terc−/− mice, but not in G4 Terc−/− mice (Supplemental Table 1) probably also as a result of the diminished regenerative capacity in these mice.
Histopathology of Kidney
Tissue sections were cut in 3-μm sections with a Leica RM 2165 microtome (Leica Instruments, Nussloch, Germany) and stained with hematoxylin and eosin, periodic acid-Schiff, or Masson Trichrome. High-power field (HPF) pictures (×200 magnification) of the whole cortex (>20) and corticomedullary junction (>10) were taken of each mouse kidney using a Leica DM LB2 digitizing microscope and a Leica DFC 320 camera (Leica Instruments) with QWin V3 software (Leica). Both cortex and corticomedullary junction were analyzed, and results (“total damage”) as shown in the Results section were obtained on a prorata basis.
Acute and chronic renal damage was quantified using ImageJ 1.37c software (National Institutes of Health, Bethesda, MD). Briefly, damaged tubular area and total tubular area were marked on each HPF screen, and ratio of damaged tubules to total tubular area was determined. Acute damage was mainly reflected by tubular necrosis, whereas chronic damage consisted of tubular deterioration including reduced tubular diameter, thickened tubular basement membrane, and loss of tubular nuclei.
The degree of renal interstitial fibrosis was measured using Masson Trichrome stainings and a semiquantitative scoring system. The area of blue-stained interstitial fibrosis was detected using QWin 3 software package and related to total tubulointerstitial area. Fibrosis was evaluated by analysis of 20 HPFs (×200 magnification) for each animal.
Telomere Q-FISH Analysis
Q-FISH was performed as described.40 Nuclei were stained with DAPI, and image z stacks were taken using a Leica DM5500B (Leica Instruments) with a ×100 objective, capturing fluorescence images with a DFC360FX camera using LASAF software (Leica). Untreated mice (Terc+/+, n = 6; G4 Terc−/−, n = 6) and mice 30 d after renal ischemia (Terc+/+, n = 6; G4 Terc−/−, n = 7) were analyzed with three images per animal. Images were generated as projected, deconvolved z stacks (Huygens, SVI). TFI per nucleus in tubular cells (100 nuclei per mouse) were quantitatively analyzed using ImageJ 1.42 software (National Institutes of Health). Results are given as frequency histograms.
Real-Time Reverse Transcriptase–PCR
Total RNA was extracted from murine renal tissue samples using Trizol reagent (Invitrogen, Karlsruhe, Germany), RNA integrity was verified by agarose gel electrophoresis, and cDNA was obtained by reverse transcription of 1 μg total using MMLV reverse transcriptase and random primers (Invitrogen). For quantitative PCR of p16INK4a, p53, and p21, the following intron-spanning primers and probes were used: p16INK4a forward 5′-GGGCACTGCTGGAAGCC-3′, reverse 5′-AACGTTGCCCATCATCATC-3′, and probe 5′-CCGAACTCTTTCGGTCGTA-3′; p21 forward 5′-CAGCCACAGGCACCATGTC-3′, reverse 5′-ACGGCGCAACTGCTCACT-3′, and probe 5′-ATGTCCGACCTGTTCCGCACAGGA-3′; and p53 forward 5′-CCGACCTATCCTTACCATCATCA-3′, reverse 5′-AGGCACAAACACGAACCTCAA-3′, and probe 5′-CCAGAAGGTTCCCACTGGAGTCTTCCA-3′. For the TERT component of telomerase, we purchase a predeveloped assay reagent (TaqMan Gene Expression Assays, Assay ID: 707048; Applied Biosystems, Weiterstadt, Germany).
Immunohistochemistry for Ki-67, CD3, and CD68
Immunoperoxidase staining for Ki-67 and CD3 was performed on paraffin-embedded tissue. Antigen retrieval was performed in citrate buffer (pH 6.0) using a pressurized heating chamber (Pascal Pressurized Heating Chamber; Dako, Cambridgeshire, United Kingdom). The sections then were immersed in 3% H2O2 in methanol, blocked with 1× Universal Blocking Reagent (BioGenex, San Ramon, CA), and then incubated at room temperature with the primary antibody clone MIB-9 (Dako; 190 mg/ml, at a dilution of 1:25) for Ki-67 or CD3-12 (Serotec, Martinsried, Germany; 1 mg/ml, at a dilution of 1:50) for CD3 or appropriate isotype control antibodies, and rinsed with PBS. After 30 min of incubation with the Envision Monoclonal System (Dako) for Ki-67 or with a biotinylated goat anti-rat (Southern Biotech, Birmingham, AL) for CD3, sections were washed again in PBS. In case of CD3 staining, 30 min of incubation with ABC reagent (Vectastain, Vector Laboratories, Burlingame, CA) was performed, and sections were rinsed with PBS. Visualization was performed using the DAB substrate kit (Dako). The slides were counterstained with hematoxylin for Ki-67 or methylene blue for CD3.
