

Abstract
Bacteria of the genus Shewanella can thrive in different environments and demonstrate significant variability in their metabolic and ecophysiological capabilities including cold and salt tolerance. Genomic characteristics underlying this variability across species are largely unknown. In this study, we address the problem by a comparison of the physiological, metabolic, and genomic characteristics of 19 sequenced Shewanella species. We have employed two novel approaches based on association of a phenotypic trait with the number of the trait-specific protein families (Pfam domains) and on the conservation of synteny (order in the genome) of the trait-related genes. Our first approach is top-down and involves experimental evaluation and quantification of the species’ cold tolerance followed by identification of the correlated Pfam domains and genes with a conserved synteny. The second, a bottom-up approach, predicts novel phenotypes of the species by calculating profiles of each Pfam domain among their genomes and following pair-wise correlation of the profiles and their network clustering. Using the first approach, we find a link between cold and salt tolerance of the species and the presence in the genome of a Na+/H+ antiporter gene cluster. Other cold-tolerance-related genes include peptidases, chemotaxis sensory transducer proteins, a cysteine exporter, and helicases. Using the bottom-up approach, we found several novel phenotypes in the newly sequenced Shewanella species, including degradation of aromatic compounds by an aerobic hybrid pathway in Shewanella woodyi, degradation of ethanolamine by Shewanella benthica, and propanediol degradation by Shewanella putrefaciens CN32 and Shewanella sp. W3-18-1.
Electronic supplementary material
The online version of this article (doi:10.1007/s10142-009-0142-y) contains supplementary material, which is available to authorized users.
Keywords: Phenotypic trait, Bacteria, Molecular mechanisms of cold tolerance, Shewanella, Protein families
Introduction
Shewanella species are known for their ability to utilize a diverse set of electron acceptors for anaerobic respiration. This capability makes the bacterium important for remediation of environments contaminated with nitroaromatic compounds chromium, plutonium, uranium, and other radionuclides (Gralnick and Hau 2006; Fredrickson et al. 2008). Members of the genus typically inhabit chemically stratified sedimentary and aquatic systems with differences in available sources of nutrients and electron acceptors as well as in physical characteristics such as temperature, salinity, pH, and pressure (Gralnick and Hau 2006). To live in these diverse environments and to compete with other microorganisms, the Shewanellae have evolved different physiological and metabolic characteristics that are fine-tuned to their environmental niches. At present, 19 Shewanella strains from diverse environments have been sequenced providing a unique opportunity for comprehensive comparative analyses of their genomes and for revealing genomic determinants of the various phenotypes of the species. Objectives of the study were (1) to link complex ecophysiological traits of Shewanella species, such as cold and salt tolerance, to specific genes and molecular mechanisms, and (2) to reveal phenotypic and metabolic capabilities of the newly sequenced species from their genomic characteristics.
Current computational technologies are limited in their ability to associate a specific phenotypic characteristic of an organism with its genomic signature in a comprehensive manner. Traditional linkage analysis (Lander and Kruglyak 1995) allows mapping of phenotypic differences between individuals to their genotypic differences but the analysis was applied to eukaryotes and investigated intra-individual genetic differences within the same species. Other known approaches include phylogenetic profiling based on correlations between the presence/absence of phenotypes and the presence/absence of genomic characteristics (e.g., specific enzymes, Pfam domains, clusters of orthologous groups of proteins (COGs), or orthologous genes) in a set of species (Venkateswaran et al. 1999; Liu et al. 2006; Ranea et al. 2007; Date and Peregrin-Alvarez 2008). More flexible, although less-specific, strategies are phylogenetic pattern analysis (Makarova et al. 2003) and natural language processing approaches (Korbel et al. 2005), methods used to find associations between COGs and phenotypes. To our knowledge, none of the current computational methods that address genotype and phenotype associations compare genomes in terms of synteny (a genomic locus with a conserved order of similar genes) at the protein family level calculating the number of each protein family instead of its binary characteristic (present/absent) and correlating this number with a quantitative outcome of a complex phenotype. Feasibility of the approach stems from knowledge about molecular mechanisms involved in development of new phenotypic traits in living organisms (Fig. 1S). It is believed that the enormous interspecies phenotypic variation in both eukaryotes and prokaryotes is caused by elaboration on the existing gene structures rather than by de novo invention of genes. For example, combining existing domains in novel gene architectures, also known as domain shuffling, has been estimated to have involved up to 20% of eukaryotic genes (Long et al. 1995). In addition to domain shuffling, new gene structures may be created by duplication of a gene with subsequent accumulation of mutations (Long et al. 2003) or by acquisition of genes by horizontal transfer, a well-known mechanism in prokaryotes (Gogarten and Townsend 2005). Duplication of protein domains, if they are important for a particular phenotypic trait, in combination with deletion of domains, if they are not needed, leads to an enrichment in the genome of domains specific for phenotypic traits and to a conserved domain synteny in the microorganisms implementing the trait (Apic et al. 2001; Methe et al. 2003; Madera et al. 2004; Orengo and Thornton 2005).
Based on the genomic mechanisms described above, we propose to link genotype and phenotype of the Shewanella species using (1) correlation between the number of trait-specific protein domains as defined by the Pfam database (also referred as domain enrichment) and a quantitative characteristic of the phenotypic trait and (2) conserved synteny of the trait-specific genes (Fig. 1). The first, a top-down approach, involves quantification and comparison of a higher-level characteristic, the bacterial cold tolerance, to reveal genomic characteristics and specific genes related to the phenotype (Fig. 1a). The second, a bottom-up approach, predicts novel phenotypes of the Shewanella species by quantification of molecular functions in their genomes and by following pair-wise comparison of enriched molecular functions across the species (Fig. 1b). The approach is implemented using network analysis tools. Using the top-down approach, we are able to identify specific genomic clusters related to cold tolerance of the Shewanella species, to predict novel cellular mechanisms underlying the bacterial cold tolerance, and to link this trait to salt tolerance of the species. By using the second approach, we reveal novel, environmentally important phenotypes in the species.
Fig. 1.
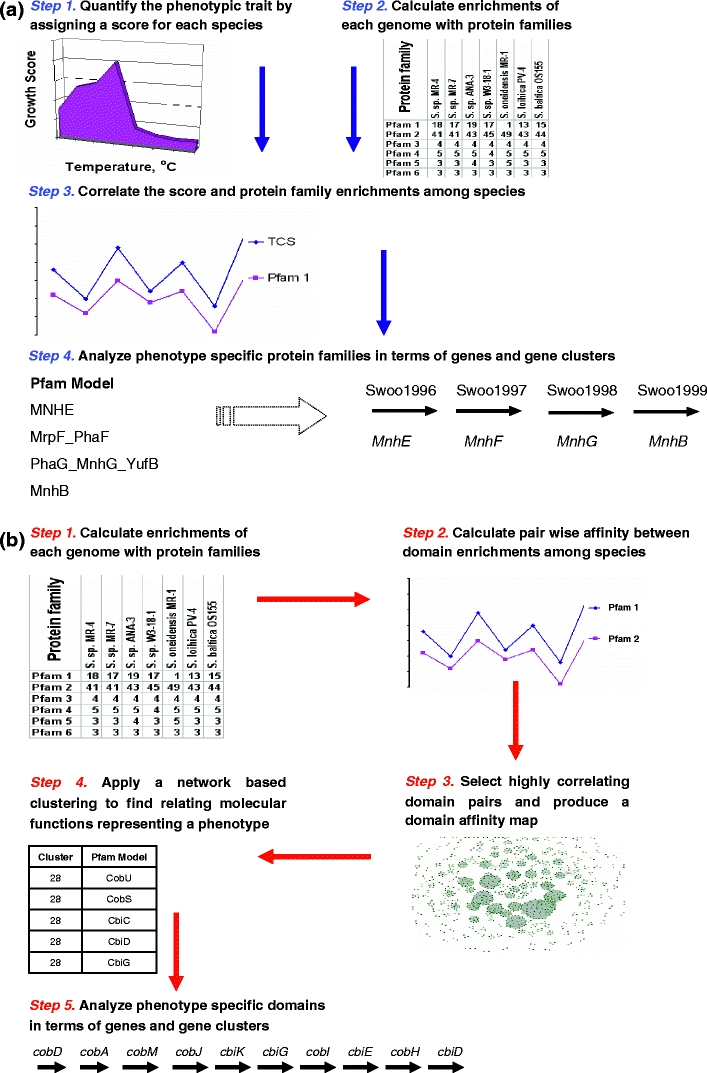
Top-down (a) and bottom-up (b) computational frameworks for relating phenotypic and genomic characteristics of the Shewanella species based on quantification of molecular functions in the bacterial genomes
Materials and methods
Bacterial strains and culture conditions for testing Shewanella species cold and salt tolerance
The following 15 Shewanella strains were used in testing of the phenotypic traits: Shewanella oneidensis MR-1; Shewanella amazonensis SB2B; Shewanella putrefaciens CN-32; Shewanella sp. PV-4; Shewanella denitrificans OS217; Shewanella baltica OS155; Shewanella baltica OS195; Shewanella woodyi ATCC 51908; Shewanella pealeana ATCC 700345; Shewanella putrefaciens 200; Shewanella sp. ANA-3; Shewanella sp. MR-4; Shewanella sp. MR-7; Shewanella sp. W3-18-1; and Shewanella frigidimarina NCIMB400.
