

Abstract
During adipocyte differentiation, the expression of C/EBPα is activated, which in turn serves to transcriptionally activate numerous adipocyte genes. A previous search for cis elements that regulate transcription of the C/EBPα gene led to the identification of a potential repressive element within the proximal 5′ flanking region of the gene. Nuclear extracts from 3T3-L1 preadipocytes, but not adipocytes, were found to contain a factor, CUP (C/EBPα undifferentiated protein), that binds to this site (the CUP-1 site). In the present investigation, we show that C/EBPα promoter-luciferase constructs containing both the proximal 5′ flanking and the entire 5′ untranslated regions of the gene exhibit an expression pattern during adipocyte differentiation comparable to that of the endogenous C/EBPα gene. Mutation of the CUP-1 site in these constructs had little effect on reporter gene expression; however, when this mutation was combined with deletion of the 5′ untranslated region, reporter gene expression by preadipocytes was dramatically up-regulated. Consistent with this finding, a second CUP binding site (the CUP-2 site) was identified in the 5′ untranslated region. Although mutation of either CUP element in constructs containing both the 5′ flanking and 5′ untranslated region had little effect on reporter gene transcription, mutation of both CUP elements markedly activated transcription. Thus, it appears that dual CUP regulatory elements repress transcription of the C/EBPα gene prior to induction of the adipocyte differentiation program.
Keywords: 3T3-L1 cells, preadipocyte, adipocyte, differentiation adipose, C, EBPα undifferentiated protein
A large body of evidence has implicated C/EBPα as a pleiotropic transcriptional activator of adipocyte genes during adipogenesis (1–3). The C/EBPα gene is expressed just prior to the coordinate expression of numerous adipocyte genes (4–8). Moreover, the proximal promoters of many of these genes have been shown to possess C/EBP binding sites that mediate transactivation by C/EBPα (4–8). Consistent with these findings, ectopic expression of C/EBPα, either under the control of an inducible Lac-switch promoter (9) or by retroviral transduction (10), is sufficient to activate differentiation of 3T3-L1 preadipocytes into adipocytes without the exogenous hormonal stimulation usually required. Conversely, the expression of antisense C/EBPα RNA in 3T3-L1 preadipocytes blocks the adipocyte differentiation program (11, 12). In support of these findings C/EBPα knockout mice fail to accumulate adipose triacylglycerol (13), the hallmark of this tissue.
Once it became evident that C/EBPα plays a key role in adipogenesis, studies were initiated to identify cis regulatory elements (and their cognate transacting factors) in the C/EBPα gene promoter with the goal of gaining insight into events that occur earlier in the differentiation program. DNase I footprinting of the proximal promoter of the mouse C/EBPα gene identified several binding sites for transacting factors that are differentially regulated during adipogenesis. These included a C/EBP binding site and a potential repressor binding site (14, 15). Several lines of evidence suggest that the C/EBP binding site mediates activation of transcription by C/EBPβ and C/EBPδ (2) early in the differentiation program and autoactivation of transcription by C/EBPα as cells enter the terminally differentiated state (16). In the 5′ flanking region about 40 bp 5′ of the C/EBP site is a DNA sequence that is footprinted by a nuclear factor, referred to as CUP (C/EBP undifferentiated protein), that is expressed by preadipocytes but not by adipocytes (14, 17). During adipocyte differentiation, CUP binding activity decreases concomitant with the activation of transcription of the C/EBPα gene (17), suggesting that CUP might act as a repressor of the gene. As shown in this report, however, mutation of this site (referred to as the CUP-1 site) in promoter-reporter constructs containing both the proximal 5′ flanking and 5′ untranslated regions (UTRs) of the gene had little effect on reporter gene expression. These findings prompted a search for and the discovery of an additional CUP binding site (referred to as the CUP-2 site) in the 5′-UTR of the gene that, when mutated, also has little effect on promoter activity. However, when both CUP sites are mutated together, promoter activity is dramatically activated or “derepressed.”
EXPERIMENTAL PROCEDURES
Cell Culture and Induction of Differentiation.
