

Abstract
Patterns in sequences of amino acid hydrophobic free energies predict secondary structures in proteins. In protein folding, matches in hydrophobic free energy statistical wavelengths appear to contribute to selective aggregation of secondary structures in “hydrophobic zippers.” In a similar setting, the use of Fourier analysis to characterize the dominant statistical wavelengths of peptide ligands’ and receptor proteins’ hydrophobic modes to predict such matches has been limited by the aliasing and end effects of short peptide lengths, as well as the broad-band, mode multiplicity of many of their frequency (power) spectra. In addition, the sequence locations of the matching modes are lost in this transformation. We make new use of three techniques to address these difficulties: (i) eigenfunction construction from the linear decomposition of the lagged covariance matrices of the ligands and receptors as hydrophobic free energy sequences; (ii) maximum entropy, complex poles power spectra, which select the dominant modes of the hydrophobic free energy sequences or their eigenfunctions; and (iii) discrete, best bases, trigonometric wavelet transformations, which confirm the dominant spectral frequencies of the eigenfunctions and locate them as (absolute valued) moduli in the peptide or receptor sequence. The leading eigenfunction of the covariance matrix of a transmembrane receptor sequence locates the same transmembrane segments seen in n-block-averaged hydropathy plots while leaving the remaining hydrophobic modes unsmoothed and available for further analyses as secondary eigenfunctions. In these receptor eigenfunctions, we find a set of statistical wavelength matches between peptide ligands and their G-protein and tyrosine kinase coupled receptors, ranging across examples from 13.10 amino acids in acid fibroblast growth factor to 2.18 residues in corticotropin releasing factor. We find that the wavelet-located receptor modes in the extracellular loops are compatible with studies of receptor chimeric exchanges and point mutations. A nonbinding corticotropin-releasing factor receptor mutant is shown to have lost the signatory mode common to the normal receptor and its ligand. Hydrophobic free energy eigenfunctions and their transformations offer new quantitative physical homologies in database searches for peptide-receptor matches.
The importance of the sequential arrangements of amino acid side chain hydrophobicities in the determination of peptide and protein secondary structures has been established knowledge in protein biology and physics for many decades (1, 2). Determinants of polypeptide interactions, such as those between peptide segments in protein folding, are coded in the one-dimensional sequence space of physical characteristics (3), most prominently the hydrophobic free energies of their individual amino acids (4). These chemical potentials have been approximated as relative solubility in organic vs. aqueous solvents in which glycine = 0 and quantified as hydrophobic free energies in kcal/mol (5, 6). The hydrophobic attractive forces in newtons between hydrophobic moieties as a function of surface area and radius of curvature are measured by using techniques such as atomic force microscopy (7) and are up to two orders of magnitude larger than those predicted by van der Waals theory and extend spatially in a slower-than-exponential decay to beyond 20 nanometers (8). Complete substitution of hydrophobically equivalent amino acids in peptides maintains and sometimes increments their peptide-receptor mediated physiological potency (9, 10). Helical secondary structures of differing turn lengths can be designed with sequences of amino acids of high and low hydrophobicities, independent of the specific amino acids chosen within each hydrophobicity class (11).
Secondary structures with matching hydrophobic amplitude variational frequencies, such as in the β-strands of interleukin 1β, have been shown to bind together and initiate protein folding (12) in a process called the “hydrophobic zipper” (13). Two long, helical secondary structures with congruent hydrophobic frequencies bind to create the central “hydrophobic knot” that stabilizes the structure of phospholipase A2 (14). Recent studies of the binding of extracellular domains of growth hormone receptor by polyclonal antibodies to ovine growth hormone was shown to be related to common helical, loop, and/or disordered secondary structure (15).
