

Abstract
The instability of common fragile sites (CFSs) contributes to the development of a variety of cancers. The ATR-dependent DNA damage checkpoint pathway has been implicated in maintaining CFS stability, but the mechanism is incompletely understood. The goal of our study was to elucidate the action of the ATR protein in the CFS-specific ATR-dependent checkpoint response. Using a chromatin immunoprecipitation assay, we demonstrated that ATR protein preferentially binds (directly or through complexes) to fragile site FRA3B as compared to non-fragile site regions, under conditions of mild replication stress. Interestingly, the amount of ATR protein that bound to three regions of FRA3B peaked at 0.4 μM aphidicolin (APH) treatment and decreased again at higher concentrations of APH. The total amounts of cellular ATR and several ATR-interacting proteins remained unchanged, suggesting that ATR binding to the fragile site is guided initially by the level of replication stress signals generated at FRA3B due to APH treatment and then sequestered from FRA3B regions by successive signals from other non-fragile site regions, which are produced at the higher concentrations of APH. This decrease in ATR binding to fragile site FRA3B at the higher concentrations of APH may account for the increasing number of chromosome gaps and breaks observed under the same conditions. Furthermore, inhibition of ATR kinase activity by treatment with 2-aminopurine (2-AP) or by over-expression of a kinase-dead ATR mutant showed that the kinase activity is required for the binding of ATR to fragile DNAs in response to APH treatment. Our results provide novel insight into the mechanism for the regulation of fragile site stability by ATR.
Keywords: Fragile site, FRA3B, ATR
1. Introduction
The ATR-dependent DNA damage checkpoint pathway plays a central role in regulating common fragile site (CFS) stability [1-5]. CFSs are regions of the genome that are prone to forming gaps and breaks on metaphase chromosomes following partial inhibition of DNA synthesis. CFSs are found in all individuals and comprise regions of DNA between 0.5 and 9 Mb in size. The implications of CFS instability in the development of cancers have been well documented. CFSs have been found near or coincident with the positions of cancer-related translocation breakpoints [6,7] and are associated with amplification of proto-oncogenes [8-11], deletion of tumor suppressor genes [12-14], and integration of oncogenic viruses [15-20]. To date, about 80 CFSs have been identified in the human genome and are highly conserved among mammals [6].
One of these, FRA3B, is the most inducible CFS in the human genome. FRA3B is located within the chromosomal band 3p14.2 and is deleted or rearranged in many cancers [21]. This site is normally stable in cultured human cells. However, when cells are cultured under conditions of replication stress induced by inhibitors of DNA synthesis such as aphidicolin (APH), FRA3B displays visible gaps and breaks on metaphase chromosomes [22] and exhibits submicroscopic deletions resembling those seen in cancer cells [23]. APH-induced fragility of FRA3B was reported to extend over about 4 Mb including five genes—SCA7, CADPS, HT021, PTPRG, and FHIT [24] (Fig. 1). Although the boundaries of FRA3B are not well defined, in tumor-derived cell lines, most deletions and rearrangements observed within FRA3B map specifically to a region of ∼500 kb spanning from intron 3 to intron 5 of the FHIT gene [12,13,25]. The FHIT gene encodes a tumor suppressor, and these disruptions result in its inactivation [26]. These studies have implicated FRA3B instability in the development of a variety of cancers.
Fig. 1.
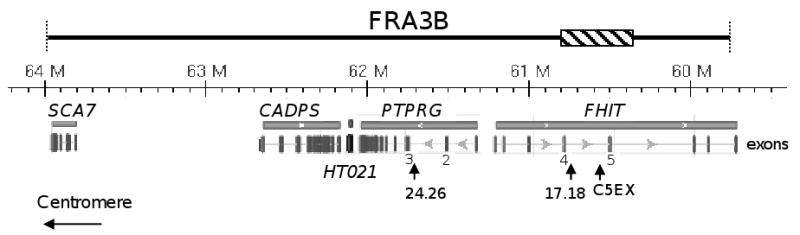
Genomic organization of the FRA3B region. Although the boundaries of FRA3B are not well defined, APH-induced fragility of FRA3B has been reported to span ∼4 Mb, including five genes - SCA7, CADPS, HT021, PTPRG, and FHIT [24]. The genomic coordinates (Human Genome GRch37 assembly) including distance scale are listed above each gene. Exons of each gene are listed as vertical bars below, and exons 4 and 5 of FHIT and exons 2 and 3 of PTPRG are numbered. The ∼500 kb unstable region of FRA3B which has been investigated thoroughly [12,13,25], extends from intron 3 (near exon 4) to intron 5 (∼150 kb distal to exon 5) of the FHIT gene, and is indicated by a hatched bar. The positions of the three examined regions: FRA3B_C5EXR (chr3:60516490-60516900), FRA3B_24.26 (chr3: 61742231-61742422), and FRA3B_17.18 (chr3:60719769-60719981) are indicated by vertical arrowheads.
Sequence analysis has revealed that common fragile sites have high A/T content and exhibit significantly higher DNA flexibility compared to non-fragile regions [27,28]. Thus, CFSs have the potential to form stable secondary structures that could disrupt replication. This notion is supported by evidence that CFSs are inherently difficult to replicate, and low doses of replication inhibitors further delay replication at the CFSs [29-31]. In addition, studies of the fragile site FRA16D in yeast have shown that an AT-rich region contributes to stalling of the replication fork and causes increased chromosomal breakage [32].
As a major DNA damage sensor protein, ATR, along with downstream target molecules, responds to stalled and collapsed replication forks and, in turn, inhibits further firing of replication origins, blocks entry into mitosis, and promotes DNA repair, recombination or apoptosis [33,34]. Cells lacking functional ATR are defective in their checkpoint response to agents that block replication fork progress, including APH and hydroxyurea, and to conditions of hypoxia. In ATR-deficient cells, fragile site expression increases up to 20-fold with APH induction and, even in the absence of APH, the frequency of fragile sites is at a level similar to APH-treated ATR wild-type cells [1]. Furthermore, cells from individuals with Seckel syndrome, which contain a hypomorphic mutation in the ATR gene and a low level of ATR protein expression, display increased chromosomal breakage at CFSs when compared to control cells [2]. This difference in fragile site instability increases as the concentration of APH increases, suggesting that the amount of ATR is critical to respond to replication stress and signal repair mechanisms. In addition, down-regulation of several other DNA damage response proteins, including BRCA1 [3], Chk1 [4], SMC1 [35], FANCD2 [5], HUS1 [36], and WRN [37] also increases fragile site instability, establishing their role in control of common fragile site integrity. Components of both homologous recombination and nonhomologous end-joining pathways have been shown to participate in maintenance of fragile site stability [38].