The 3T3-L1 preadipocytes were maintained and propagated in DMEM containing 10% (vol/vol) calf serum. Two-day postconfluent (designated day 0) cells were induced to differentiate (18) with DMEM containing 10% (vol/vol) fetal bovine serum (FBS), 1 μg of insulin per ml, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methyl-xanthine until day 2. Cells were then fed DMEM supplemented with 10% FBS and 1 μg of insulin per ml for 2 days, after which they were fed every other day with DMEM containing 10% FBS. Expression of adipocyte genes and acquisition of the adipocyte phenotype begins on day 3 and is maximal by day 8.
Gene Constructs and Mutations.
A 1,575-bp 5′ segment of the C/EBPα gene (from nucleotides −1450 to +125) containing 1,450 bp of 5′ flanking sequence and the entire (125 bp) 5′-UTR was excised from pC/EBPα9.7 (14), with BamHI and NcoI, and cloned into the BglII and NcoI sites in the pGL3-BA luciferase expression vector (Promega) giving rise to p1575. A 468-bp 5′ segment of the C/EBPα gene (from nucleotides −343 to +125) containing 343 bp of 5′ flanking sequence and the entire 5′-UTR was excised from pC/EBPα9.7 (14), with SmaI and NcoI, and inserted into the same sites of pGL3-BA luciferase expression vector giving rise to p468. Constructs p1455 and p348, lacking the 5′-UTR, were prepared by cutting p1575 and p468, respectively, with NruI and NcoI followed by filling in and religation. Two single and one double CUP-site mutant constructs, i.e., p468M1, p468M2, and p468M1/M2, respectively, were generated by the PCR method as described (19); the core CUP-1 site was mutated from GCCGCCG to GGATCCG and the core CUP-2 site was mutated from GCCGCCG to GAATTCG (where the mutated bases are underlined). The authenticity of the mutations was verified by sequencing.
Transfections and Reporter Gene Analysis.
Proliferating (70% confluent) 3T3-L1 preadipocytes or day 5 differentiated 3T3-L1 adipocytes were transiently transfected with 2 μg of promoter-reporter constructs along with 8 μg of carrier DNA by using the calcium phosphate precipitation method (8). After 48 hours in culture, cell extracts were prepared and assayed for luciferase activity. To produce cell lines harboring the wild-type (wt) or 5′-UTR-deleted constructs, 3T3-L1 preadipocytes were cotransfected with 10 μg of p1575, p468, p1455, or p348 along with 3 μg of SV40Neo as above by using the calcium phosphate precipitation method. Five G418-resistant cell lines harboring each 5′ flanking region/5′-UTR-driven luciferase construct were selected and subjected to the differentiation protocol. At various times after induction of differentiation, cell lysates were prepared and assayed for luciferase activity. To assess the effect of insulin on reporter gene expression mediated by the 5′ flanking region/5′-UTR of the C/EBPα gene, day 9 fully differentiated adipocytes harboring the p1575 and p468 constructs were switched to medium containing insulin after which lysates for luciferase assays were prepared at various times over the next 24 h.
Western Blotting and Electrophoretic Mobility Shift Assays (EMSA).
Nuclei were isolated from 3T3-L1 cells and nuclear extracts were prepared by using 1× NUN solution (20) containing 0.3M NaCl, 1M urea, 1% Nonidet P-40, 25 mM Hepes (pH 7.9), and 1 mM DTT. Protein concentration was determined by using the Bradford protein assay (Bio-Rad). Equal amounts of nuclear protein were subjected to Western blot analysis with antibody directed against a peptide corresponding to the C-terminal amino acid sequence of C/EBPα as described (11).
EMSA analysis was performed essentially as described (21, 22) with the following modifications. Reaction mixtures containing 0.25 ng of 32P-labeled oligonucleotide, 1 μg of poly(dA-dC⋅dG-dT), and 10 μg of nuclear extract protein in 30 μl of buffer (10 mM Hepes/0.1 mM EDTA/5% glycerol/100 mM NaCl/0.3 M urea/0.3% Nonidet P-40) were incubated at 4°C for 15 min and then were separated in TBE/5% polyacrylamide gels [30:1 acrylamide/N,N′-methylenebisacrylamide in 0.5× TBE (44.5 mM Tris/44.5 mM boric acid/1 mM EDTA at pH 8.3)] by electrophoresis in 0.5× TBE buffer. For competition experiments, a 50-fold excess of unlabeled competitor oligonucleotide was added to the reaction mixtures prior to addition of labeled probe. The wt CUP-1, CUP-2, and mutated CUP-1 site oligonucleotides were double-stranded and included the following sequences: CUP-1 site, GATC/CAGCGCCGCCGGGG/; CUP-2 site, GATC/GGAGGCCGCCGAGG/; and CUP-1* (mutated CUP-1 site), GATC/CAGCGTTTAAAGGG/(where a slash indicates the delimiting linker sequence).