Staining for CD68 was performed on frozen tissue. Fresh-frozen sections were briefly fixed in acetone at 4°C. The sections then were immersed in 3% H2O2 in methanol and blocked with 100% FCS. Slides were then incubated with the primary rat anti-mouse antibody against CD68 (clone FA-11; Serotec) at a dilution of 1:100 or the appropriate isotype control antibody. Next, slides were exposed to a biotinylated goat anti-rat IgG (Vector Laboratories) and then incubated with ABC reagent (Vector Laboratories). Visualization was performed using DAB substrate kit (Vector Laboratories). The slides were counterstained with hematoxylin.
Analysis for Ki-67 was done by counting 10 randomly photographed HPFs (×200 magnification) within the cortex by a blinded observer. Tubules, glomeruli, and interstitium were analyzed separately for days 3 and 30. Percentage of positively stained nuclei was related to total nucleus number for each compartment. For Ki-67, a subgroup of animals for days 3 (Terc+/+, n = 7; G1 Terc−/−, n = 6; G4 Terc−/−, n = 6) and 30 (Terc+/+, n = 8; G1 Terc−/−, n = 9; G4 Terc−/−, n = 7) as well as sham-operated controls (Terc+/+, n = 3; G1 Terc−/−, n = 3; G4 Terc−/−, n = 3) and nonoperated controls (Terc+/+, n = 6; G1 Terc−/−, n = 7; G4 Terc−/−, n = 7) were evaluated.
Characterization of infiltrating cells was done by counting all CD3+ and CD68+ cells, respectively, within the corticomedullary zone of each kidney. Depending on kidney size, 10 to 20 HPFs (×200 magnification) were photographed and analyzed by a blinded observer. For each kidney section, the mean number of CD3+ and CD68+ cells was calculated. Cell counts were done in a subgroup of animals for days 3 (Terc+/+, n = 5; G1 Terc−/−, n = 5; G4 Terc−/−, n = 5), 7 (Terc+/+, n = 5; G1 Terc−/−, n = 5; G4 Terc−/−, n = 5), and 30 (Terc+/+, n = 5; G1 Terc−/−, n = 5; G4 Terc−/−, n = 5).
TUNEL Assay
The ApopTag Peroxidase Kit-In situ Apoptosis Detection Kit (Chemicon Europe Ltd., Hofheim, Germany) was used for TUNEL staining. Three-micrometer sections were deparaffinized and rehydrated in ethanol followed by incubation with proteinase K (Sigma-Aldrich, Taufkirchen, Germany). After quenching, equilibration buffer was applied, followed by working-strength enzyme. Cells were regarded as TUNEL-positive when their nuclei were stained brown and displayed typical apoptotic morphology. Ten HPFs at ×200 magnification were evaluated, and the percentage of positively stained nuclei was related to total nuclei number for tubules, glomeruli, and interstitium. For TUNEL, a subgroup of animals for days 3 (Terc+/+, n = 5; G1 Terc−/−, n = 5; G4 Terc−/−, n = 5) and 30 (Terc+/+, n = 7; G1 Terc−/−, n = 7; G4 Terc−/−, n = 7) and sham-operated controls (Terc+/+, n = 3; G1 Terc−/−, n = 3; G4 Terc−/−, n = 3) were evaluated.
Western Immunoblot
Approximately 50 mg of snap-frozen kidney tissue was lysed in 250 μl of ice-cold Triton extraction buffer (1% Triton X 100, 100 mM Tris [pH 7.4], 100 mM Na4P2O3, 100 mM NaF, 10 mM EDTA, and a cocktail of protease inhibitors [complete mini; Roche, Mannheim Germany]) using a tissue lyser (Qiagen, Hilden, Germany). The homogenized samples were agitated for 30 min at 4°C, vortexed, and centrifuged for 30 min at 30,000 × g. The protein content of the supernatants was measured using the Bradford method (Protein Assay Kit; BioRad, Munich, Germany).