These studies suggest a working hypothesis for the mechanism of instability at CFSs in which the AT-rich sequences present difficulties during replication which are further exacerbated by APH or other replication stresses. Long stretches of single-stranded DNA could be generated due to incomplete replication, activating the ATR-dependent DNA damage checkpoint response. These unreplicated regions of DNA within CFSs, if left unrepaired, could result in DNA breakage. However, there is no information that ATR protein is directly recruited to the fragile site, if it can distinguish fragile and non-fragile site regions under mild replication stress, or how increasing replication stress affects this association and results in increased DNA breakpoints. We addressed these questions in the current study, initially showing that treatment with low doses of APH leads to preferential binding of ATR protein (directly or through complexes) to fragile site FRA3B, as compared to non-fragile site regions. Further examination revealed that ATR binds to all three fragile regions of FRA3B in a similar manner, with peak binding at 0.4 μM APH treatment and decreased binding at higher concentrations of APH. This binding pattern could reflect the initial replication stress-induced association of ATR with FRA3B, and subsequent sequestration by other non-fragile site regions, leading to DNA breaks at fragile sites. In addition, we demonstrate that the kinase activity of the ATR protein is required for binding to fragile DNAs in response to APH treatment. This study provides mechanistic insight into the CFS-specific ATR-dependent checkpoint response.
2. Materials and Methods
2.1. Cell culture and chemical treatments
HeLa cells and GM13069 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) and RPMI 1640 medium (Gibco) respectively, both supplemented with 10% (v/v) fetal bovine serum (Gibco). Cultures were incubated at 37°C in a humidified incubator with 5% CO2. The apparently healthy lymphoblastoid cell line GM13069 transformed by Epstein-Barr-virus was obtained from Coriell Cell Repositories (Camden, NJ). Cells were treated with either APH (Sigma) or APH plus 2-AP (Sigma) for 24 hrs at various concentrations as indicated in the figure legends. For fragile site analysis, cells were analyzed at the University Diagnostic Laboratories (RWJMS-UMDNJ) by the GTG banding method [39].
2.2. Chromatin Immunoprecipitation assay (ChIP)
ChIP assays were performed as described in Jiang and Sancar [40] with the following modifications. Briefly, 1.4 × 107 GM13069 or HeLa cells were grown in T75 flasks and subjected to either no treatment or treatments with APH or APH plus 2-AP before cross-linking with formaldehyde. A fixing solution (11% formaldehyde, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 50 mM Tris-Cl, pH 8.0) was mixed with the cells to a final concentration of 1% formaldehyde, and the mixture was incubated at room temperature for 20 minutes. The cross-linking reaction was terminated by adding glycine to a final concentration of 125 mM and incubating for 5 minutes. Chromatin was recovered by centrifugation and washed first with wash solution 1 (0.25 % Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris-HCl, pH 8.0) and then with wash solution 2 (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM Tris-HCl, pH 8.0). The cross-linked chromatin was dissolved in 0.5 ml TEE buffer (1 mM EDTA, 0.5 mM EGTA, 10 mM Tris- HCl, pH 8.0). The samples were sonicated using a Fisher Model 60 Sonic Dismembrator (Fisher Scientific). The supernatants were collected by centrifugation, and 1% of chromatin was aliquoted to check the amount of input DNA present in each sample. To reduce nonspecific background, the remaining solutions were incubated with salmon sperm DNA/protein A agarose slurry (Upstate Biotechnology) for 2 hours at 4°C. The pre-cleared supernatants were obtained by centrifugation, and 2 μg of rabbit polyclonal antibodies against human ATR (Affinity Bioreagents) were added to the supernatant and incubated overnight at 4°C. The DNA-protein-antibody complexes were collected by incubating with salmon sperm DNA/protein A agarose slurry for 45 min at 4°C followed by centrifugation. The agarose beads were washed with low salt, high salt, and LiCl immune complex wash buffers (Upstate Biotechnology) three times each. The DNA-protein complexes were eluted in freshly made 0.2% SDS/0.1 M NaHCO3 solution, and then reverse-crosslinked at 65 °C for 6 hours. DNA was further purified by phenol/chloroform extraction and ethanol precipitation. For each ChIP preparation, a no-antibody control, in which the same procedure was carried out in parallel but without the addition of anti-ATR antibody, was included.
2.3. PCR detection
The immunoprecipitated DNAs were analyzed by PCR, along with 0.1% of input DNA, for three regions of FRA3B and four non-fragile site regions. Three fragments of FRA3B: FRA3B_C5EXR (nt 60516530∼60516940 of GenBank accession number NT_022517), FRA3B_17.18 (nt 60719809∼60720021 of NT_022517), and FRA3B_24.26 (nt 61742271∼61742462 of NT_022517), and the four non-CFS fragments: 12p12.3 (nt 9314740∼9315076 of NT_009714), G6PD (nt 4595743∼4596222 of NT_011726), TFF1 (nt 781097∼781472 of NT_030188), and DHFR (30545737∼30546017 of NT_006713) were amplified by the primers listed in Supplementary Fig. 1. PCR products were separated on 2% agarose gels and detected by the FluorChem 8900 gel imaging system (Alpha Innotech). For each PCR reaction, 20 ng genomic DNA and a no-DNA control served as positive and negative PCR controls. The amount of PCR product in each band was quantitatively analyzed by AlphaeaseFC software (Alpha Innotech), and normalized against that of a genomic DNA control. For each primer set, a titration of genomic DNA was performed to ensure that the intensities of the bands were proportional to the amount of input DNA.