RESULTS
Differentiation-Induced Up-Regulation and Insulin-Induced Down-Regulation of Reporter Gene Expression Mediated by the 5′ Flanking and 5′-UTRs of the C/EBPα Gene.
Before assessing the effect of mutating the putative CUP repressive binding site in the proximal 5′ flanking region of the C/EBPα gene (14, 17), it was necessary to verify that the wt promoter of the gene could mediate differentiation-induced reporter gene expression. Previous attempts to demonstrate differentiation-dependent expression of C/EBPα promoter-reporter constructs with kinetics comparable to those of the endogenous C/EBPα gene had been unsuccessful. In those studies, constructs contained up to 6,000 bp of 5′ flanking sequence but lacked the complete 5′-UTR, and the kinetics of reporter gene expression did not parallel expression of the endogenous C/EBPα gene. Rather, high levels of reporter gene expression occurred in preadipocytes and fell to lower levels in adipocytes (ref. 23 and R. J. Christy, O. A. MacDougald, Q.-Q.T., and M.D.L., unpublished results).
Recently, however, we discovered that C/EBPα promoter-reporter constructs, containing the proximal 1,450-bp or 343-bp 5′ flanking region and the entire 125-bp 5′-UTR, exhibited a pattern of expression closely approximating that of the endogenous gene. As shown in Fig. 1A, reporter gene expression mediated by these 5′ flanking sequences in combination with the 5′-UTR increased markedly during the course of differentiation. Moreover, the kinetics of differentiation-induced reporter gene expression were similar to expression of the 42- and 30-kDa isoforms of C/EBPα (Fig. 1B) and were inversely correlated to the expression of CUP binding activity as measured by EMSA (Fig. 1C). These results are consistent with our previous findings (14) that showed that the differentiation-induced increase in expression of C/EBPα mRNA is the result of transcriptional activation (and/or derepression) of the C/EBPα gene. Thus, it appeared that a regulatory sequence(s) in the 5′-UTR of the C/EBPα gene is required to maintain the promoter in the inactive, possibly repressed, state prior to differentiation.
Figure 1.
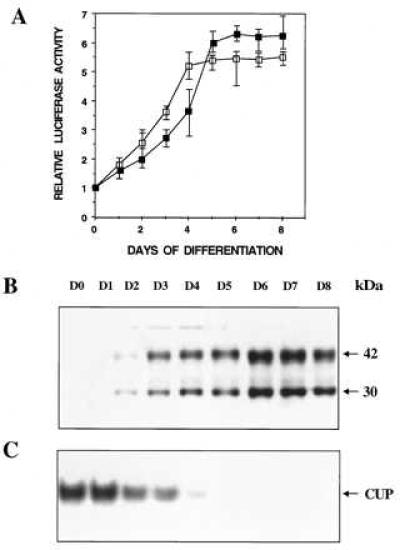
Comparison during adipocyte differentiation of reporter gene expression mediated by the 5′ flanking and 5′-UTRs of the C/EBPα gene (A), expression of endogenous C/EBPα (B), and CUP binding activity (C). A representative 3T3-L1 preadipocyte line harboring either a 1,575-bp (▪) or a 468-bp (□) 5′ flanking/5′-UTR-luciferase construct was subjected to the differentiation protocol. In A, luciferase assays were conducted on cell lysates at the times indicated. In B and C, control 3T3-L1 preadipocytes were subjected to the differentiation protocol and nuclear extracts prepared (at the times indicated) and subjected to either SDS/PAGE and Western blotting with antibody directed against a peptide corresponding to the C-terminal amino acid sequence of C/EBPα or EMSA using a 32P-labeled probe corresponding to the nucleotide sequence of the CUP-1 binding site (C). With all cell lines, cytoplasmic triacylglycerol began to accumulate on day 3 after induction of differentiation and the levels accumulated by day 8 were similar.