Proteins were separated on polyacrylamide gels and blotted onto nitrocellulose membranes. After blocking for 1 h in TBS-T (10 mM Tris [pH 7.4], 138 mM NaCl, and 0.05% Tween-20) containing 3% BSA, blots were incubated with Ctgf primary antibody (1:200; Santa Cruz Biotechnology, Heidelberg, Germany) in 1% BSA at 4°C overnight. Blots were washed three times for 15 min with TBS-T; and incubated with horseradish peroxidase–conjugated anti-goat secondary antibody (1:50,000; Santa Cruz Biotechnology) for 1 h at room temperature, and washed again three times. Immune complexes were detected using enhanced chemiluminescence (ECL; GE Healthcare, Munich, Germany). Blots were exposed to ECL films (GE Healthcare), and protein bands were quantified densitometrically using Image J 1.42 software (National Institutes of Health). Blots were stripped and reprobed with β-actin antibody (Abcam, Cambridge, UK), which served as a loading control.
Western blot analysis was performed only for a subgroup of animals for days 7 (Terc+/+, n = 3; G1 Terc−/−, n = 3; G4 Terc−/−, n = 3) and 30 (Terc+/+, n = 3; G1 Terc−/−, n = 3; G4 Terc−/−, n = 3). Results are given as fold expression on the basis of the expression of two samples from kidneys of Terc+/+ nonoperated controls.
Statistical Analysis
Data were evaluated using the SPSS 16.0 (SPSS Inc., Chicago, IL). Means among treatment groups were compared using ANOVA, and t tests with Bonferroni correction were applied for multiple pair-wise comparisons. Differences with regard to the number of critically shortened telomeres within the groups were analyzed using χ2 test. All data are shown as means ± SEM.
Disclosures
None.
Supplementary Material
Acknowledgments
This study was supported by grants from Roche Organ Transplantation Research Foundation and an Astellas Study and Research Grant of the European Society for Organ Transplantation to A.M. This work was also supported by grants from the German Federal Ministry of Education and Research (reference 01EO0802). J.H.W. was supported by a postdoctoral fellowship from the University of Heidelberg.
We thank M. Überheide and J. Lünig for technical assistance.
Note Added in Proof
Shortly after the online appearance of this article, the authors were made aware of a discrepancy in the number of lanes for the Western blot shown in Figure 2B. The difference came from a 13th lane on the far right of the gel (shown for β-actin and removed for Ctgf), which contained a sample unrelated to the content of the current article and which should have been removed from both blots. The mistake has been corrected. Figure 2B now only shows the 12 lanes that have been analyzed by densitometry, for which the data are presented in Figure 2A. The authors apologize for the error.
Footnotes
The original and corrected versions of this manuscript have been published online ahead of print. Publication dates available at www.jasn.org.
The contents of this article are the sole responsibility of the authors.
See related editorial, “Telomere Shortening and Regenerative Capacity after Acute Kidney Injury,” on pages 202–204.
Supplemental information for this article is available online at http://www.jasn.org/.
References
- 1.Lindeman RD, Goldman R: Anatomic and physiologic age changes in the kidney. Exp Gerontol 21: 379–406, 1986 [DOI] [PubMed] [Google Scholar]
- 2.Goyal VK: Changes with age in the human kidney. Exp Gerontol 17: 321–331, 1982 [DOI] [PubMed] [Google Scholar]
- 3.Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF: Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int 65: 510–520, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Schmitt R, Coca S, Kanbay M, Tinetti ME, Cantley LG, Parikh CR: Recovery of kidney function after acute kidney injury in the elderly: A systematic review and meta-analysis. Am J Kidney Dis 52: 262–271, 2008 [DOI] [PubMed] [Google Scholar]
- 5.US Renal Data System: USRDS 2007 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, Bethesda, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, 2007 [Google Scholar]
- 6.de Fijter JW, Mallat MJ, Doxiadis II, Ringers J, Rosendaal FR, Claas FH, Paul LC: Increased immunogenicity and cause of graft loss of old donor kidneys. J Am Soc Nephrol 12: 1538–1546, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Blasco MA: Telomere length, stem cells and aging. Nat Chem Biol 3: 640–649, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Choudhury AR, Ju Z, Djojosubroto MW, Schienke A, Lechel A, Schaetzlein S, Jiang H, Stepczynska A, Wang C, Buer J, Lee HW, von Zglinicki T, Ganser A, Schirmacher P, Nakauchi H, Rudolph KL: Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat Genet 39: 99–105, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Greider CW, Blackburn EH: Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43: 405–413, 1985 [DOI] [PubMed] [Google Scholar]