In earlier studies, we found that when the amino acid sequences of neuropeptides and peptide hormones were transformed into their individual hydrophobic free energies, functional families demonstrated similarities in hydrophobic free energy power spectral modes. We refer to the statistically dominant power spectral (inverse frequency) wavelengths in amino acid residues as h(ω). Family members shared the same h(ω), though differing in their ordered amino acid content by as much as 60% (16, 17). The range of h(ω) included the well known h(ω) = 3.6 and h(ω) = 2.0 of the α-helix and β-strand, respectively (18), but many others as well, ranging from the h(ω) = 13.10 amino acid residue mode of acid fibroblast growth factor to the h(ω) = 2.18, which dominates the hydrophobic free energy power spectrum of corticotropin releasing factor.
The AIDS coat protein manifests a waxing and waning h(ω) = 7–9 [observed by sliding a 50 residue windowed Fourier transform (19) along its sequence], which appears to be conserved across many of its mutations (17). Fibroblast growth factor (FGF; at the time, of unknown molecular mechanism) was predicted and confirmed to have a regulatory influence on ribonuclease A, with which it was found to share dominant hydrophobic modes (20). The specific amino acid sequences of the calcitonins, the peptide hormone family that regulates the rate of enzymatic bone catabolism, vary by ≈60% across species, but all are dominated by a h(ω) = 3.6 (21). Our system of analysis demonstrates the expected hydrophobic mode invariance under translation and reflection that has been studied experimentally in new peptide design by using nucleotide complementation (22).
Among the difficulties encountered in our previous work is that the usual Fourier, power spectral transformation is at best a crude approximation when used on these short and irregular data series with aliasing, end effects and broad-band multimodality. This study reports the results of the new use of three consecutive linear decompositions and transformations, making hydrophobic mode isolation and identification less ambiguous. Their successful application to examples from a peptide and ligand receptor sequence database‖ support the claim that these peptide ligand modes are functionally relevant.
The component of the peptide-receptor ligand binding process of interest here is the initial, long-range, faster-than-diffusive, selective aggregation (“hydrophobic zippering”) of peptides with extramembranous transmembrane receptor segments. This process precedes their internalization and equilibrium charge or ionic binding within the membrane (23–25). Inside the low dielectric constant environment of the lipid membrane, amino acid side chains with charged and/or ionic groups are protected from charge-screening by the aqueous and ionic extramembranous fluid, and dominate the binding mechanism. In contrast, hydrophobic mode matches in peptide ligand-receptor pairs are assumed to play a significant role in the extramembranous, high dielectric constant, charge-screened, aqueous portions of the receptor sequences.
de Gennes’ work (26) on the statistical mechanics of constrained polymers and polymeric reptation and models of the behavior of one-dimensional polymers with immobilized ends making “trains” and/or “loops” on a surface or in a membrane (27) predict that extramembranous ligand-receptor hydrophobic mode aggregation would increase the local hydrophobic free energy density above some critical threshold for internalization. In the vicinity of the metastable boundaries between the intramembranous and extramembranous portions of the receptor sequence, this would favor the internalization of that ligand-receptor segment into the membrane where charge and ion-dependent equilibrium binding can occur more easily.
Construction of Hydrophobic Free Energy Sequences and Their Eigenfunctions from Orthogonal Decompositions
Among the many amino acid hydrophobicity scales (28, 29), we have consistently used the results of partitions in binary, aqueous-organic solvents begun in the pioneering work of Edsall in the 1930s (5, 6). We chose this system because of its long history of successful use and our finding that it had a statistically significant correlation (R2 = 0.723, P = 0.00475) with a measurable physical property, amino acid partial specific volume (in ml/gm) (30), in the physiological circumstance for which we wish to make predictive inferences. Tanford and coworkers (31) demonstrated that amino acid free energy relations were associated with aqueous cavity surface area derived from these measures made by using the aqueous partition. Our results are consonant with those achieved with the currently more popular, but less physiological, method of condensed vapor partition (32). This agreement in the patterns of the hydropathy plots of the seven transmembrane segments is seen, for example, in graphs of the very well studied rhodopsin I (see Fig. 1, upper left).
Figure 1.