Cells were grown aerobically on Shewanella Federation (SF) medium (pH 7.0) containing 3 mM PIPES, 2.8 mM NH4Cl, 0.4 mM NaH2PO4, 3 mM NaCl, 0.1 mM KCl, 1 μM Na2SeO4, 10 μM l-arginine hydrochloride, 13 μM l-glutamate, 19 μM l-serine, and 10 ml each of 100× Wolfe’s vitamin and 100× mineral solutions prepared as described previously (Kieft et al. 1999) (Supplementary Protocol). Then, 10 mM of lactate was added as the carbon and energy source. Additional sodium chloride (2.5%) was added to the medium of S. woodyi. For S. denitrificans OS217, experiments were conducted with Luria–Bertani (LB) broth because of its inability to grow on SF medium supplemented with lactate. Starter cultures were initiated by overnight growth in 5 ml of LB or Marine broth 2216 (S. pealeana and S. woodyii only) on a VWR shaker (speed = 125 rpm) at 22°C. Cells were collected by centrifugation at 4,000 rpm for 10 min, washed twice in SF medium lacking lactate, and resuspended in 1 ml of SF medium to inoculate 4 ml SF medium in 10-ml tubes. Temperatures tested ranged from 0 to 55°C (4, 12, 18, 24, 30, 37, 42, and 55°C), and the salinities tested were 0, 0.5%, 1.0%, 2.5%, 5.0%, and 10% NaCl. At least two cultures of each species were grown at each temperature and salinity tested (Supplementary Tables 1S and 5S). Growth was monitored by measuring absorbance at 600 nm with a visible light spectrophotometer (Spec 20). Optical density (OD660) readings were recorded throughout the growth period.
Quantification of phenotypic traits
In order to capture the most distinct features of the phenotypes and diminish the effect of the culturing conditions, the traits were scored. The effect of temperature on growth was characterized by the cold tolerance score (CTS), and the effect of salinity on growth was estimated by the salt tolerance score (STS).
To calculate the cold tolerance score we first determined the Growth Scores (GS) for each characterized temperature (T
j) from the growth profiles, namely, values of OD660 (ODi) against time points (t
i). GS values were calculated as the area under the growth profile normalized to 100 h of the growth: 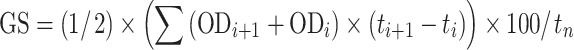 where i=0,…, n−1, n is the number of time points, and t
i is the time values during the bacterial growth when OD was monitored. The cold tolerance scores (CTS) were then calculated for each species as the ratio of GS at 4°C to that at 24°C (optimal temperature) and expressed as percentage; thus, the greater the CTS value, the greater the cold tolerance of the species.
where i=0,…, n−1, n is the number of time points, and t
i is the time values during the bacterial growth when OD was monitored. The cold tolerance scores (CTS) were then calculated for each species as the ratio of GS at 4°C to that at 24°C (optimal temperature) and expressed as percentage; thus, the greater the CTS value, the greater the cold tolerance of the species.
The calculation of the salt tolerance score was also based on comparison of the GS values calculated as described above. The STS was calculated as the ratio of GS at 5% of NaCl versus growth without NaCl; thus, the greater the score, the greater salt tolerance or salt requirement of the species.
Quantification of molecular functions and their mapping to gene annotations
Nineteen Shewanella strains were analyzed either as complete genomes (S. amazonensis, S. baltica strains OS155 and OS195, S. denitrificans, S. frigidimarina, S. loihica, S. oneidensis, S. pealeana, S. woodyi, Shewanella sp. strains ANA-3, MR-4, MR-7, and W3-18-1) or as draft genomes with gene annotations (S. baltica strains OS185 and OS223, S. putrefaciens strains 200 and CN-32, S. benthica, and S. sediminis). The number of protein domains in each genome was used as a quantitative parameter to characterize the importance of the molecular function of the protein domain. A list of Pfam domains and gene annotations were downloaded from the US Department of Energy Joint Genome Institute (JGI) http://www.jgi.doe.gov/. Since S. oneidensis and S. benthica KT99 were not sequenced by JGI, their sequences were downloaded from the J. Craig Venter Institute (http://www.jcvi.org) and annotated using the JGI pipeline. Briefly, genes were modeled using software packages Critica 1.05 and Glimmer 3.1 (Badger 1999; Delcher et al. 1999). A combination of BLAST (1e−5 cutoff), HMMER (1e−10 cutoff), and PRIAM (1e−30 cutoff) were used to assign functions to the sequences. In addition, the revised gene/protein set was searched against the KEGG GENES and InterPro databases. The number and locations of the protein domains were mapped to the genes in each genome using SQL queries and MS Access software.
Computational framework for relating phenotypic and genomic characteristics
The top-down approach for relating phenotypic and genomic characteristics (Fig. 1a) is based on quantification of cold and salt tolerance from growth curves of 15 species as described above. Pearson correlation coefficients (R) are calculated between the CTS and STS and the domain enrichments among the species. To find a threshold for the coefficient, the correlations with the cold tolerance score were calculated by permutation of the domain numbers among species. The permutations gave a distribution of the correlations that was close to normal with the average value of the coefficient −0.0012 and the standard deviation 0.27 indicating that ~90% of random correlations are expected to be less than 0.44. This value was used for initial selection of the domains correlated with the cold tolerance score. The domains were further analyzed using the synteny evidence as described below.
The bottom-up approach (Fig. 1b) does not involve a quantification of bacterial phenotypes and is solely based on quantification of molecular functions in the genomes of 19 sequenced Shewanella strains. Molecular functions were quantified by the number of Pfam domains or domain enrichments as described above. Affinities between pairs of domains were estimated using the Pearson correlation. Correlated domain pairs (the Pearson coefficient is less than a threshold) were selected to produce an affinity graph, where the Pfam domains are the nodes connected by the edges if they are correlated. The threshold for correlating domains was found by testing different R values (0.80, 0.85, 0.90, and 0.99). WebR environment was used for the statistical calculations and VisANT 3.04 system for visualization of the networks (Hu et al. 2005). To find subsets of pair-wise correlating domains, which potentially reveal relating molecular functions in the genome, we applied Markov clustering algorithm (Enright et al. 2002). This algorithm is remarkably robust to graph alterations and shows superiority for the extraction of clusters from interaction networks (Brohee and van Helden 2006). The inflation value was set to 1.8 as recommended by the software. The automatic cluster annotation was made by a Perl script. The script assigned a name to each cluster comprised of “+”/“−” followed by the concatenated nicknames of the microorganisms. If the number of microorganisms with overrepresented domains was less or equal 10, then the cluster name had “+” and concatenated nicknames of the microorganisms with overrepresented number of domains in the cluster, Otherwise the cluster name had “−” and concatenated nicknames of the microorganisms with underrepresented number of domains (Supplementary Table 6S). Synteny evidence for identified clusters was extracted considering the conserved gene order in the organisms having an overrepresented number of domains as described below.