2.4. Western blot analysis
The cells from various treatments were collected prior to performing ChIP assays, and whole cell lysates were prepared and separated by SDS-PAGE gel electrophoresis. The Western blotting was carried out by transferring the proteins to pre-wetted PVDF membranes, and probing with antibodies for ATR (Affinity Bioreagents), phospho-ATR (Ser 428) (Cell Signaling), ATRIP (Abcam), TopBP1 (Abcam), RPA (Calbiochem), phospho-Chk1 (Ser 345) (Cell Signaling), and actin (Sigma). Proteins were detected using the Immun-Star HRP Chemiluminescent Kit (Bio-Rad).
2.5. Cell transfection
Two FLAG-tagged ATR pcDNA3 constructs containing the full-length cDNA clone of human ATR and the kinase-dead mutant (K2327R) [41,42] were provided by Aziz Sancar (UNC-Chapel Hill). Transfections of HeLa cells were carried out using Fugene6 Kits (Roche) according to the manufacturer's protocol with a DNA/lipid ratio of 3:1. After 30 h, the cells were subjected to treatment with various amounts of APH for an additional 24 h. ChIP and western blot analyses were performed as described above using an anti-FLAG antibody (Sigma).
3. Results
3.1. ATR preferentially binds to DNA regions of common fragile site FRA3B
To test whether ATR directly associates with CFSs, ChIP assays in conjunction with semi-quantitative PCR were employed to examine three regions of FRA3B: FRA3B_C5EXR, FRA3B_17.18, and FRA3B_24.26 (Fig. 1). FRA3B_C5EXR and FRA3B_17.18 are located within intron 4 of the FHIT gene [39]. In APH-treated hybrid cells, these regions represent two major clusters of APH-induced breakpoints in FRA3B [43]. Both are highly AT-rich, and contain the vertebrate topoisomerase II consensus sequences and the core autonomously replicating sequences [43]. The FRA3B_24.26 region is about 1 Mb centrometric from FRA3B_17.18, is located within intron 2 of the PTPRG gene, and has been analyzed as a highly flexible AT-rich region [28]. Two non-CFS DNAs located within the 12p12.3 chromosomal band and the G6PD gene, respectively, were also examined. The 12p12.3 region contains a high A/T content (76%), and has been analyzed as a non-flexible and non-fragile region [28], while the G6PD region is GC-rich (73%). The cross-linked chromatin samples were prepared from HeLa cells treated with varying amounts of APH (0, 0.2, 0.4, 0.7, and 1.0 μM) and then immunoprecipitated with anti-ATR antibody. The immunoprecipitated DNAs were subjected to PCR analysis.
We found that in HeLa cells, ATR bound to all three regions of FRA3B but did not bind to either of the non-CFS DNA regions 12p12.3 or G6PD, regardless of the concentration of APH (Fig. 2A). This result suggests that these three fragile DNA regions contain distinctive features recognized by ATR protein, and that this “binding signal” is not present in the two non-CFS DNA regions. It is worth noting that the binding of ATR to fragile DNAs was detected in the samples with no APH treatment, implying that the “binding signal” for ATR protein is already present in these fragile regions even without replication stress.
Fig. 2.
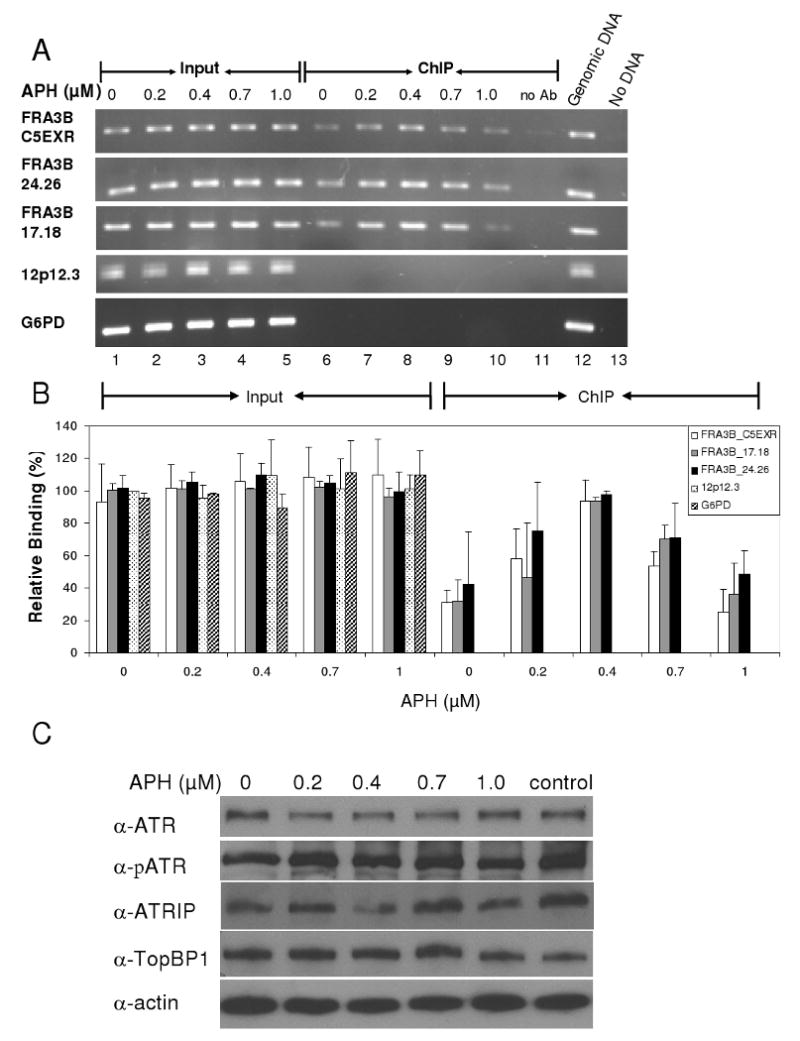
ATR protein preferentially binds to FRA3B fragile site in HeLa cells upon APH treatment. (A) A representative PCR analysis of DNA immunoprecipitated from HeLa cells with anti-ATR antibody. HeLa cells were treated with various APH concentrations (0, 0.2, 0.4, 0.7, and 1.0 μM). The 0.1% of input DNAs (lanes 1-5) and chromatin immunoprecipitated (ChIP) DNAs (lanes 6-10) were amplified by PCR at the following DNA regions: FRA3B_C5EXR, FRA3B_24.26, FRA3B_17.18, 12p12.3, and G6PD. For each ChIP assay, a no-antibody control (lane 11) was included. For each PCR reaction, there was a PCR positive control in which 20 ng genomic DNA served as PCR template (lane 12) and a PCR negative control in which no DNA template was added (lane 13). (B) Quantitation of the level of ATR protein binding to various DNA regions in HeLa cells. The amount of DNA in either input or ChIP samples, relative to that of the input of the untreated 12p12.3 sample (which was assigned a value of 1), is an average from three separate but identical experiments. Average with standard deviation is shown. (C) The amount of total cellular ATR, phosphorylated ATR, ATRIP, TopBP1 proteins during APH induction. Whole cell lysates from HeLa cells treated with various amounts of APH were separated by SDS-PAGE, and analyzed with the respective antibodies and an anti-actin antibody.