(Left) Graphic results of asymptotically smoothing, 32 iterated nearest neighbor averaging computations, the “hydropathy plots” of the hydrophobic free energy series, Hi, of the amino acid sequences, Ai, of rhodopsin I, the kappa and mu opioid, the corticotropin releasing factor, and the cholecystokinin B seven-transmembrane receptors. (Right) Graphs of the leading (transmembrane) eigenfunction, ΨT, of the same Hi. Note that for the same Hi, the hydropathy plots and those of ΨT are quite similar.
To assign hydrophobic free energy values, Hi, to each amino acid, Ai, of the peptide ligand and its transmembrane receptor, we used the results of studies in which each amino acid’s H was determined from its relative concentration at equilibrium in the two phases of a binary solvent, and quantified as a free energy of transfer, in kcal/mol, from a nonpolar hydrocarbon solvent to water at 37°C, with glycine = 0 as a reference (5, 6). This results in the following set of amino acid hydrophobic free energy equivalents, Hi, in kcal/mol: G, Q = 0.0; S, T = 0.07; N = 0.09; D = 0.66; E = 0.67; R = 0.85; A, H = 0.87; C = 1.52; K = 1.65; M = 1.67; V = 1.87; L = 2.17; Y = 2.76; P = 2.77; F = 2.87; I = 3.15; and W = 3.77.
Let each amino acid sequence of length N, A1, A2, … , AN, be represented by a sequence of hydrophobic free energy values, H1, H2, … , HN, where Hi represents the hydrophobic free energy of the amino acid Ai in the ith place in the amino acid sequence, using the H values listed above.
From the lagged vectors, V1 = (H1, H2, … , HM), V2 = (H2, H3, … , HM+1), … , VN−M+1 = (HN−M+1, HN−M+2, … , HN), with M ≈ 16, an M × M covariance matrix, CM, is formed in which K = N − M + 1 and N ranges along the receptor sequence lengths from ≈400 to 1,200 residues. M is chosen to maximize the least square fits of the first eigenfunctions (ΨT, Fig. 1 Right) with the standard hydropathy plots (Fig. 1 Left) used to demonstrate the (seven) transmembrane segments (32).
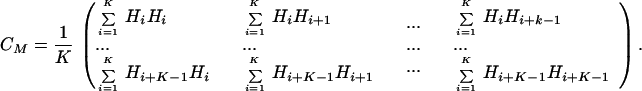 |
We compute the eigenvalues, {νi}i=1M, and the associated eigenvectors, Xi(j), of CM, where i =1 … M labels the eigenvector, and j = 1 to M refers to the jth component of the eigenvector Xi(j). The {νi}i=1M are ordered largest to smallest as are the corresponding Xi(j). The ordered Xi(j) are then used as multiplicative “weights” to transform the H1, H2, … , HN into M statistically weighted eigenfunctions, Ψi(j), where i = 1… M labels the eigenvector and j = 1… N − M indexes its jth component. The Ψi(j), for j − k + 1 > 0, are given by
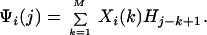 |
In effect, CM contains a scan for hydrophobic modes across a range of covariance/correlation lengths from 1 to M. Because CM is by definition real, symmetric, and normal, its {νi}i=1M are real, nonnegative, and distinct, and its associated Xi(j) constitute natural bases for orthonormal projections on H1, H2, … , HN. The set of Ψi(j) are orthonormally decomposed sequences of moving average values (33–35). We designate the leading eigenfunction representing the transmembrane segments of G-coupled receptor proteins as ΨT, the secondary eigenfunction containing the peptide receptor modes as ΨR, and the leading peptide ligand eigenfunction as ΨL, when the peptide is long enough to permit its construction.