Synteny evidence
A program written in R (Ihaka and Gentleman 1996) was used to simulate the relationship between (1) the probability for two and more neighboring genes to cluster together and (2) the number of randomly sampled genes. The strand orientation of the neighboring genes was not taken into account during simulation. The probability was estimated empirically by repeating the random sampling of m numbers, where m simulated the cluster size changing from 2 to 250, from the ordered array of numbers from 1 to 4,000, where 4,000 indicated the average number of coding sequences in the Shewanella genome. The probability was calculated as a ratio of positive results from 100,000 repeating samplings for each cluster size. Results indicate a low probability of finding two adjacent genes in the cluster by chance, if the cluster size is less than 10. The probability significantly decreases when the cluster size decreases or when the number of identified neighbors increases. For small clusters (less than five genes), even two neighbors with a gap have a small chance to be found by the random gene selection. Based on the empirical probability simulations we decided to search three types of synteny in the identified clusters as additional evidence that the cluster assembly does not occur by chance alone and is likely driven by a common phenotype. The synteny types included (1) two neighboring genes (N), (2) two neighboring genes with a gap (G), and (3) a synteny loci (L) with three and more neighboring genes. After mapping domains to genes, we used a Perl script to identify and characterize the syntenies in each cluster (Supplementary Table 7S). This information was used to select clusters for validation based on known phenotypes in the MR1 strain (Table 1), which is the most studied organism, and for discovery of new phenotypes in recently sequenced species (Table 2).
Table 1.
Domain clusters representing known phenotypes in S. oneidensis MR-1
| Cluster IDs | Domains comprised the clusters (Pfam models) | Synteny evidence | Related Genomic cluster (locus tags or gene names) | Known Phenotypes | Ref. | Flanking transposases | Other species inferred from the cluster annotation that likely express the same phenotype |
|---|---|---|---|---|---|---|---|
| 22, 172a | Complex1_24kDa; Complex1_30kDa; Complex1_49kDa; Complex1_51K; NADHdh; Oxidored_q1; Oxidored_q1_N; Oxidored_q3; Oxidored_q4; Oxidored_q1; Oxidored_q1_N; Oxidored_q2 | SO_1011; SO_1013; SO_1015; SO_1017; SO_1018; SO_1019; SO_1021; SO_1009–SO_1012 | nqr-2 operon: nuoABCDIFGHIJKLMN | Na(+)-transporting NADH: ubiquinone oxidoreductase nqr-2 operon | (Mrazek et al. 2006) | SO_1008 is ISSod20 transposase, TnpA_ISSod20 | S. benthica and S. woodyi |
| 5, 68a | DNA_circ_N; GP46; Phage_Mu_Gp45; Phage_Mu_P; DUF935; Phage_Mu_F; DUF1320; DUF1018 | SO_0667; SO_0668; SO_2680; SO_2681; SO_2698–SO_2701 | Prophage loci SO_2667–SO_2701 | Prophage MuSo2 | (Chourey et al. 2006; Qiu et al. 2006) | ||
| 105 | BATS; Fe_hyd_SSU; Fe_hyd_lg_C; Radical_SAM | SO_3920–SO3923 | Formate dehydrogenase Fdh, periplasmic [Fe–Fe] hydrogenase HydAB, and assembly proteins HydGXEF | Hydrogen metabolism | (Meshulam-Simon et al. 2007) | Shewanella spp. MR4 and ANA3 | |
| 150 | FdhE; SelA | SO_0104–SO_0105 | Formate dehydrogenase fdnGHIE and l-seryl-tRNA selenium transferases SelA, MetY, MdeA | Formate utilization | (Romine et al. 2004) | Shewanella spp. MR4, MR7, ANA3, S. baltica strains OS223, OS195, and OS185, and S. pealeana |
aThe phenotype was represented by two clusters
Table 2.
Phenotypes of the newly sequenced Shewanella species predicted by the bottom-up approach and their characteristics inferred from the identified genomic clusters (see Supplementary Table 8S for more details)
| Predicted phenotypes | Species | Phenotype characteristics predicted from the genomic cluster content |
|---|---|---|
| Degradation of aromatic compounds by an aerobic hybrid pathway | S. woodyi | The cluster is similar to the paa gene cluster in Pseudomonas sp. (Ferrandez et al. 1998; Nogales et al. 2007). The aerobic degradation involves a novel “hybrid” principle of the catabolism of phenylacetic acids with the involvement of CoA derivatives. It resembles the conventional strategy of anaerobic degradation of aromatic compounds |
| Bioluminescence (known phenotype) | S. woodyi | The cluster represents the lux operon responsible for luminescence (Nelson et al. 2007). The order of genes in the S. woodyi operon is different from the other known marine bioluminescent bacteria |
| Ethanolamine utilization | S. benthica (draft genome) | The cluster is similar to the eut operon in Salmonella enterica (Brinsmade et al. 2005) |
| Anaerobic biosynthesis of vitamin B12 | S. benthica and S. sediminis | Products encoded by the clusters are similar to enzymes involved in the cobalamin synthesis under anaerobic conditions (Roth et al. 1993). In S benthica, the aerobic and anaerobic pathways of B12 biosynthesis coexist. The anaerobic route is likely employed when ethanolamine is utilized considering co-localization of the eut and cob loci and the requirement of B12 for the growth on ethanolamine |
| Propanediol degradation | S. putrefaciens CN32 and Shewanella sp. W3181 | The genomic clusters are similar to the pdu operon in Salmonella (Bobik et al. 1999). The propanediol degradation in S. putrefaciens CN32 and Shewanella sp. W3181 presumably occurs only aerobically, because propanediol is utilized in a coenzyme B12-dependent fashion and neither organism has the phenotype of anaerobic biosynthesis of B12 identified in S. benthica and S. sediminis |
| Arsenic resistance | S. putrefaciens strains CN-32 and 200, Shewanella sp. W3-18-1 and ANA-3 | The clusters include arsD and arsA genes that confer an increased rate of extrusion and resistance to arsenite (Lin et al. 2007). A recent study of the ars operon in strain ANA-3 provided evidence of the operon function under either aerobic or anaerobic conditions (Saltikov et al. 2003) |
| Copper resistance | S. benthica KT99, S. denitrificans OS217, S. frigidimarina NCIMB 400, Shewanella sp. ANA-3, S. putrefaciens CN-32 | The clusters are similar to the cop operon in Pseudomonas syringae (Maher et al. 2006). In all five genomes, the first gene in the cluster is a conserved hypothetical protein (DUF411), which likely encodes the copper-responsive repressor CopR that is often transcribed as a part of the cop operon (Magnani et al. 2008) |
| Mating pair formation to transfer DNA and other macromolecules | S. baltica strains OS223, OS195 and OS185, Shewanella sp. W3-18-1 and ANA-3 (surface-exposed sex pilus only) | The cluster encodes products of the type IV secretion system (Lawley et al. 2003). Interestingly, this system is implemented in the Shewanella species with different levels of complexity. It is clearly indicated by the number of Pfam domains |
| Uptake and release of H2 | Absent from S. denitrificans and S. woodyi only | The genomic clusters are similar to the periplasmic [Ni–Fe] hydrogenase hyaB (Armstrong and Albracht 2005). The absence of the hydrogenase in S. denitrificans and S. woodyi reveals that these two species may be deficient in their metabolic capabilities compared to the other sequenced Shewanella species |
Results and discussion
Revealing molecular functions involved in bacterial adaptation to cold temperatures by the top-down approach
Cold tolerance phenotype of Shewanella species
Although all tested Shewanella species have similar optimal growth temperatures with an average value about 24°C and must be classified as mesophilic (Vieille et al. 2001), they show significant differences in their ability to support growth when the temperature dropped to 4–12°C (Supplementary Tables 1S, Supplementary Fig. 2S). Some species show only small changes in growth (accumulated biomass) at the lower temperatures indicating a striking adaptability to the cold conditions. Other species are less tolerant and show a significant impairment of growth. To characterize the phenotypic difference among species, we compared changes in the biomass accumulated by each species at 4°C versus 24°C. By using this ratio, we made the parameter less dependent on the absolute value of the biomass or on the organism’s ability to utilize specific resources and more dependent on its ability to respond to lower temperatures. A higher score reflects a greater the cold tolerance. We referred to the calculated parameter as the cold tolerance score and to the phenotypic trait as cold tolerance. Results of the cold tolerance score calculations (Fig. 2a) indicated significant differences in the adaptability of the Shewanella strains to low temperature. S. pealeana, a strain isolated from the nidamental gland of the squid Loligo pealei, showed the greatest cold tolerance. A significantly lower cold tolerance was observed for S. amazonensis SB2B and S. loihica PV-4, strains isolated from the Amazon River delta and the microbial mat at a hydrothermal in the Pacific Ocean, respectively. It is possible that these two latter strains more frequently encounter higher temperatures in their environments and are therefore less adapted to growth in the cold. The optimal growth temperatures at 35°C (S. amazonensis SB2B), 25°C (S. pealeana), and 18°C (S. loihica PV-4) did not reflect their cold tolerance. There also appeared not to be a correlation between phylogenetic distance and cold tolerances reflected by the difference observed for the two S. putrefaciens strains, CN-32 and 200.