We also sought to determine whether cis elements, located within the proximal 5′ flanking/5′-UTR, are responsible for other aspects of regulation of the C/EBPα gene. For example, MacDougald et al. showed that insulin (24) and glucocorticoid (25) down-regulate transcription of the C/EBPα gene in fully differentiated 3T3-L1 adipocytes. As illustrated in Fig. 2, insulin treatment of 3T3-L1 adipocytes, stably transfected with the 1,575- or 468-bp C/EBPα 5′ flanking region/5′-UTR-luciferase constructs, rapidly (t½ ∼ 7 h) down-regulated reporter gene expression. Moreover, the down-regulation process was reversed within 6–8 hours upon withdrawal of insulin. Similarly, glucocorticoid, which down-regulates transcription of the C/EBPα gene (25), also down-regulates reporter gene expression mediated by the 1,575- or 468-bp promoter/UTR (results not shown). Thus, these findings showed that 468 bp of C/EBPα promoter are sufficient to mediate differentiation-induced up-regulation and insulin- and glucocorticoid-induced down-regulation of reporter gene expression.
Figure 2.

Insulin-induced down-regulation of reporter gene expression mediated by the 5′ flanking and 5′-UTRs of the C/EBPα gene. Representative 3T3-L1 cell lines harboring either a 1,575-bp (□) or a 468-bp (▪) 5′ flanking/5′-UTR-luciferase construct were induced to differentiate as in Fig. 1. On day 9, differentiated adipocytes were exposed to 1 μg of insulin per ml, cell lysates were prepared, and luciferase assays were conducted at the times indicated. At 24 hours, other cell monolayers were washed once and then incubated with insulin-free medium. At the times indicated, cell lysates were prepared and luciferase assays were conducted.
Apparent Repressive Role of the 5′-UTR of the C/EBPα Gene.
Initially, experiments were conducted to determine whether the CUP site (CUP-1 site; Fig. 3) in the 5′ flanking region of the C/EBPα gene that is footprinted by preadipocyte (14) but not adipocyte nuclear extract functions as a repressor binding site in preadipocytes. Therefore, the CUP-1 sites in both the 1,450- or 343-bp proximal 5′ flanking segments were mutated (from GCCGCCG to GGATCCG, where mutated bases are underlined) and these segments were inserted into a luciferase expression vector, either with or without the full-length 5′-UTR. EMSA analysis verified that the preadipocyte nuclear factor does not bind to an oligonucleotide corresponding to the mutated site (data not shown). The mutant and wt luciferase constructs were then transiently transfected into 3T3-L1 preadipocytes and after 48 hours luciferase assays were conducted. As shown in Fig. 4, mutation of the CUP-1 site caused only a small increase of reporter gene expression in constructs containing the 5′-UTR. However, mutation of the CUP-1 site in constructs lacking the 5′-UTR caused a marked, i.e., 8- to 10-fold, increase in reporter gene expression.
Figure 3.

Location of CUP and C/EBP binding sites in the 5′ flanking and 5′-UTRs of the C/EBPα gene. The transcriptional start site is indicated by a bent arrow and the translational start site by an ATG in the upper left of the boxed intronless coding region of the gene. The numbers refer to the number of nucleotides 5′ and 3′ (− and +, respectively) from the transcriptional start site. The open boxes in the 5′ flanking region and 5′-UTR represent CUP binding sites and the darkened box represents the C/EBP binding site. BamHI, SmaI, NruI, and NcoI indicate restriction enzyme cleavage sites.
Figure 4.

Effect of deleting the 5′-UTR and mutating the CUP-1 binding site on reporter gene expression mediated by C/EBPα promoter/5′-UTR-reporter gene constructs. Duplicate monolayers of 3T3-L1 preadipocytes were transiently transfected with luciferase reporter gene constructs containing both the 5′ flanking region and the entire 5′-UTR (nucleotides −343 to +125 or nucleotides −1,450 to +125) or only the 5′ flanking region lacking the 5′-UTR (nucleotides −343 to +5 or nucleotides −1,450 to +5) without (wt) or with a mutated CUP-1 site (Mut). Luciferase assays were conducted on cell lysates 48 hours after transfection. The results shown are representative of three experiments.