Best Bases, Discrete Trigonometric Wavelet Transformation of Hydrophobic Free Energy Eigenfunctions
Wavelet transformations of the receptor and ligand hydrophobic free energy eigenfunctions generate wavelet graphs, W(ΨR) ≡ WR and W(ΨL) ≡ WL. Wavelet transformation, W(a,b), consists of decomposing the ΨR values into translated W(n) → W(n − b) and scaled W(n) → [W(n)/a] (scale is analogous to the inverse radian frequency of a trigonometric function) versions of the mother wavelet, w, a waveform with an average value of zero (∫−∞∞ w(n)dn = 0), of finite length, arbitrary regularity and symmetry, and which is composed with data series, Hn=1, Hn=2, … , Hn=N, as
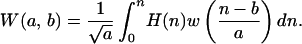 |
For w we chose the family of windowed, Gaussian (the dilate-location product bounded from below by finite Gaussian variance), discrete sine transformations globally maximized with respect to the trade-off in resolution between sequence frequency (dilation, l) and location (translation, a)
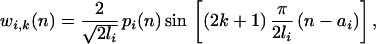 |
in which pi(n) is a “partition-like” window function supported in [ai − li/2, ai+1 + (li+1)/2] such that Σpi2(n) = 1. The wavelet plane portrays discrete steps of dilation of the local trigonometric wave graphed along the ordinate. The sequence location of these windowed dilates is indicated along the abscissa.
“Best basis” refers to optimization of the Heisenberg trade-off between specifying the locations in the sequence vs. those in dilate (wavelength) space, such that the “Shannon entropy of the expansion,” a Pythagorian distance in a probability Hilbert-like space between the chosen basis and the function, is minimized (36). Discrete, rather than continuous wavelet techniques, allow cutting smooth windows of differing lengths while preserving orthogonality during pattern identification in W (37). The graphs of the wavelet transformations of the receptor and ligand eigenfunctions, WR and WL, are represented as isopotential plots of their interpolated moduli.
Maximum Entropy, Complex Poles Power Spectral Transformation of Hydrophobic Free Energy Sequences and Their Eigenfunctions
The ΨR and ΨL, as well as the undecomposed hydrophobicity sequences, Hi, of the shorter peptide ligands, were transformed into their dominant inverse hydrophobic frequencies, ωi,
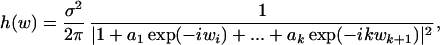 |
by using maximum entropy, complex poles, power spectra: h(ω). The Fourier coefficients, ai, match a very small set of k known autocorrelations, chosen so that the entropy of the spectral estimate, H(ω) = ∫ln h(ω)dω, is maximal. Beyond the limited information of a small set of autocorrelation-matched Fourier coefficients (38), the process is extended into a Gaussian process such that H(ω) is maximized (39). In these studies k ≤ 8 for receptor eigenfunctions of sequence lengths of several hundred amino acids, to avoid “splitting” h(ω) into spurious modes. The minimum, k = 2, was used for the hydrophobic free energy sequences of the short peptide ligands.
h(ω) is much like an autoregressive, maximum-likelihood spectral estimate in that it is not model-dependent but is derived directly from the data (40) and behaves like a filter that yields the one or two leading complex poles of discrete hydrophobic variational frequency in undistorted form from hydrophobic free energy sequences and/or their eigenfunctions.
Locating the TMH1−7 in G Protein-Coupled Receptor Sequences by Using the Graphs of Their Leading Hydrophobic Free Energy Transmembrane Eigenfunctions, ΨT
The locations of the transmembrane segments, TMH1−7, along the sequences of the G protein-coupled receptors for hormones, neurotransmitters, odorants, and light can be predicted graphically with reasonable accuracy by using hydropathy plots of the n-block averages of their amino acid sequences as hydrophobic free energies (32, 41–43). The hydropathy plots and the associated electron microscopic, electron diffraction studies, and projection maps of bacteriorhodopsin and rhodopsin I are generic for the seven-transmembrane receptor family and define four extracellular domains: the extracellular N-terminal chain, the first extracellular loop called e1 between TMH2−3, e2 between TMH4−5, and e3 between TMH6−7. The top row of Fig. 1 compares the graphs of the 32 times iterated, asymptotically smoothed, nearest neighbor averages (called an hydropathy plot) (Left) and the leading, transmembrane, hydrophobic free energy eigenfunction, ΨT (Right) derived from the Hi of rhodopsin I. The seven peaks characteristic of the G protein-coupled receptor family are easily recognizable. Structural analogies with rhodopsin proteins are made by comparing the results of site mutagenic studies, chimeric exchanges, and fluorescent emission spectroscopy of membrane loop binding domains (23–25).