Fig. 2.
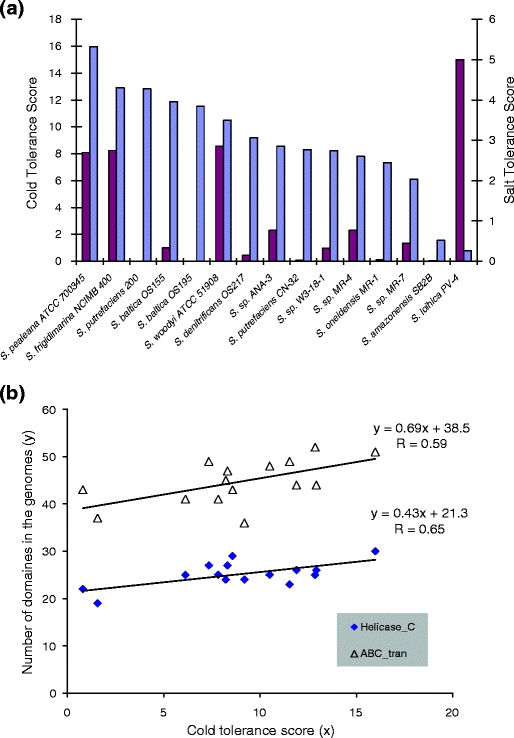
Cold (blue) and Salt (violet) tolerance scores for tested Shewanella strains and two examples (a) of correlation between the cold tolerance score among the strains and the number of domains that represent helicase conserved C-terminal domain (Helicase_C) and ABC transporter (ABC_tran) (b)
Selection of Pfam domains related to Shewanella cold tolerance
To find Pfam domains related to the cold tolerance, we calculated correlations between the number of Pfam domains and the cold tolerance score using the Pearson coefficient (Supplementary Table 2S, Fig. 2b). The threshold for selecting significant correlations was estimated by permutating the number of domains among species and calculating correlations of the permuted values with the cold tolerance score as described in “Materials and methods”. Although the selected domains are likely important, either directly or indirectly, for Shewanella growth at low (<12°C) temperatures, they also may contain false positive correlations. A typical approach to remove false positive correlations and to estimate their rate is to use a benchmark, or a set of known Pfam domains related to the studied phenotype. When studying complex phenotypes, it is difficult to establish a benchmark. There were no previous attempts to quantify cold tolerances of the species, and the biological mechanisms underlying the cold response are largely unknown and may also differ between psychrophiles and mesophiles. To solve the benchmark problem, we used the conserved gene neighborhood of the identified domains among species as a filter. There is very low probability that a random sampling of a small number of genes (less than 10) into a cluster will result in two to three consecutive genes. According to our empirical modeling (see “Materials and methods” and Fig. 3), an alternative hypothesis (three consecutive genes will not cluster together as a result of random sampling of 30 genes out of 4,000 genes in the genome) has a p value less than 0.0015. Thus, finding three to four consecutive genes in the domain cluster provides very stringent criteria for revealing domains that correlate with the cold tolerance score.
Fig. 3.
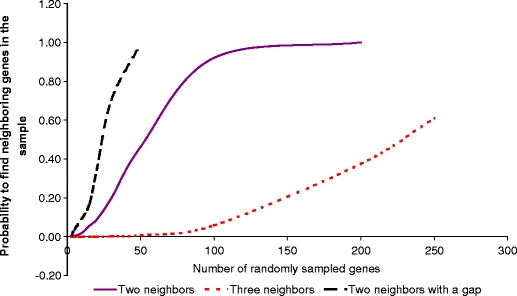
The empirically calculated probability for finding neighboring genes among a randomly sampled cluster of genes in the genome comprised 4,000 genes. The probability was calculated by random sampling genes in the clusters
Information on conserved gene neighborhoods, also referred to as synteny, was obtained by mapping Pfam domains to genes in the S. pealeana genome (Supplementary Tables 3S and 4S) as this organism has the greatest cold tolerance of the tested species. The search indicated a set of four domains in S. pealeana, S. woodyi, and S. frigidimarina that mapped to four consecutive genes representing hydrophobic subunits of an Na+/H+ antiporter. This gene cluster was followed by six genes encoding subunits of a respiratory NADH-ubiquinone oxidoreductase (Fig. 4). Both of these clusters likely belong to a single operon, considering the very short intergenic regions and the overlap of coding sequences in the organisms. The presence of this operon in the three most cold-tolerant species suggests that the Na+-based pump is involved in cold tolerance. Indeed, the use of sodium ions instead of protons for generation of the motive force is a known microbial strategy in response to changes in the fluidity of the cytoplasmic membrane (Chattopadhyay 2006; Konings 2006). Many marine and pathogenic bacteria couple the Na+ pump to the respiratory chain (Kogure 1998; Hase et al. 2001; Dibrov et al. 2004). The gene clusters we have identified in cold-tolerant Shewanella species are similar to the Na+/H+ antiporter clusters found in the halotolerant bacterium Staphylococcus aureus (Hiramatsu et al. 1998). Thus, the cold tolerance of Shewanella species may also entail their salt tolerance or possibly a sodium requirement for growth.
Fig. 4.