These results and the fact that both the proximal 5′ flanking and 5′-UTR were required to obtain a pattern of differentiation-induced reporter gene expression comparable to that of the endogenous C/EBPα gene, i.e., repressed expression in the preadipocyte and increased expression in the adipocyte (Fig. 1 A and B), suggested that a repressor element might be located in the 5′-UTR. Close inspection of the nucleotide sequence of the 5′-UTR revealed a potential CUP regulatory element located between nucleotides +83 and +96 (designated the CUP-2 site; see Fig. 3) that possesses a core sequence, GCCGCCG, identical to that of the upstream CUP binding site (the CUP-1 site) located between nucleotides −252 and −246. EMSA was performed with oligonucleotide probes corresponding to the CUP-1 and CUP-2 sites to ascertain whether the CUP-2 sequence constitutes a functional binding site. As shown in Fig. 5, preadipocyte nuclear extract (lanes 2 and 6), but not adipocyte nuclear extract (lanes 1 and 5), gave rise to protein–oligonucleotide complexes of similar mobility with either a CUP-1 or a CUP-2 probe. In both cases, binding was specific because most of the CUP–oligonucleotide probe complexes were competed away by the corresponding unlabeled CUP oligonucleotide but not by a mutated CUP-1 oligonucleotide. These findings and the fact that their core nucleotide sequences are identical suggested that the same (or similar) preadipocyte nuclear factor(s) interacts with both CUP sites. Competitive EMSA experiments showed that the CUP protein(s) binds more tightly to the CUP-1 binding site than to the CUP-2 binding site (results not shown). This difference in affinity may be due to differences in nucleotide sequence in the region adjacent to the 7-bp core sequence.
Figure 5.

EMSAs of CUP-1 and CUP-2 binding site probes with preadipocyte and adipocyte nuclear extracts. Nuclear extracts from 3T3-L1 adipocytes (lanes A) or preadipocytes (lanes P) were incubated either with a 32P-labeled CUP-1 site or CUP-2 site oligonucleotide in the absence or presence of unlabeled competitor CUP-1, CUP-2, or mutated CUP-1* site oligonucleotides. After incubation, reaction mixtures were subjected to TBE/polyacrylamide gel electrophoresis and radioautography.
Effect of Mutating the CUP-1 and CUP-2 Binding Sites on Promoter Activity.
To determine whether both CUP binding sites in the C/EBPα gene repress reporter gene transcription, the effects of mutating these sites (individually or in combination) on luciferase reporter gene expression were compared. That mutation of these sites prevents CUP binding was verified by EMSA (Fig. 5, lanes 4 and 9). The wt and mutated 468-bp C/EBPα 5′ flanking region/5′-UTR-luciferase constructs were transiently transfected into 3T3-L1 preadipocytes or adipocytes and luciferase assays conducted on cell lysates 48 hours later. As shown in Fig. 6A, mutation of either CUP site alone had little effect on reporter gene expression. However, mutation of both CUP sites produced a marked activation (or derepression) of reporter gene expression in 3T3-L1 preadipocytes. In contrast, mutation of both CUP sites had no effect on reporter gene expression when the same constructs were transfected into fully differentiated 3T3-L1 adipocytes (Fig. 6B). These findings indicate that repression of transcription of the C/EBPα gene by CUP in preadipocytes is mediated by dual repressor elements, CUP-1 and CUP-2, located in the 5′ flanking region and the 5′-UTR of the gene, respectively. The fact that the CUP-1 and CUP-2 site mutations have no effect on reporter gene expression in adipocytes is consistent with the observed loss of CUP binding activity (probably due to a decrease in CUP expression) during adipocyte differentiation (Fig. 1C and ref. 17).
Figure 6.