The second to fifth rows of Fig. 1 demonstrate the close similarity between the paired graphs of hydropathy and the ΨT values for the kappa and mu opioid, corticotropin releasing factor, and cholecystokinin B seven transmembrane (TMH1−7) receptors. In obtaining information about transmembrane structures, the hydropathy technique eliminates other mode structures of the receptor sequence, whereas the extraction of ΨT leaves ΨR intact, allowing further analyses for peptide ligand binding mode(s).
Characterizing and Localizing the Dominant Hydrophobic Modes of Peptide Ligand Hi Sequences and Their G Protein-Coupled Receptor Eigenfunctions, ΨR
Fig. 2A Left is a graph of the hydrophobic free energy sequence of Hi values of the delta opioid peptide ligand, β-endorphin. Fig. 2A Right shows its maximum entropy, complex poles power spectrum, in which the dominant hydrophobic free energy inverse frequency, h(ω) = 4.44 amino acids. The shortness of the β-endorphin sequence, Hi, i = 32, broadened the band of its conventional power spectral transformation and included a larger wavelength peak, but the selection of the leading poles k = 2 in the maximum entropy method minimizes these effects.
Figure 2.

(A Left) Graph of the Hi of β-endorphin, the definitional peptide ligand for the delta opioid receptor. (Right) Plot of its maximum entropy, complex poles power spectrum indicating an h(ω) = 4.44 wavelength in amino acid residues. (B Upper Left) Graph of the wavelet transform, WR = 4.4, of the ΨR (Lower Left) of the delta opioid receptor. A plot of the h(ω) = 4.43 of its extracellular, N-terminal Hi (Upper Right) is identifiable in the isopotential plot of the WR (Upper Left) and the global h(ω) = 4.44 of its ΨR is portrayed in its graph (Lower Right).
Fig. 2B Upper Left portrays the WR of the second leading, β-endorphin-activating, delta opiate receptor eigenfunction, ΨR (Lower Left). The h(ω) = 4.43 residues of the extracellular N-terminal chain, Hi, i = 1–72 (Upper Right) is clearly located in the isopotential plot of the WR(Upper Left). The more opaque isopotential densities in the modular graph of WR signify the expression of the dominant dilate (wavelength) across larger variations in hydrophobic free energies. For example, the broad modular density on the left end of WR representing the N-terminal extracellular portion of the delta opiate receptor’s ΨR results from the dominant statistical wavelength expressed in both relatively low average hydrophobic values, M, R, T, L, N, T, S, A, M, D, G, T, and G = 1.67, 0.85, 0.07, 2.17, 0.09, 0.07, 0.07, 0.87, 1.67, 0.66, 0.1, 0.07, and 0.1, respectively, and relatively higher average hydrophobic values, I, L, T, A, C, F, L, S, L, L, I, and L = 3.15, 2.17, 0.07, 0.87, 1.52, 2.87, 2.17, 0.07, 2.17, 2.17, 3.15, and 2.17, respectively. To the right of the N-terminal, extracellular segment, WR also portrays modular peaks in sequence positions roughly corresponding to the cytoplasmic loops, e1, e2, and e3 of ΨR. The distributed character of this hydrophobic free energy mode is consistent with the finding that h(ω) = 4.44 (Fig. 2B Right Lower) when computed on the entire sequence of ΨR (Fig. 2B Left Lower). The hydrophobic mode matches between β-endorphin and the N-terminal domain, and the extracellular loops of the delta opiate receptor are consistent with results of site-directed mutagenesis studies performed on opiate receptors, which suggest a “discriminatory role” for these receptor extracellular loop-peptide ligand interactions (44). In addition, chimeric exchange experiments involving the delta opiate receptor have suggested that e2 and e3 (and a portion of TMH5−7) are required for opioid peptide ligand (45, 46) but not necessarily for small molecule binding (47, 48).