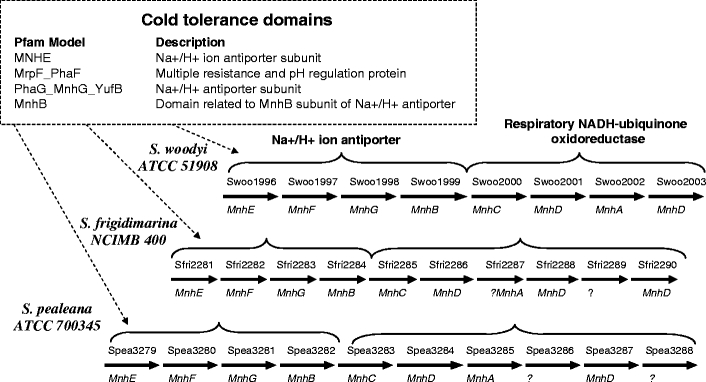
Schematic map of the gene regions in Shewanella sp. with the cold tolerance domains representing Na+/H+ antiporter and NADH-ubiquinone oxidoreductase
Phenotypic correlation between cold and salt tolerance of the species
Although growing evidence associates bacterial cold tolerance with adaptation to other stresses (Chattopadhyay 2006), a link between cold and salt tolerance and an association with the Na+/H+ antiporter gene clusters has not been previously reported. To further evaluate this phenotypic association, we experimentally tested the effect of different NaCl concentrations on the growth of the Shewanella species (Supplementary Table 5S). Salt tolerance scores were calculated for each strain as the ratio of OD660 accumulated after 1 week of incubation of the strain with 5% of NaCl versus culturing without NaCl. Thus, the greater the value of the score, the greater salt tolerance of the species and its salt requirement. Figure 2a shows a comparison of the species according to their salt tolerance scores, and it indicates that the three most cold-tolerant species, S. pealeana, S. woodyi, and S. frigidimarina, with a predicted Na-based respiration have very similar salt tolerances. Their salt tolerance scores are significantly greater (more than three times) than those of the rest of the tested Shewanella species. The only exception is S. loihica PV-4. The salt tolerance score of S. loihica PV-4 is two times greater than the scores of the Shewanella species with the Na+/H+ antiporter. This suggests that the genetic mechanism underlying the exceptional salt tolerance and requirement of S. loihica PV-4 differs from that of S. pealeana, S. woodyi, and S. frigidimarina NCIMB 400.
The top-down approach identified many known protein functions involved in bacterial cold tolerance
The synteny analysis allowed us to establish a more conservative threshold for correlation of the cold tolerance score with number of Pfam domains. We revealed 109 domains that likely relate to cold tolerance (Supplementary Tables 2S and 3S). Indeed, 24 of these domains represent known mechanisms of cold tolerance including (1) maintenance of the membrane fluidity (Phadtare 2004) by introduction of double bonds into fatty acids via desaturases (FA_hydroxylase), (2) enhanced translation and transcription (Nakaminami et al. 2006; Wilson 2007) indicated by the presence of unique tRNA synthetases and chaperons (DALR_2, TruB-C_2, tRNA-synt_1g, Elong-fact-P_C, HAS-barrel), (3) increased proteolysis (Peptidase_C39, Peptidase_U32, Leuk-A4-hydro_C, Creatinase_N), and (4) transport of ions (ABC_tran, Aa_trans, Peripla_BP_2, OpuAC, Sec_GG, Glyphos_transf, Multi_Drug_Res, DcuA_DcuB, MotA_ExbB, UAA, MnhB, MrpF_PhaF, MNHE, PhaG_MnhG_YufB) (Erdal et al. 2003; Susana et al. 2004; Vezzi et al. 2005; Auman et al. 2006; Chattopadhyay 2006; Konings 2006). Several of the domains (OmpA_membrane, DHHA2, DHH, Aldedh, Oxidored_FMN, Elong-fact-P_C, Surf_Ag_VNR) are among the genes and proteins activated by cold shock in Shewanella piezotolerans WP3 and Shewanella livingstonensis Ac10 (Venkateswaran et al. 1999; Kawamoto et al. 2007).
Revealing novel genomic loci of bacterial cold tolerance by the proposed approach
We identified 10 S. pealeana loci with three or more consecutive genes that have a very high probability of being linked to cold tolerance (Supplementary Table 4S). These loci are believed to represent novel mechanisms of cold tolerance. In addition to the Na+/H+ antiporter considered above, one loci (Spea_0901–Spea_0922) includes genes encoding two helicases, a thiol-specific antioxidant, a valyl-tRNA synthetase, and two peptidases. Another cold tolerance locus (Spea_3491–Spea_3502) includes two chemotaxis sensory transducer proteins, a cysteine exporter, a helicase, and several hypothetical proteins. Similarity searches show that the chemotaxis sensory transducer proteins (Spea_3491 and Spea_3492) have close homologs in the barophilic and psychrophilic bacterium Photobacterium profundum SS9 (Vezzi et al. 2005) and that the cysteine exporter is homologous to cysteine transporters (Ping_1298 and Ping_3454) of the psychrophilic bacterium Psychromonas ingrahamii isolated from Arctic polar sea ice (Auman et al. 2006). The presence of proteins with DEAD/DEAH box helicase domains in three of the identified loci strongly links RNA metabolism to resistance to low temperature. It is known that these helicases loosens RNA secondary structure and promotes folding of RNA (Polissi et al. 2003). This may be important at lower temperatures, because they stabilize the RNA secondary structures, and this stabilization is a major detrimental physiological effect of temperature downshift.
Predicting phenotypes of Shewanella species from comparative analysis of molecular functions using the bottom-up approach
The approach (Fig. 1b) was applied to 19 sequenced Shewanella strains. Similar to the top-down analysis, the selection of clusters related to phenotypes was initially based on the correlation coefficient threshold and then further augmented by synteny information inside each cluster by mapping domains to genes and searching for neighboring genes and loci.
The analysis included three main computational steps: (1) calculate a profile for each domain comprised of numbers of the domain in the tested genomes (Supplementary Table 6S), (2) calculate Pearson correlations for each pair of the domain profiles, and (3) find disconnected networks by clustering the correlated profiles according to their pair-wise correlations using a threshold. To find an optimal threshold for the correlation, the clustering step was repeated using different thresholds for the correlation coefficient. The ultimate goal of repeating the calculation was to find the correlation coefficient that maximizes the number of the identified networks and minimizes the networks/clusters connectivity. By maximizing the number of networks, we increase the number of phenotypes we can reveal, and by minimizing the connectivity between networks we increase specificity of domain attribution to each network. The selected threshold, therefore, provided a reasonable tradeoff between specificity and sensitivity of the phenotype identification. Results of the threshold selection are presented in Fig. 5 and indicate the optimal value for the correlation coefficient R = 0.90. The affinity map includes 201 disconnected networks comprised of two and more domains, which after the Markov clustering separated into 217 clusters. The clusters and networks mainly represent sets of domains specific to one or several Shewanella species. They were annotated automatically (with manual validation) by nicknames of the organisms having overrepresented or underrepresented numbers of domains (Supplementary Table 6S). To assign each cluster to a phenotype and to augment validity of the assignment, we extracted synteny information from each cluster by mapping domains to genes (Supplementary Table 7S). We found that 84 of the 217 identified clusters have additional synteny evidence, i.e., two or more adjacent genes in the genome. Thirty-five clusters consisted of loci with three to eight conserved genes. We further analyzed the clusters to validate the bottom-up approach and to discover novel phenotypes in the Shewanella strains. The synteny information in the clusters included (1) neighboring genes; (2) neighboring genes with a gap, which is one gene between neighbors; and (3) a genomic locus of three or more neighboring genes.
Fig. 5.
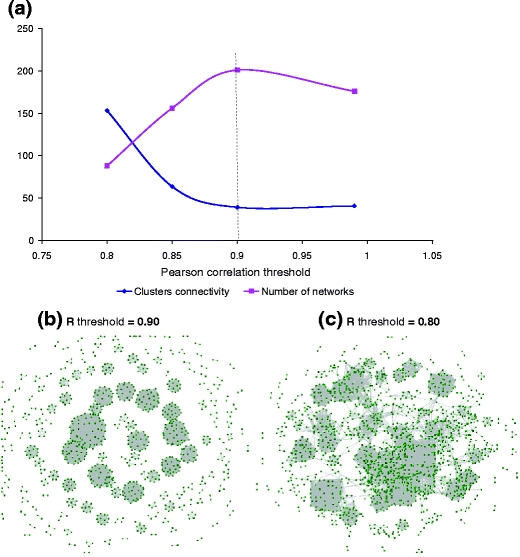
Selection of the domain affinity threshold. The domain affinity is estimated by the Pearson correlation coefficient (R) and different thresholds of the coefficient were tested to visualize the obtained domain affinity maps and to calculate the number of pairs that are greater than the thresholds, the number of networks (disconnected clusters), and the cluster connectivity (the ratio of selected pairs to the number of network). The optimum threshold (dashed line) for R was selected to maximize the number of the identified networks and to minimize the cluster connectivity (a). Figures (b) and (c) demonstrate difference between networks based on different thresholds
For validation of the approach, we selected seven clusters with orthologs in S. oneidensis MR-1. We reasoned that since S. oneidensis MR-1 is an experimentally well-studied environmental bacterium, the clusters are more likely to be linked to a phenotype. Indeed, we find that the clusters can be linked to four known MR1 phenotypes, and that the expression of genes related to each phenotype has the experimental support in the literature (Table 1). In addition to validating the approach, the enrichment of these domains allows us to infer similar phenotypes for the newly sequenced genomes containing these clusters.