For all three fragile DNA regions, interaction with ATR depended upon the level of APH. The recruitment of ATR protein to the fragile DNA regions increased at up to 0.4 μM APH and decreased during treatment with 0.7 μM to 1.0 μM (Fig. 2B). Increased recruitment is likely to reflect the response to increasing replication stress, because ATR is a major sensor for stalled replication forks. Cytogenetic studies from our lab (data not shown) and others [2,44,45], have shown that APH concentrations above 0.4 μM, in folic acid-containing medium, induce a significantly higher number of breaks and gaps at fragile sites. Our observation, that ATR recruitment to fragile regions decreases when replication stress is increased beyond 0.4 μM APH, could provide an explanation for this phenomenon.
We also evaluated the amount of total cellular ATR protein following APH treatment, using whole cell lysates collected immediately prior to performing ChIP and subjected to Western blots. We found that there were no apparent differences in the amount of ATR or phospho-ATR (Ser 428) protein at the various concentrations of APH (Fig. 2C). Two ATR-interacting proteins, ATRIP and TopBP1, that respond to DNA damage by binding to ATR, also remained unchanged at the total protein level. This observation supports the notion of ATR redistribution during APH treatment, with initial binding at “sensitive” sites such as fragile sites in response to low doses of APH, but subsequent sequestration from fragile sites by other regions which react to replication stress at a higher concentration of APH.
We also evaluated ATR binding to fragile DNAs in a lymphoblastoid cell line GM13069, derived from an apparently healthy individual (Fig. 3). ATR bound to all three regions of FRA3B and had a binding profile similar to that of HeLa cells, with peak binding at 0.4 μM APH and decreased binding at higher concentrations. Interestingly, ATR also bound to the non-fragile 12p12.3 region in GM13069 cells. This discrepancy between HeLa and GM13069 cells could be due to the highly AT-rich sequence of the 12p12.3 region, which is a characteristic of fragile sites and may have allowed some proteins in the relatively “normal” GM13069 cells to promote ATR recognition. Notably, no binding occurred to G6PD, and we subsequently examined two additional non-CFS regions – TFF1 and DHFR. The TFF1 gene encodes the trefoil factor 1 precursor, a gastrointestinal mucosa protein, while the DHFR gene encodes dihydrofolate reductase. The AT content of the TFF1 and DHFR regions is 48 and 59%, respectively. We found there was no detectable ATR binding to these two regions (Fig. 3). Therefore, these results suggest that in both cell lines, ATR protein displays preferential binding to fragile site FRA3B regions.
Fig. 3.
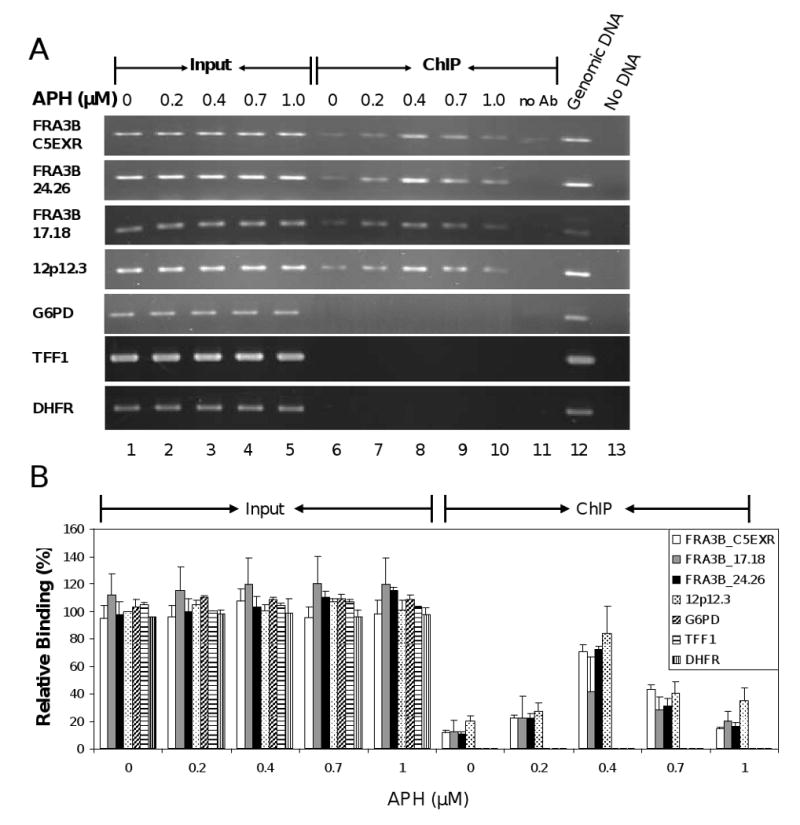
ATR protein binds FRA3B fragile site in GM13069 cells upon APH treatment. (A) A representative PCR analysis of DNA immunoprecipitated from GM13069 cells with anti-ATR antibody. GM13069 cells were treated with various APH concentrations (0, 0.2, 0.4, 0.7, and 1.0 μM). The 0.1% of input DNAs (lanes 1-5) and chromatin immunoprecipitated DNAs (lanes 6-10) were amplified by PCR at the following DNA regions: FRA3B_C5EXR, FRA3B_24.26, FRA3B_17.18, 12p12.3, G6PD, TFF1 and DHFR. For each ChIP assay, a no-antibody control (lane 11) was included. For each PCR reaction, there was a PCR positive control in which 20 ng genomic DNA served as PCR template (lane 12) and a PCR negative control in which no DNA template was added (lane 13). (B) Quantitation of the level of ATR protein binding to various DNA regions in GM13069 cells. The amount of DNA in either input or ChIP samples, relative to that of the input of the untreated 12p12.3 sample (which was assigned a value of 1), is an average from four separate but identical experiments. Average with standard deviation is shown.