Fig. 3 summarizes comparisons of the dominant hydrophobic free energy modes of several representative peptide ligand-receptor pairs. Each panel contains three subsections: (Left in each) a graph of the discrete wavelet transformation, WR, of each peptide receptor’s ΨR; (Upper Right in each) a graph of the maximum entropy, complex poles power spectral transformation, h(ω), of the peptide ligand sequences as H′i values; and (Lower Right in each) is the h(ω) of the peptide receptor’s ΨR. The dominant hydrophobic mode in the amino acid sequence of dynorphin A, h(ω) = 3.46 residues, is relatively close to the global h(ω) = 3.31 of the ΨR of the kappa opioid receptor. The peptide ligand does not manifest the h(ω) = 8.8 of the ΨR, which is also seen as a set of distributed not necessarily receptor-related wavelets modes of W ≅ 8.88. The WR ≅ 3.48, however, is shared with dynorphin A and is particularly well localized in the e2 region between TMH4−5, which chimeric exchange studies have shown to be required for kappa receptor binding of the dynorphins (45, 46).
Figure 3.

Summaries of the graphs of the discrete wavelet transforms WR of each ΨR (Left of each panel), the maximum entropy, complex poles power spectra h(ω) values of each peptide ligand (Upper Right of each panel), and the h(ω) of its receptor ΨR (Lower Right of each panel) for a representative set of peptide ligands and their G protein-coupled, transmembrane receptor sequences. The sequence lengths along the abscissa of the WR plots (receptor length in amino acid residues minus the lag of the autocovariance matrix) are as follows: KAPPA OPIOID RECEPTOR (REC). = 362, MU OPIOID REC. = 402, ORPHAN OPIOID REC. = 352, NEUROPEPTIDE-Y REC. = 368; CRF-1 REC. = 307, CRF-1 REC. MUTANT = 307, SOMATOSTATIN-5 REC. = 373, and BOMBESIN-3 REC. = 486. See text for details of each peptide-receptor match and the relevant experiments in chimeric exchanges and point mutations. Note particularly the top two panels on the right that associate the loss of the peptide ligand-matched dominant hydrophobic free energy receptor mode in a nonbinding mutant receptor for ovine corticotropin releasing factor.
The dominant hydrophobic free energy wavelength, h(ω) = 3.02, of the ΨR of the mu opioid receptor evidences high absolute-valued wavelet coefficients at WR ≅ 2.96 in the N-terminal region and more diffusely distributed in e2 and e3. The results of these studies have varied from those suggesting the importance of segments in the N-terminal region (49, 50), to those pointing to e3 (47). The generic pentapeptide agonists for the mu opioid receptor, the enkephalins, are too short, Ai, i = 1–5, to justify an analysis by even the high resolution, maximum entropy, complex poles power spectra, as are the recently discovered, very high affinity mu receptor ligands (the endomorphins) with Ai, i = 1–4 (51).
The ΨR of the orphan opioid receptor, OLR1 (52) (which does not manifest high affinity, equilibrium binding of the delta, kappa, or mu peptide opioid ligands, though it has a high sequence homology with the opioid receptor family) and its recently sequenced endogenous ligand (54), share a h(ω) = 2.99–3.09 dominant mode. Its WR ≅ 3.13 manifests a well localized density at the C-terminal end of the ΨR of the receptor sequence; but of greater potential physiological significance is the graphically diffuse mode, roughly corresponding to e2, that OLR1 shares with the more densely marked e2 region in kappa opioid receptor’s WR, the extracellular loop required for binding with the dynorphins (54).