Most of the identified clusters did not include S. oneidensis MR-1 and represented phenotypes that are novel for other Shewanella species. The clustering augmented by synteny information facilitated the linking of clusters to phenotypes and highlighted loci of phenotype related genes in two and more genomes. We selected 15 clusters with different types of synteny evidence for manual evaluation of phenotypes (Supplementary Table 7S) using literature-based information. We find that the selected clusters represent nine phenotypes in Shewanella species, including degradation of aromatic compounds by an aerobic hybrid pathway in S. woodyi, degradation of ethanolamine by S. benthica, and propanediol degradation by S. putrefaciens CN32 and Shewanella sp. W3-18-1. Arsenic and copper resistance phenotypes were detected for some Shewanella species, as well as differing abilities of the species to transfer macromolecules and H2. Table 2 and Supplementary Table 8S provide some characteristics of the phenotypes and their specificities discovered from the cluster content of the organisms. A comparison of the clusters with metabolic reconstructions provided by the Shewanella Knowledgebase (http://shewanella-knowledgebase.org:8080/Shewanella/shewcyc.jsp) indicated that none of the phenotype could be predicted based on the enzymatic annotation.
Applications and limitations of the proposed approaches
The results of our study indicate that quantifying complex phenotypes and combining statistical and synteny criteria are valuable novel computational tools to study the molecular mechanisms behind the adaptation of an organism’s physiology to its environment. As a rule, the adaptation has a continuing nature and cannot be characterized simply as present or absent. The quantification of the molecular function in terms of the number of domains instead of a binary characterization (presence/absence) is equally important. Although, in many cases, domains that are unique to an organism indicate a specific phenotypic trait, the enriched domains may reveal additional molecular functions that, in combination with the organism-specific domains, more accurately map domain clusters to gene loci underlying the phenotype. One example in Table 2 is the identification of two additional gene clusters encoding enzymes involved in anaerobic biosynthesis of B12 in S. benthica. This vitamin is an essential cofactor for the utilization of ethanolamine, a specific phenotype of this bacterium. A combination of enriched and specific domains was mapped to a gene cluster in the genome that was attributed to a specific phenotypic trait of the bacterium. The other example is an increased number of the BMC domains relating to carboxysome-like structural proteins that was found in three of the species. These proteins are structural components of bacterial microcompartments used for utilization of ethanolamine in S. benthica and propanediol in S. putrefaciens CN32 and Shewanella sp. W3-18-1. In both cases, the microcompartments are believed to diminish aldehyde toxicity.
A noticeable feature of most genomic loci attributed to phenotypes is the presence of transposases and integrases immediately upstream or downstream of the mapped genes (Table 1, Supplementary Table 8S). This suggests that many phenotypes are acquired by horizontal gene transfer and that this mechanism plays an important role in the innovation and evolution of the Shewanella genomes. Known phylogenetic methods for identification gene transfer events look for unusual distributions of genes across organisms. Phylogeny-independent methods look for genes that appear abnormally in their genomic context by examining nucleotide frequencies or codon usage bias. Both methods often produce inconsistent results (Gogarten et al. 2002). The proposed algorithm automatically highlights clusters in a genome that have both abnormal sequences and patchy distribution across the genomes and provides a novel method for collecting evidence of potential gene transfer events.
Our proposed approaches provide valuable tools not only for prediction of phenotypes and revealing genomic content underlying complex physiological characteristics but also for prediction of orthologs in the genomes, and functions of unknown domains and hypothetical proteins. A genome-scale search for orthologs is usually performed by a reciprocal-best-BLAST-hits approach (Altschul et al. 1990). Such predictions, however, are based solely on sequence similarity between two genes and do not take into consideration the nearby genes or the domain structure of the sequences. The domain clustering and subsequent mapping to genes reveal not only individual orthologs in the genomes but sets of orthologs that are common across organisms and relate to shared biological processes or even phenotypes. The approach automatically integrates genomic content into the analysis, thus improving the specificity of the ortholog predictions across organisms without a sophisticated phylogenetic analysis. The prediction of function for unknown domains and hypothetical proteins is another potential application of the bottom-up approach. A good example is DUF59 (PF01883) a domain of unknown function that is found in the cluster representing phenylacetic acid degradation. DUF59 is found in the missing PaaI subunit of phenylacetate-CoA oxygenase, and the domain is encoded adjacent to the genes for the PaaG and PaaJ subunits of the oxygenase. This finding clearly attributes the molecular function “PaaI subunit of phenylacetate-CoA oxygenase” to DUF59. Another example is DUF411, a domain found in a hypothetical protein of the Shewanella cop operons. The protein is likely a regulator of the MerR family based on its sequence similarity to a transcriptional regulator in Saccharophagus degradans 2-40. Interestingly, we find that Shewanella sp. ANA-3 and S. putrefaciens CN-32 have an additional common synteny with the DUF411 domain. A comparison of all proteins with the DUF411 domain in Shewanella species (Supplementary Fig. 3S) indicate a higher similarity among the proteins of the same synteny in different species and a lower similarity for proteins belonging to different syntenies within the same genome. This is probably reflective of divergence of paralogs and demonstrates feasibility of the approaches from an evolutionary standpoint (Supplementary Fig. 1S) considered in the introduction in more detail.