- 10.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW: Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Flores I, Benetti R, Blasco MA: Telomerase regulation and stem cell behaviour. Curr Opin Cell Biol 18: 254–260, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Melk A, Ramassar V, Helms LM, Moore R, Rayner D, Solez K, Halloran PF: Telomere shortening in kidneys with age. J Am Soc Nephrol 11: 444–453, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA: Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361: 393–395, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Melk A, Kittikowit W, Sandhu I, Halloran KM, Grimm P, Schmidt BM, Halloran PF: Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int 63: 2134–2143, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA: Essential role of mouse telomerase in highly proliferative organs. Nature 392: 569–574, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA: Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96: 701–712, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Satyanarayana A, Wiemann SU, Buer J, Lauber J, Dittmar KE, Wustefeld T, Blasco MA, Manns MP, Rudolph KL: Telomere shortening impairs organ regeneration by inhibiting cell cycle re-entry of a subpopulation of cells. EMBO J 22: 4003–4013, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melk A: Senescence of renal cells: Molecular basis and clinical implications. Nephrol Dial Transplant 18: 2474–2478, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Melk A, Halloran PF: Cell senescence and its implications for nephrology. J Am Soc Nephrol 12: 385–393, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Melk A, Schmidt BM, Braun H, Vongwiwatana A, Urmson J, Zhu LF, Rayner D, Halloran PF: Effects of donor age and cell senescence on kidney allograft survival. Am J Transplant 9: 114–123, 2009 [DOI] [PubMed] [Google Scholar]
- 21.Schmitt R, Marlier A, Cantley LG: Zag expression during aging suppresses proliferation after kidney injury. J Am Soc Nephrol 19: 2375–2383, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA: Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 287: 1253–1258, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Lechel A, Satyanarayana A, Ju Z, Plentz RR, Schaetzlein S, Rudolph C, Wilkens L, Wiemann SU, Saretzki G, Malek NP, Manns MP, Buer J, Rudolph KL: The cellular level of telomere dysfunction determines induction of senescence or apoptosis in vivo. EMBO Rep 6: 275–281, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hemann MT, Rudolph KL, Strong MA, DePinho RA, Chin L, Greider CW: Telomere dysfunction triggers developmentally regulated germ cell apoptosis. Mol Biol Cell 12: 2023–2030, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim KH, Park GT, Lim YB, Rue SW, Jung JC, Sonn JK, Bae YS, Park JW, Lee YS: Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-beta-mediated signaling pathway. Biochem Biophys Res Commun 318: 819–825, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Campisi J: Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 120: 513–522, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Flores I, Blasco MA: A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS One 4: e4934, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T: DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8: 311–323, 2009 [DOI] [PubMed] [Google Scholar]
- 29.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP: A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Megyesi J, Udvarhelyi N, Safirstein RL, Price PM: The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol 271: F1211–F1216, 1996 [DOI] [PubMed] [Google Scholar]
- 31.Brown JP, Wei W, Sedivy JM: Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277: 831–834, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA: p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538, 1999 [DOI] [PubMed] [Google Scholar]
- 33.Rodier F, Campisi J, Bhaumik D: Two faces of p53: Aging and tumor suppression. Nucleic Acids Res 35: 7475–7484, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collado M, Blasco MA, Serrano M: Cellular senescence in cancer and aging. Cell 130: 223–233, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC: Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A 93: 13742–13747, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro GI, Edwards CD, Ewen ME, Rollins BJ: p16INK4A participates in a G1 arrest checkpoint in response to DNA damage. Mol Cell Biol 18: 378–387, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs JJ, de Lange T: p16INK4a as a second effector of the telomere damage pathway. Cell Cycle 4: 1364–1368, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Cao I, Garcia-Cao M, Tomas-Loba A, Martin-Caballero J, Flores JM, Klatt P, Blasco MA, Serrano M: Increased p53 activity does not accelerate telomere-driven ageing. EMBO Rep 7: 546–552, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, Meyer M, Sadick M, Pill J: Plasma creatinine determination in mice and rats: An enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int 71: 74–78, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Lechel A, Holstege H, Begus Y, Schienke A, Kamino K, Lehmann U, Kubicka S, Schirmacher P, Jonkers J, Rudolph KL: Telomerase deletion limits progression of p53-mutant hepatocellular carcinoma with short telomeres in chronic liver disease. Gastroenterology 132: 1465–1475, 2007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.