3.2. Kinase activity of ATR protein is required for binding to fragile DNA in response to APH treatment
The kinase activity of ATR protein is essential to the ATR-dependent DNA damage checkpoint pathway. It phosphorylates substrates such as p53, BRCA1, Chk1, and Rad17 to extend checkpoint signaling, resulting in the inhibition of DNA replication and mitosis, and supporting DNA repair and recombination [34,46]. However, it is not clear whether kinase activity is required for binding to DNA, especially in the case of CFSs. To examine the role of kinase activity in the binding of ATR protein to CFS DNA in response to APH treatment, we investigated the binding of ATR to DNA using ChIP analysis of cells treated with various amounts of 2-AP, a general kinase inhibitor, along with 0.4 μM APH. A concentration of 0.4 μM APH was chosen to maximize ATR binding and improve the chances of detecting reduced binding caused by 2-AP.
Treatment of GM13069 cells with increasing amounts of 2-AP decreased the level of ATR protein binding to all three regions of FRA3B (Fig. 4A and B) without altering the amount of total cellular ATR protein (Fig. 4C), indicating that kinase activity is essential for the binding of ATR to CFS FRA3B regions. A similar outcome was observed in HeLa cells (data not shown). These results are consistent with previous studies demonstrating that 2-AP in combination with APH significantly increases overall chromosomal gaps and breaks, and also increases the expression of specific fragile sites such as FRA3B [2]. Presumably, the reduced binding of ATR protein due to the 2-AP treatment causes a less active ATR-dependent DNA damage response. Chk1, a major effector in the ATR-dependent pathway, requires direct phosphorylation by ATR to activate its function. Therefore, the observation that the level of phosph-Chk1 (Ser 345) decreases as the concentration of 2-AP increases (Fig. 4C) reveals the effect of 2-AP/APH treatment on ATR kinase and on the ATR-dependent DNA damage response.
Fig. 4.
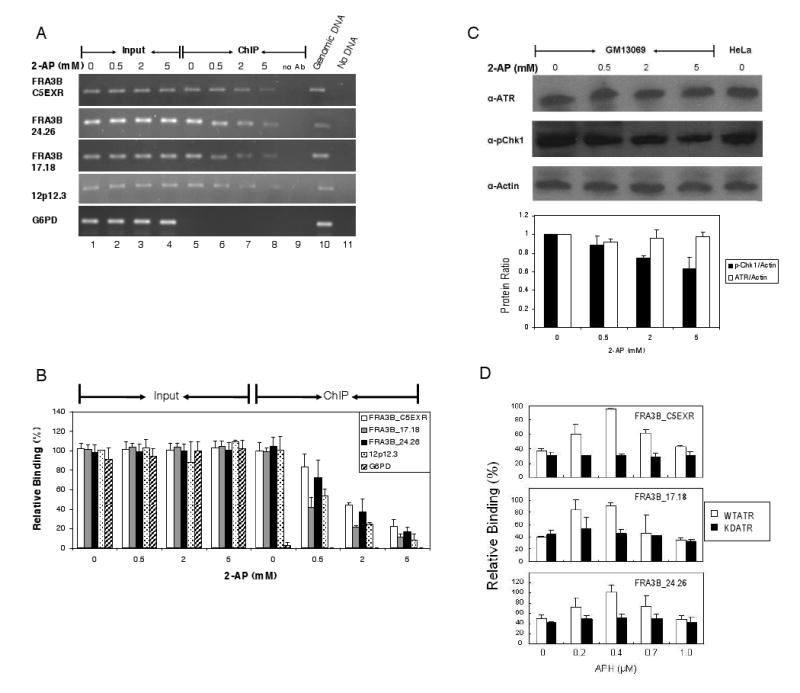
Kinase activity of ATR protein is required for binding to fragile DNA in response to APH treatment. (A) A representative PCR analysis of DNAs immunoprecipitated from GM13069 cells with anti-ATR antibody. GM13069 cells were treated with various 2-AP concentrations (0, 1, 2, and 5 mM), an inhibitor of ATR kinase activity, along with 0.4 μM APH. The 0.1 % of input DNA (lanes 1-4) and chromatin immunoprecipitated DNA (lanes 5-8) were amplified by PCR at the following DNA regions: FRA3B_C5EXR, FRA3B_24.26, FRA3B_17.18, 12p12.3, and G6PD. For each ChIP assay, a no-antibody control (lane 9) was included. For each PCR reaction, there was a PCR positive control in which 20 ng genomic DNA served as PCR template (lane 10) and a PCR negative control in which no DNA template was added (lane 11). (B) Quantitation of the level of ATR protein binding to various DNA regions in GM13069 cells. The amount of DNA in either input or ChIP samples, relative to that of the input of the untreated 12p12.3 sample (which was assigned a value of 1), is an average from four separate but identical experiments. Average with standard deviation is shown. (C) The amount of total cellular ATR protein during 2-AP treatment. Whole cell lysates from GM13069 cells, that were treated with various 2-AP concentrations along with 0.4 μM APH, were separated by SDS-PAGE, and analyzed with anti-ATR, anti-phospho-Chk1 (Ser345), or anti-actin antibodies. Quantitative analysis of protein ratios for ATR/actin (white bar) and p-Chk1/actin (black bar) was plotted. Average with standard deviation is shown. (D) ATR protein binding to three FRA3B regions in HeLa cells transfected with FLAG-tagged wild type (open bars) or kinase-dead (black bars) ATR constructs. Thirty hours after transfection, the cells were treated with APH (0, 0.2, 0.4, 0.7, and 1.0 μM) for an additional 24 h. ChIP assays were carried out with anti-FLAG antibody, and chromatin immunoprecipitated DNAs were amplified by PCR at the FRA3B_C5EXR, FRA3B_24.26, FRA3B_17.18, and 12p12.3 regions. The amount of DNA in ChIP samples, relative to the DNA input of the 12p12.3 sample from untreated, wild-type ATR transfected cells (which was assigned a value of 1), is an average of three separate but identical experiments. Average with standard deviation is shown. Both types of ATR showed no binding to the non-fragile 12p12.3 region (data not shown).