Although our proposed approaches provide valuable tools for large-scale linking of phenotypic traits to genetic elements, it should be emphasized that the relationships are predicted computationally. Additional experimental studies are necessary to validate the putative relationships. Our analysis also does not include some important aspects of phenotypes, such as regulation, and is limited to the functions characterized by Pfam domains. Anaerobic respiration in Shewanella species, for example, involves various multi-heme cytochromes. These proteins are not characterized by Pfam domains and are therefore excluded from the analysis. The integration of different data types in the computational analysis will make it more robust. Nonetheless, we believe that the proposed approaches can be used to provide a molecular-level understanding of complex ecophysiological characteristics and predict phenotypes of different organisms that can be evaluated experimentally.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Experimental characterization of the Shewanella species cold tolerance. (XLS 114 kb)
Number of Pfam domains in the tested species and their correlations with the cold tolerance score. (XLS 853 kb)
Mapping cold tolerance domains (assignment to specific genes) in the S. pealeana genome. (XLS 86 kb)
Genomic loci in the S. pealeana genome related to cold tolerance. (XLS 20 kb)
Experimental characterization of the Shewanella species salt tolerance (XLS 25 kb)
Conserved synteny in the identified clusters (A) and their mapping to locus tags in the organisms (B) (XLS 136 kb)
Predicted phenotypes of the newly sequenced Shewanella species including related domain clusters and location of the genomic clusters. (XLS 25 kb)
Recipe of the medium used for culturing of the Shewanella strains. (PDF 45 kb)
Evolutional relationship between phenotypic traits and a variety of protein domains in the genome. Changes in the environment are the driving force for changes in the genome (1) leading to changes in the number and variety of protein domains (2) and to the evolution of species with novel phenotypic traits (3). (PDF 39 kb)
Growth scores of the tested Shewanella strains as functions of their growth temperature. Growth score for each temperature was calculated as the area under the growth profile normalized to 100 h of the growth as described in “Materials and methods”. (PDF 33 kb)
Acknowledgments
We are very grateful to Arcady R. Mushegian and other reviewers of the paper for thoughtful suggestions and comments on the study. The sequence data for the Shewanella species except S. oneidensis MR-1 and S. benthica KT99 were produced by the US Department of Energy Joint Genome Institute http://www.jgi.doe.gov/. The sequence data for S. oneidensis MR-1 and S. benthica KT99 were produced by J. Craig Venter Institute (http://www.tigr.org/). This research was supported by the U.S. Department of Energy (DOE) Office of Biological and Environmental Research under the Genomics: GTL Program via the Shewanella Federation consortium.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Apic G, Gough J, Teichmann SA. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J Mol Biol. 2001;310:311–325. doi: 10.1006/jmbi.2001.4776. [DOI] [PubMed] [Google Scholar]
- Armstrong FA, Albracht SPJ. [NiFe]-hydrogenases: spectroscopic and electrochemical definition of reactions and intermediates. Phil Trans Math Phys Eng Sci. 2005;363:937–954. doi: 10.1098/rsta.2004.1528. [DOI] [PubMed] [Google Scholar]
- Auman AJ, Breezee JL, Gosink JJ, Kampfer P, Staley JT. Psychromonas ingrahamii sp. nov., a novel gas vacuolate, psychrophilic bacterium isolated from Arctic polar sea ice. Soc General Microbiol. 2006;56:1001–1007. doi: 10.1099/ijs.0.64068-0. [DOI] [PubMed] [Google Scholar]
- Badger JH (1999) CRITICA: coding region identification tool invoking comparative analysis. SMBE, pp 512–524 [DOI] [PubMed]
- Bobik TA, Havemann GD, Busch RJ, Williams DS, Aldrich HC. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B12-dependent 1, 2-propanediol degradation. J Bacteriol. 1999;181:5967. doi: 10.1128/jb.181.19.5967-5975.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinsmade SR, Paldon T, Escalante-Semerena JC. Minimal functions and physiological conditions required for growth of Salmonella enterica on ethanolamine in the absence of the metabolosome. J Bacteriol. 2005;187:8039–8046. doi: 10.1128/JB.187.23.8039-8046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohee S, van Helden J. Evaluation of clustering algorithms for protein–protein interaction networks. BMC Bioinformatics. 2006;7:488. doi: 10.1186/1471-2105-7-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay MK. Mechanism of bacterial adaptation to low temperature. J Biosci. 2006;31:157–165. doi: 10.1007/BF02705244. [DOI] [PubMed] [Google Scholar]
- Chourey K, Thompson MR, Morrell-Falvey J, Verberkmoes NC, Brown SD, Shah M, Zhou J, Doktycz M, Hettich RL, Thompson DK. Global molecular and morphological effects of 24-hour chromium(VI) exposure on Shewanella oneidensis MR-1. Appl Environ Microbiol. 2006;72:6331–6344. doi: 10.1128/AEM.00813-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date SV, Peregrin-Alvarez JM. Phylogenetic profiling. Methods Mol Biol. 2008;453:201–216. doi: 10.1007/978-1-60327-429-6_9. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibrov P, Dibrov E, Pierce GN, Galperin MY. Salt in the wound: a possible role of Na gradient in chlamydial infection. J Mol Microbiol Biotechnol. 2004;8:1–6. doi: 10.1159/000082075. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdal UG, Erdal ZK, Randall CW. A thermal adaptation of bacteria to cold temperatures in an enhanced biological phosphorus removal system. Water Sci Technol. 2003;47:123–128. [PubMed] [Google Scholar]
- Ferrandez A, Miñambres B, Garcia B, Olivera ER, Luengo JM, Garcia JL, Diaz E. Catabolism of phenylacetic acid in Escherichia coli characterization of a new aerobic hybrid pathway. J Biol Chem. 1998;273:25974–25986. doi: 10.1074/jbc.273.40.25974. [DOI] [PubMed] [Google Scholar]
- Fredrickson JK, Romine MF, Beliaev AS, Auchtung JM, Driscoll ME, Gardner TS, Nealson KH, Osterman AL, Pinchuk G, Reed JL, Rodionov DA, Rodrigues JL, Saffarini DA, Serres MH, Spormann AM, Zhulin IB, Tiedje JM. Towards environmental systems biology of Shewanella. Nat Rev Microbiol. 2008;6:592–603. doi: 10.1038/nrmicro1947. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3:679–687. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19:2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- Gralnick JA, Hau HH. Ecology and biotechnology of the genus Shewanella. Annu Rev Microbiol. 2006;61:237–250. doi: 10.1146/annurev.micro.61.080706.093257. [DOI] [PubMed] [Google Scholar]
- Hase CC, Fedorova ND, Galperin MY, Dibrov PA. Sodium ion cycle in bacterial pathogens: evidence from cross-genome comparisons. Microbiol Mol Biol Rev. 2001;65:353. doi: 10.1128/MMBR.65.3.353-370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramatsu T, Kodama K, Kuroda T, Mizushima T, Tsuchiya T. A putative multisubunit Na+/H+ antiporter from Staphylococcus aureus. J Bacteriol. 1998;180:6642. doi: 10.1128/jb.180.24.6642-6648.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Mellor J, Wu J, Yamada T, Holloway D, DeLisi C. VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res. 2005;33:W352–W357. doi: 10.1093/nar/gki431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. doi: 10.2307/1390807. [DOI] [Google Scholar]
- Kawamoto J, Kurihara T, Kitagawa M, Kato I, Esaki N. Proteomic studies of an Antarctic cold-adapted bacterium, Shewanella livingstonensis Ac10, for global identification of cold-inducible proteins. Extremophiles. 2007;11:819–826. doi: 10.1007/s00792-007-0098-6. [DOI] [PubMed] [Google Scholar]
- Kieft TL, Fredrickson JK, Onstott TC, Gorby YA, Kostandarithes HM, Bailey TJ, Kennedy DW, Li SW, Plymale AE, Spadoni CM. Dissimilatory reduction of Fe (III) and other electron acceptors by a thermus isolate. Appl Environ Microbiol. 1999;65:1214. doi: 10.1128/aem.65.3.1214-1221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure K. Bioenergetics of marine bacteria. Curr Opin Biotechnol. 1998;9:278–282. doi: 10.1016/S0958-1669(98)80059-1. [DOI] [PubMed] [Google Scholar]