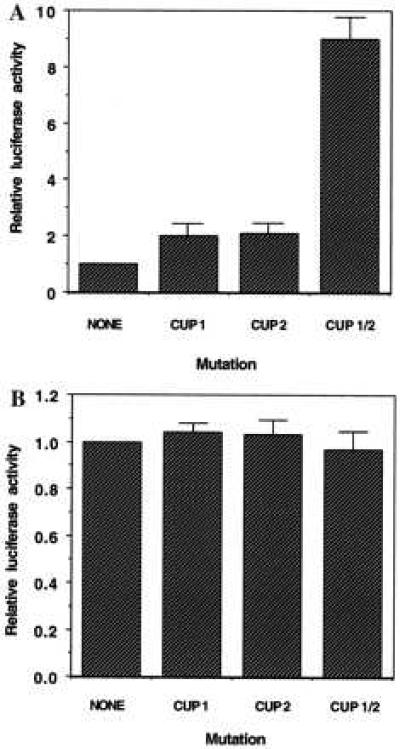
Effect of mutations in the CUP-1 and CUP-2 binding sites on reporter gene expression mediated by C/EBPα 5′ flanking region/5′-UTR-luciferase constructs in 3T3-L1 preadipocytes (A) and 3T3-L1 adipocytes (B). Duplicate monolayers of 3T3-L1 preadipocytes (A) or adipocytes (B) were transiently transfected with the 468-bp C/EBPα 5′-flanking/5′-UTR-luciferase construct without (wt) or with mutations in one or both CUP-1 and CUP-2 binding sites. Luciferase assays were conducted on cell lysates 48 hours after transfection. The results shown are representative of five experiments.
DISCUSSION
Previous investigations in this laboratory (14, 17) identified a factor, CUP, present in nuclei of preadipocytes but not adipocytes, that binds to a site in the proximal 5′ flanking region of the C/EBPα gene. It was suggested that CUP might act as a repressor of the gene in preadipocytes prior to differentiation and be responsible, at least in part, for activation (via derepression) of the gene when CUP binding activity decreased with the onset of differentiation (17). Because C/EBPα is a plieotropic transactivator of adipocyte genes (1, 3), a differentially expressed repressor acting on the C/EBPα gene could play an important role in the differentiation program. Although CUP had been partially purified and characterized (17), difficulties were encountered in our initial attempts to assign function to the CUP binding site in the C/EBPα promoter. Cell lines harboring C/EBPα promoter-reporter constructs that lack the 5′-UTR failed to exhibit an expression pattern (during differentiation) like that of the endogenous C/EBPα gene, thus precluding a definitive test of mutations in the CUP binding site. Only when it was discovered that there is a second CUP binding site in the 5′-UTR of the gene was it possible to obtain an expression pattern with C/EBPα promoter-reporter constructs that mimic that of the endogenous gene (Fig. 1A).
The present report shows that CUP, in addition to binding at the CUP-1 site in the 5′ flanking region of the C/EBPα gene, binds at a similar site in the 5′-UTR. Importantly, only when both CUP sites are mutated in promoter-reporter gene constructs does “derepression” of the reporter gene expression occur (Fig. 6A). Thus, it appears that both sites function together to ensure that the C/EBPα gene is strongly repressed prior to differentiation.
It is of interest and possibly important that the two CUP elements are located on opposite sides of the C/EBP binding site and the transcriptional start site. If, as has been shown for many other transcription factors, CUP exists as a dimer, it is possible that in the preadipocyte CUP might bind to both sites simultaneously and cause looping of this segment of DNA. This action could render the C/EBP binding and the transcriptional start sites inaccessible to factors that activate and/or initiate transcription and, thereby, block expression of the gene. To address this and other mechanistic questions concerning the action of CUP and to determine whether the constitutive expression of CUP can derail the differentiation process, it will be necessary to clone and express the gene encoding this nuclear factor. Work is in progress to clone the CUP cDNA and to express and characterize the protein.
Acknowledgments
We thank Dr. Tom Loftus for critically reviewing this paper. This work was supported by a research grant (DK-38418) from the National Institutes of Health (National Institute of Diabetes and Digestive and Kidney Diseases).