Because 2-AP is a general kinase inhibitor and not ATR specific, we next employed transient transfection of a FLAG-tagged wild type or kinase-dead mutant (K2327R) [41,42] ATR construct into HeLa cells to assess whether ATR kinase activity was specifically required for binding to common fragile sites. The expression levels of the recombinant wild-type and mutant ATR protein were similar (Supplementary Fig. 2). In the absence of APH, the kinase-dead mutant protein bound to all three FRA3B regions to an extent which was similar to that of wild type ATR (Fig. 4D), and was comparable to the ATR binding at high doses of 2-AP (Fig. 4B). Upon APH treatment, the level of binding of the kinase-dead mutant did not change significantly, in contrast to the APH-induced binding pattern of wild-type ATR (Fig. 4D) which was in agreement with our previous observations of endogenous ATR in un-transfected cells (Fig. 2). In all three FRA3B regions, at a treatment of 0.4 μM APH, the increased binding of wild-type ATR was statistically significant compared to that of the kinase-dead mutant (P<0.01). These results make an important distinction by indicating that ATR kinase activity is not required for this protein to bind to FRA3B regions under normal conditions, but that it is required for ATR binding to FRA3B under conditions of replication stress caused by APH.
4. Discussion
The ATR-dependent DNA damage checkpoint pathway is important in maintaining CFS stability, especially under conditions of replication stress [1-5]. In this study, we provide direct evidence that ATR binds to the three FRA3B regions spanning over ∼1.2 Mb in both HeLa and GM13069 cells. Binding is enhanced under mild replication stress caused by low doses of APH up to 0.4 μM. It has been shown that at the stalled replication fork, ATR associated with ATR-interacting protein (ATRIP) can be loaded onto RPA-coated single-stranded DNA [47,48]. Also, TopBP1, localized by the Rad9-Hus1-Rad1 complex at the stalled replication fork, was shown to bind and recruit ATR to DNA in response to DNA damage [49]. Therefore, the ATR binding of fragile sites that we observed is likely to be mediated through these protein complexes. However, we cannot exclude the possibility that ATR binding could be due to direct recognition of certain features of fragile DNAs, as shown in the case of direct ATR binding to UV-damaged DNA [41]. Regardless of whether ATR acts through complex or direct binding, the evidence of the recruitment of ATR at FRA3B regions upon APH treatment supports a possible local role of ATR in stabilizing the stalled replication forks at FRA3B [50].
Perhaps more importantly, our studies also demonstrate that ATR displays preferential binding to fragile site FRA3B DNA as compared to non-fragile DNA, suggesting that fragile DNA regions are more sensitive to ATR activation and may form a stronger “checkpoint signal” than non-fragile DNA regions. It is known that, in addition to RPA-coated single-stranded DNA, a primer/template-like structure is needed for ATR checkpoint activation [51-53]. Delayed replication is a general feature of the fragile sites examined to date including FRA3B [22,29], and this could result in a longer, unreplicated single-stranded DNA in fragile site regions. In combination with APH treatment, that inhibits DNA polymerases α, δ, and ε but not helicase/primase complexes [44,54], lagging strand regions of the DNA replication fork at fragile sites can perhaps have a higher number of primer/template-like structures to create a stronger “checkpoint signal” for ATR recognition. Furthermore, other characteristic features of CFS sequences are their high AT-content with AT islands or AT-rich minisatellite repeats and their propensity to form stable secondary structure [28], which could provide both sequence [55,56] and structural elements [57,58] for topoisomerase II binding. In fact, all three FRA3B regions that we examined are highly flexible and AT-rich. Further, the FRA3B_C5EXR and FRA3B_17.18 regions contain the vertebrate topoisomerase II consensus sequences [43], and the presence of topoisomerase II/TopBP1 at the fragile site regions could increase the recruitment of ATR at these locations. Irrespective of the specific features that fragile sites are presenting, our results strongly suggest that ATR can distinguish fragile site FRA3B from non-fragile site regions.
As the concentration of APH treatment increased from 0.4 to 1.0 μM, which only partially inhibits replication and does not arrest the cell cycle [44,45], the binding of ATR to fragile site regions decreased. Because the total amounts of ATR and phosphorylated ATR were unchanged during APH treatment, as were the amounts of ATRIP and TopBP1, the decrease in the binding of ATR to FRA3B regions may have resulted from the sequestration of ATR protein by other non-fragile site regions, and this could account for the increasing number of chromosome gaps and breaks at fragile sites which are observed under the same conditions [2,44,45]. Also, in comparing Seckel syndrome affected patient and normal cells, not only is there a more significant increase in fragile site breaks at APH concentrations ranging from 0.5∼0.9 μM than at lower concentrations, but the presence of metaphase chromosomes with a “shattered” morphology was observed only at higher APH concentrations in patient cells, which indicates widely scattered chromosome damage [2]. These studies support the idea that redistribution of ATR from fragile sites to other genome regions occurs at higher APH concentrations, and with a shortage of ATR such as in Seckel syndrome cells, breaks at fragile sites and non-fragile sites are both elevated at the higher APH concentrations.