- Konings WN. Microbial transport: adaptations to natural environments. Antonie van Leeuwenhoek. 2006;90:325–342. doi: 10.1007/s10482-006-9089-3. [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lawley TD, Klimke WA, Gubbins MJ, Frost LS. F factor conjugation is a true type IV secretion system. FEMS Microbiol Lett. 2003;224:1–15. doi: 10.1016/S0378-1097(03)00430-0. [DOI] [PubMed] [Google Scholar]
- Lin YF, Yang J, Rosen BP. ArsD: an As (III) metallochaperone for the ArsAB As (III)-translocating ATPase. J Bioenerg Biomembranes. 2007;39:453–458. doi: 10.1007/s10863-007-9113-y. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li J, Sam L, Goh CS, Gerstein M, Lussier YA. An integrative genomic approach to uncover molecular mechanisms of prokaryotic traits. PLoS Comput Biol. 2006;2:e159. doi: 10.1371/journal.pcbi.0020159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Rosenberg C, Gilbert W. Intron phase correlations and the evolution of the intron/exon structure of genes. Proc Natl Acad Sci U S A. 1995;92:12495. doi: 10.1073/pnas.92.26.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Betran E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003;4:865–875. doi: 10.1038/nrg1204. [DOI] [PubMed] [Google Scholar]
- Madera M, Vogel C, Kummerfeld SK, Chothia C, Gough J. The SUPERFAMILY database in 2004: additions and improvements. Nucleic Acids Res. 2004;32:D235–239. doi: 10.1093/nar/gkh117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani D, Barré O, Gerber SD, Solioz M. Characterization of the CopR regulon of Lactococcus lactis IL1403? J Bacteriol. 2008;190:536–545. doi: 10.1128/JB.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher MJ, Wedd AG, Koay M. Intermolecular transfer of copper ions from the CopC protein of Pseudomonas syringae. Crystal structures of fully loaded Cu I Cu II forms. J Am Chem Soc. 2006;128:5834–5850. doi: 10.1021/ja058528x. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Koonin EV. Potential genomic determinants of hyperthermophily. Trends Genet. 2003;19:172–176. doi: 10.1016/S0168-9525(03)00047-7. [DOI] [PubMed] [Google Scholar]
- Meshulam-Simon G, Behrens S, Choo AD, Spormann AM. Hydrogen metabolism in Shewanella oneidensis MR-1. Appl Environ Microbiol. 2007;73:1153–1165. doi: 10.1128/AEM.01588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methe BA, Nelson KE, Eisen JA, Paulsen IT, Nelson W, Heidelberg JF, Wu D, Wu M, Ward N, Beanan MJ. Genome of Geobacter sulfurreducens: metal reduction in subsurface environments. Science. 2003;302:1967–1969. doi: 10.1126/science.1088727. [DOI] [PubMed] [Google Scholar]
- Mrazek J, Spormann AM, Karlin S. Genomic comparisons among gamma-proteobacteria. Environ Microbiol. 2006;8:273–288. doi: 10.1111/j.1462-2920.2005.00894.x. [DOI] [PubMed] [Google Scholar]
- Nakaminami K, Karlson DT, Imai R. Functional conservation of cold shock domains in bacteria and higher plants. Proc Natl Acad Sci U S A. 2006;103:10122. doi: 10.1073/pnas.0603168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EJ, Tunsjø HS, Fidopiastis PM, Sørum H, Ruby EG. A novel lux operon in the cryptically bioluminescent fish pathogen Vibrio salmonicida is associated with virulence? Appl Environ Microbiol. 2007;73:1825–1833. doi: 10.1128/AEM.02255-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales J, Macchi R, Franchi F, Barzaghi D, Fernandez C, Garcia JL, Bertoni G, Diaz E. Characterization of the last step of the aerobic phenylacetic acid degradation pathway. Microbiology. 2007;153:357. doi: 10.1099/mic.0.2006/002444-0. [DOI] [PubMed] [Google Scholar]
- Orengo CA, Thornton JM. Protein families and their evolution—a structural perspective. Annu Rev Biochem. 2005;74:867–900. doi: 10.1146/annurev.biochem.74.082803.133029. [DOI] [PubMed] [Google Scholar]
- Phadtare S. Recent developments in bacterial cold-shock response. Curr Issues Mol Biol. 2004;6:125–136. [PubMed] [Google Scholar]
- Polissi A, De Laurentis W, Zangrossi S, Briani F, Longhi V, Pesole G, Dehò G. Changes in Escherichia coli transcriptome during acclimatization at low temperature. Res Microbiol. 2003;154:573–580. doi: 10.1016/S0923-2508(03)00167-0. [DOI] [PubMed] [Google Scholar]
- Qiu X, Daly MJ, Vasilenko A, Omelchenko MV, Gaidamakova EK, Wu L, Zhou J, Sundin GW, Tiedje JM. Transcriptome analysis applied to survival of Shewanella oneidensis MR-1 exposed to ionizing radiation. J Bacteriol. 2006;188:1199–1204. doi: 10.1128/JB.188.3.1199-1204.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranea JA, Yeats C, Grant A, Orengo CA. Predicting protein function with hierarchical phylogenetic profiles: the Gene3D Phylo-Tuner method applied to eukaryotic genomes. PLoS Comput Biol. 2007;3:e237. doi: 10.1371/journal.pcbi.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romine MF, Elias DA, Monroe ME, Auberry K, Fang R, Fredrickson JK, Anderson GA, Smith RD, Lipton MS. Validation of Shewanella oneidensis MR-1 small proteins by AMT tag-based proteome analysis. Omics. 2004;8:239–254. doi: 10.1089/omi.2004.8.239. [DOI] [PubMed] [Google Scholar]
- Roth JR, Lawrence JG, Rubenfield M, Kieffer-Higgins S, Church GM. Characterization of the cobalamin (vitamin B12) biosynthetic genes of Salmonella typhimurium. J Bacteriol. 1993;175:3303–3316. doi: 10.1128/jb.175.11.3303-3316.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltikov CW, Cifuentes A, Venkateswaran K, Newman DK. The ars detoxification system is advantageous but not required for As (V) respiration by the genetically tractable Shewanella species strain ANA-3. Appl Environ Microbiol. 2003;69:2800–2809. doi: 10.1128/AEM.69.5.2800-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susana CV, Silvia HC, Walter PM. Extracellular proteases from eight psychrotolerant Antarctic strains. Microbiol Res. 2004;159:157–166. doi: 10.1016/j.micres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Doerks T, Jensen LJ, Perez-Iratxeta C, Kaczanowski S, Hooper SD, Andrade MA, Bork P (2005) Systematic association of genes to phenotypes by genome and literature mining. PLoS Biol 3(5): e134 [DOI] [PMC free article] [PubMed]
- Venkateswaran K, Moser DP, Dollhopf ME, Lies DP, Saffarini DA, MacGregor BJ, Ringelberg DB, White DC, Nishijima M, Sano H. Polyphasic taxonomy of the genus Shewanella and description of Shewanella oneidensis sp. nov. Int J Syst Evol Microbiol. 1999;49:705–724. doi: 10.1099/00207713-49-2-705. [DOI] [PubMed] [Google Scholar]
- Vezzi A, Campanaro S, D’Angelo M, Simonato F, Vitulo N, Lauro FM, Cestaro A, Malacrida G, Simionati B, Cannata N. Life at depth: Photobacterium profundum genome sequence and expression analysis. Science. 2005;307:1459–1461. doi: 10.1126/science.1103341. [DOI] [PubMed] [Google Scholar]
- Vieille C, Epting KL, Kelly RM, Zeikus JG. Bivalent cations and amino-acid composition contribute to the thermostability of Bacillus licheniformis xylose isomerase. Eur J Biochem. 2001;268:6291–6301. doi: 10.1046/j.0014-2956.2001.02587.x. [DOI] [PubMed] [Google Scholar]
- Wilson DN. The weird and wonderful world of bacterial ribosome regulation. Crit Rev Biochem Mol Biol. 2007;42:187–219. doi: 10.1080/10409230701360843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental characterization of the Shewanella species cold tolerance. (XLS 114 kb)
Number of Pfam domains in the tested species and their correlations with the cold tolerance score. (XLS 853 kb)
Mapping cold tolerance domains (assignment to specific genes) in the S. pealeana genome. (XLS 86 kb)
Genomic loci in the S. pealeana genome related to cold tolerance. (XLS 20 kb)
Experimental characterization of the Shewanella species salt tolerance (XLS 25 kb)
Conserved synteny in the identified clusters (A) and their mapping to locus tags in the organisms (B) (XLS 136 kb)
Predicted phenotypes of the newly sequenced Shewanella species including related domain clusters and location of the genomic clusters. (XLS 25 kb)
Recipe of the medium used for culturing of the Shewanella strains. (PDF 45 kb)
Evolutional relationship between phenotypic traits and a variety of protein domains in the genome. Changes in the environment are the driving force for changes in the genome (1) leading to changes in the number and variety of protein domains (2) and to the evolution of species with novel phenotypic traits (3). (PDF 39 kb)
Growth scores of the tested Shewanella strains as functions of their growth temperature. Growth score for each temperature was calculated as the area under the growth profile normalized to 100 h of the growth as described in “Materials and methods”. (PDF 33 kb)