ABBREVIATIONS
- CUP
C/EBPα undifferentiated protein
- EMSA
electrophoretic mobility shift assay
- wt
wild type
References
- 1.MacDougald O A, Lane M D. Annu Rev Biochem. 1995;64:345–373. doi: 10.1146/annurev.bi.64.070195.002021. [DOI] [PubMed] [Google Scholar]
- 2.Mandrup S, Lane M D. J Biol Chem. 1997;272:5367–5370. doi: 10.1074/jbc.272.9.5367. [DOI] [PubMed] [Google Scholar]
- 3.Hwang, C.-S., Loftus, T. M., Mandrup, S. & Lane, M. D. (1997). Annu. Rev. Cell Biol. Dev., in press. [DOI] [PubMed]
- 4.Christy R J, Yang V W, Ntambi J M, Geiman D E, Landschulz W H, Friedman A D, Nakabeppu Y, Kelly T J, Jr, Lane M D. Genes Dev. 1989;3:1323–1335. doi: 10.1101/gad.3.9.1323. [DOI] [PubMed] [Google Scholar]
- 5.Kaestner K H, Christy R J, Lane M D. Proc Natl Acad Sci USA. 1990;87:251–255. doi: 10.1073/pnas.87.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrera R, Ro H S, Robinson G S, Xanthopoulos K G, Spiegelman B M. Mol Cell Biol. 1989;9:5331–5339. doi: 10.1128/mcb.9.12.5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheneval D, Christy R J, Geiman D E, Lane M D. Proc Natl Acad Sci USA. 1991;88:8465–8469. doi: 10.1073/pnas.88.19.8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang C-S, Mandrup S, MacDougald O A, Geiman D E, Lane M D. Proc Natl Acad Sci USA. 1996;93:873–877. doi: 10.1073/pnas.93.2.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin F-T, Lane M D. Proc Natl Acad Sci USA. 1994;91:8757–8761. doi: 10.1073/pnas.91.19.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fregtag S O, Paielli D L, Gillbert J D. Genes Dev. 1994;8:1654–1663. doi: 10.1101/gad.8.14.1654. [DOI] [PubMed] [Google Scholar]
- 11.Lin F-T, Lane M D. Genes Dev. 1992;6:533–544. doi: 10.1101/gad.6.4.533. [DOI] [PubMed] [Google Scholar]
- 12.Samuelsson L, Stromberg K, Vikman K, Bjursell G, Enerback S. EMBO J. 1991;10:3787–3793. doi: 10.1002/j.1460-2075.1991.tb04948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang N-D, Finegold M J, Bradly A, Ou C N, Abdelsayed S V, Wilde M D, Taylor L R, Wilson D R, Darlington G J. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 14.Christy R J, Kaestner K H, Geiman D E, Lane M D. Proc Natl Acad Sci USA. 1991;88:2593–2597. doi: 10.1073/pnas.88.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legraverend C, Antonson P, Flodby P, Xanthopoulos K G. Nucleic Acids Res. 1993;21:1735–1742. doi: 10.1093/nar/21.8.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin F-T, MacDougald O A, Diehl A M, Lane M D. Proc Natl Acad Sci USA. 1993;90:9606–9610. doi: 10.1073/pnas.90.20.9606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasseur-Cognet M, Lane M D. Proc Natl Acad Sci USA. 1993;90:7312–7316. doi: 10.1073/pnas.90.15.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Student A K, Hsu R Y, Lane M D. J Biol Chem. 1980;255:4745–4750. [PubMed] [Google Scholar]
- 19.Saiki R K, Gelfand D H, Stoffel S, Scharf S J, Higuchi R, Horn G T, Mullis K B, Erlich H A. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 20.Lavery D J, Schibler U. Genes Dev. 1993;7:1871–1884. doi: 10.1101/gad.7.10.1871. [DOI] [PubMed] [Google Scholar]
- 21.Raymondjean M, Cereghini S, Yaniv M. Proc Natl Acad Sci USA. 1988;85:757–761. doi: 10.1073/pnas.85.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fried M, Crothers D M. Nucleic Acids Res. 1981;9:6506–6525. doi: 10.1093/nar/9.23.6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xanthopoulos K G, Cannon P D, Robinson G S, Mirkovith J, Darnell J E., Jr Eur J Biochem. 1992;208:501–509. doi: 10.1111/j.1432-1033.1992.tb17214.x. [DOI] [PubMed] [Google Scholar]
- 24.MacDougald O A, Cornelius P, Liu R, Lane M D. J Biol Chem. 1995;270:647–654. doi: 10.1074/jbc.270.2.647. [DOI] [PubMed] [Google Scholar]
- 25.MacDougald O A, Cornelius P, Lin F-T, Chen S S, Lane M D. J Biol Chem. 1994;269:19041–19047. [PubMed] [Google Scholar]