In response to DNA damage, ATR protein phosphorylates substrates such as p53, BRCA1, and Chk1, to initiate and extend the checkpoint signal cascade [34,46]. Our observation that ATR kinase activity is essential for its binding to the FRA3B regions in response to APH, indicates that the kinase activity of ATR may be also critical for damage recognition at an early step of ATR-dependent checkpoint activation. The requirement of kinase activity for the interaction between ATR and the fragile site regions is likely due to the necessity of phosphorylation of proteins such as ATRIP and TopBP1 for stabilizing the interaction between these proteins and ATR. Indeed, it was found that phosphorylation of ATRIP by ATR kinase is required for the interaction between ATR and ATRIP [59,60]. TopBP1 is also a substrate for ATR, and its phosphorylation promotes its binding to the ATR-ATRIP complex and the activation of ATR kinase activity, resulting in a feedback loop of ATR activation at a stalled replication fork [61]. Therefore, decreasing ATR kinase activity, which could cause a reduction in the formation of the ATR-ATRIP complex, possibly results in impaired interactions between ATR and FRA3B regions.
ATR is a key protein in the DNA damage repair pathway and also plays an important role in chromosomal fragile site stability. Two models which are not mutually exclusive have been proposed for the role of ATR in fragile site instability: checkpoint failure and replication fork collapse [50]. Here, we demonstrate that under mild APH treatment, fragile site FRA3B DNAs are preferential regions for ATR protein to bind and suggest that ATR could play a local role in stabilizing the stalled replication forks at FRA3B, and also that fragile sites could indeed present a checkpoint signal to activate the ATR-dependent pathway. The observation of a decrease in ATR binding to fragile sites at relatively higher APH concentrations, which is known to produce more chromosomal breaks and gaps, supports both models in which the generation of fragile sites is caused by failure of ATR to stabilize the stalled replication forks at fragile sites, and/or their escape from checkpoint activation. Therefore, our studies provide further insight into the mechanism for the regulation of fragile site stability by ATR.
Supplementary Material
Acknowledgments
We sincerely thank Dr. Aziz Sancar (University of North Carolina, Chapel Hill) for providing both the wild type and kinase-dead mutant ATR constructs. This work was supported by the National Cancer Institute (CA085826 to Y.-H. W).
Footnotes
Conflict of interest statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 2.Casper AM, Durkin SG, Arlt MF, Glover TW. Chromosomal instability at common fragile sites in Seckel syndrome. Am J Hum Genet. 2004;75:654–660. doi: 10.1086/422701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arlt MF, Xu B, Durkin SG, Casper AM, Kastan MB, Glover TW. BRCA1 is required for common-fragile-site stability via its G2/M checkpoint function. Mol Cell Biol. 2004;24:6701–6709. doi: 10.1128/MCB.24.15.6701-6709.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006;25:4381–4388. doi: 10.1038/sj.onc.1209466. [DOI] [PubMed] [Google Scholar]
- 5.Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 6.Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–192. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 7.Burrow AA, Williams LE, Pierce LC, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009;10:59. doi: 10.1186/1471-2164-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuo MT, Vyas RC, Jiang LX, Hittelman WN. Chromosome breakage at a major fragile site associated with P-glycoprotein gene amplification in multidrug-resistant CHO cells. Mol Cell Biol. 1994;14:5202–5211. doi: 10.1128/mcb.14.8.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 10.Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, Trakhtenbrot L, Kerem B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1:89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- 11.Miller CT, Lin L, Casper AM, Lim J, Thomas DG, Orringer MB, Chang AC, Chambers AF, Giordano TJ, Glover TW, Beer DG. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene. 2006;25:409–418. doi: 10.1038/sj.onc.1209057. [DOI] [PubMed] [Google Scholar]
- 12.Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 13.Boldog F, Gemmill RM, West J, Robinson M, Robinson L, Li E, Roche J, Todd S, Waggoner B, Lundstrom R, Jacobson J, Mullokandov MR, Klinger H, Drabkin HA. Chromosome 3p14 homozygous deletions and sequence analysis of FRA3B. Hum Mol Genet. 1997;6:193–203. doi: 10.1093/hmg/6.2.193. [DOI] [PubMed] [Google Scholar]
- 14.Finnis M, Dayan S, Hobson L, Chenevix-Trench G, Friend K, Ried K, Venter D, Woollatt E, Baker E, Richards RI. Common chromosomal fragile site FRA16D mutation in cancer cells. Hum Mol Genet. 2005;14:1341–1349. doi: 10.1093/hmg/ddi144. [DOI] [PubMed] [Google Scholar]
- 15.Popescu NC, DiPaolo JA, Amsbaugh SC. Integration sites of human papillomavirus 18 DNA sequences on HeLa cell chromosomes. Cytogenet Cell Genet. 1987;44:58–62. doi: 10.1159/000132342. [DOI] [PubMed] [Google Scholar]
- 16.Smith PP, Friedman CL, Bryant EM, McDougall JK. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer. 1992;5:150–157. doi: 10.1002/gcc.2870050209. [DOI] [PubMed] [Google Scholar]
- 17.Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet. 1996;5:187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- 18.Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22:1225–1237. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 19.Bester AC, Schwartz M, Schmidt M, Garrigue A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Ben-Porat N, Von Kalle C, Fischer A, Kerem B. Fragile sites are preferential targets for integrations of MLV vectors in gene therapy. Gene Ther. 2006;13:1057–1059. doi: 10.1038/sj.gt.3302752. [DOI] [PubMed] [Google Scholar]
- 20.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Smith DI, Huang H, Wang L. Common fragile sites and cancer (review) Int J Oncol. 1998;12:187–196. [PubMed] [Google Scholar]
- 22.Wang L, Darling J, Zhang JS, Huang H, Liu W, Smith DI. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum Mol Genet. 1999;8:431–437. doi: 10.1093/hmg/8.3.431. [DOI] [PubMed] [Google Scholar]
- 23.Durkin SG, Ragland RL, Arlt MF, Mulle JG, Warren ST, Glover TW. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci U S A. 2008;105:246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becker NA, Thorland EC, Denison SR, Phillips LA, Smith DI. Evidence that instability within the FRA3B region extends four megabases. Oncogene. 2002;21:8713–8722. doi: 10.1038/sj.onc.1205950. [DOI] [PubMed] [Google Scholar]
- 25.Corbin S, Neilly ME, Espinosa R, 3rd, Davis EM, McKeithan TW, Le Beau MM. Identification of unstable sequences within the common fragile site at 3p14.2: implications for the mechanism of deletions within fragile histidine triad gene/common fragile site at 3p14.2 in tumors. Cancer Res. 2002;62:3477–3484. [PubMed] [Google Scholar]
- 26.Huebner K, Garrison PN, Barnes LD, Croce CM. The role of the FHIT/FRA3B locus in cancer. Annu Rev Genet. 1998;32:7–31. doi: 10.1146/annurev.genet.32.1.7. [DOI] [PubMed] [Google Scholar]
- 27.Mishmar D, Rahat A, Scherer SW, Nyakatura G, Hinzmann B, Kohwi Y, Mandel-Gutfroind Y, Lee JR, Drescher B, Sas DE, Margalit H, Platzer M, Weiss A, Tsui LC, Rosenthal A, Kerem B. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A. 1998;95:8141–8146. doi: 10.1073/pnas.95.14.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zlotorynski E, Rahat A, Skaug J, Ben-Porat N, Ozeri E, Hershberg R, Levi A, Scherer SW, Margalit H, Kerem B. Molecular basis for expression of common and rare fragile sites. Mol Cell Biol. 2003;23:7143–7151. doi: 10.1128/MCB.23.20.7143-7151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Beau MM, Rassool FV, Neilly ME, Espinosa R, 3rd, Glover TW, Smith DI, McKeithan TW. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: implications for the mechanism of fragile site induction. Hum Mol Genet. 1998;7:755–761. doi: 10.1093/hmg/7.4.755. [DOI] [PubMed] [Google Scholar]
- 30.Hellman A, Rahat A, Scherer SW, Darvasi A, Tsui LC, Kerem B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol Cell Biol. 2000;20:4420–4427. doi: 10.1128/mcb.20.12.4420-4427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palakodeti A, Han Y, Jiang Y, Le Beau MM. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer. 2004;39:71–76. doi: 10.1002/gcc.10290. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Freudenreich CH. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell. 2007;27:367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 34.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Musio A, Montagna C, Mariani T, Tilenni M, Focarelli ML, Brait L, Indino E, Benedetti PA, Chessa L, Albertini A, Ried T, Vezzoni P. SMC1 involvement in fragile site expression. Hum Mol Genet. 2005;14:525–533. doi: 10.1093/hmg/ddi049. [DOI] [PubMed] [Google Scholar]
- 36.Zhu M, Weiss RS. Increased common fragile site expression, cell proliferation defects, and apoptosis following conditional inactivation of mouse Hus1 in primary cultured cells. Mol Biol Cell. 2007;18:1044–1055. doi: 10.1091/mbc.E06-10-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol. 2008;180:305–314. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, le Sage C, Chen BP, Chen DJ, Agami R, Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005;19:2715–2726. doi: 10.1101/gad.340905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulvihill DJ, Wang YH. Two breakpoint clusters at fragile site FRA3B form phased nucleosomes. Genome Res. 2004;14:1350–1357. doi: 10.1101/gr.2304404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang G, Sancar A. Recruitment of DNA damage checkpoint proteins to damage in transcribed and nontranscribed sequences. Mol Cell Biol. 2006;26:39–49. doi: 10.1128/MCB.26.1.39-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci U S A. 2002;99:6673–6678. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Paradee W, Mullins C, Shridhar R, Rosati R, Wilke CM, Glover TW, Smith DI. Aphidicolin-induced FRA3B breakpoints cluster in two distinct regions. Genomics. 1997;41:485–488. doi: 10.1006/geno.1997.4690. [DOI] [PubMed] [Google Scholar]
- 44.Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet. 1984;67:136–142. doi: 10.1007/BF00272988. [DOI] [PubMed] [Google Scholar]
- 45.Glover TW, Stein CK. Chromosome breakage and recombination at fragile sites. Am J Hum Genet. 1988;43:265–273. [PMC free article] [PubMed] [Google Scholar]
- 46.Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004;3:901–908. doi: 10.1016/j.dnarep.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 47.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 48.Kumagai A, Kim SM, Dunphy WG. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J Biol Chem. 2004;279:49599–49608. doi: 10.1074/jbc.M408353200. [DOI] [PubMed] [Google Scholar]
- 49.Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cimprich KA. Fragile sites: breaking up over a slowdown. Curr Biol. 2003;13:R231–233. doi: 10.1016/s0960-9822(03)00158-1. [DOI] [PubMed] [Google Scholar]
- 51.Michael WM, Ott R, Fanning E, Newport J. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science. 2000;289:2133–2137. doi: 10.1126/science.289.5487.2133. [DOI] [PubMed] [Google Scholar]
- 52.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang DJ, Lupardus PJ, Cimprich KA. Monoubiquitination of proliferating cell nuclear antigen induced by stalled replication requires uncoupling of DNA polymerase and mini-chromosome maintenance helicase activities. J Biol Chem. 2006;281:32081–32088. doi: 10.1074/jbc.M606799200. [DOI] [PubMed] [Google Scholar]
- 55.Spitzner JR, Muller MT. A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucleic Acids Res. 1988;16:5533–5556. doi: 10.1093/nar/16.12.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spitzner JR, Chung IK, Muller MT. Eukaryotic topoisomerase II preferentially cleaves alternating purine-pyrimidine repeats. Nucleic Acids Res. 1990;18:1–11. doi: 10.1093/nar/18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zechiedrich EL, Osheroff N. Eukaryotic topoisomerases recognize nucleic acid topology by preferentially interacting with DNA crossovers. Embo J. 1990;9:4555–4562. doi: 10.1002/j.1460-2075.1990.tb07908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Howard MT, Lee MP, Hsieh TS, Griffith JD. Drosophila topoisomerase II-DNA interactions are affected by DNA structure. J Mol Biol. 1991;217:53–62. doi: 10.1016/0022-2836(91)90610-i. [DOI] [PubMed] [Google Scholar]
- 59.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 60.Bomgarden RD, Yean D, Yee MC, Cimprich KA. A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J Biol Chem. 2004;279:13346–13353. doi: 10.1074/jbc.M311098200. [DOI] [PubMed] [Google Scholar]
- 61.Burrows AE, Elledge SJ. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes Dev. 2008;22:1416–1421. doi: 10.1101/gad.1685108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.