The wavelet graph of the ΨR of the human neuropeptide Y’s cloned Y1 receptor demonstrates several modular dilate densities along the sequence of WR ≅ 3.774 residue wavelength, which correspond to the h(ω) = 3.63 of neuropeptide Y’s sequence of Hi’s and the h(ω) = 3.77 of the peptide receptor’s ΨR. Site mutagenic studies have suggested that extracellular loops e1, e2, and e3 of the Y1 receptor are essential ligand binding domains (55) and that alanine substitution for the amino acids in the region of the latter significantly reduced binding of neuropeptide Y (56).
The top two right panels of Fig. 3 consist of graphic comparisons of the mode matches among the h(ω) = 2.18 of the hydrophobic sequence of the ovine corticotropin releasing factor, the h(ω) = 2.22 dominant mode of the ΨR, and the WR ≅ 2.19 of the normal corticotropin releasing factor 1 receptor. The h(ω) = 3.15 residue wavelength of the ΨR of the nonbinding, mutant corticotropin releasing factor 1 receptor demonstrates a mismatch with the normal ligand (57). Poor localization in the putative e2 and e3 mode densities can be seen in the WR of the mutant’s ΨR in comparison with that of the normal receptor.
Site mutagenesis and chimeric exchanges involving the receptors of somatostatin 14 and somatostatin 28 (SOM-28) have suggested that their third (58, 59) and sixth (60) transmembrane segments as well as extracellular loops e2 and e3 (61) are important in peptide ligand binding and functionality. The h(ω) = 2.90 of the Hi of the peptide ligand corresponds to the h(ω) = 2.83 wavelength of ΨR of the somatostatin receptor 5, the receptor with the greatest affinity for SOM-28. Although some regions of the wavelet moduli are relatively indistinct across the sequence, there is a well defined peak at WR ≅ 2.86, in the general vicinity of TM3 and e2. The receptor’s “hydrophobic pocket,” posited to play an important role in somatostatin binding, has been located in e3 (62), which is more diffusely localized in the graph of WR.
The WR graph of the ΨR of human bombesin-gastrin releasing peptide receptor sequence suggests multiple distributed isoenergetic modular peaks of average wavelength (dilate) WR ≅ 5.714, which corresponds to the h(ω) = 5.71 residues of the bombesin peptide sequence, as well as the h(ω) = 5.60 of the receptor’s eigenfunction, ΨR. Chimeric exchanges involving e3 or the carboxyl end of the bombesin receptor (note the variegated densities on the right side of its WR graph) and the corresponding portion of the M3 muscarinic cholinergic receptor eliminated ligand-receptor internalization and recycling normally initiated by the bombesin peptide ligand (63, 64). The modular densities in the WR to the left of e3 may correspond to the second and third intracellular loops composed of hydrophilic, charged (low hydrophobic valued) amino acid residues, which chimeric studies have shown to be required for receptor coupling to phospholipase C-dependent intracellular processes (65).
Characterizing and Localizing the Dominant Hydrophobic Modes of Peptide Ligand Hi Sequences and Their Tyrosine Kinase-Coupled Receptor Eigenfunctions, ΨR
The leading eigenfunction, not shown, of the 1,188 amino acid precursor of epidermal growth factor (EGF, urogastrone) in Hi was dominated by an average h(ω) ≈ 53–54 residues, consistent with its known composition of 53 amino acid EGF repeats. Fig. 4 (top row, Left) is a graph of the second (ligand) eigenfunction of the EGF precursor, ΨL, for which h(ω) = 7.60 (Right, top row). As is generally the case with peptide precursors, this mode also dominated the maximum entropy, complex poles transformation of the Hi′s of the 53 residue EGF peptide ligand itself. The wavelet transformation of ΨL of the EGF precursor (Center, top row) manifests two bands of dilates, WL ≅ 7.40 and WL ≅ 11.38, hidden in the nonconvergent tail to the left side of the peak of the EGF propeptide ΨL values h(ω) = 7.60. The ΨR of the extracellular segment, i = 1–335 residues, of the EGF receptor sequence (Fig. 4 Left, second row) manifests a broad band h(ω)= 7.60 (Right, second row) and a diffusely localized, average WR ≅ 7.77 residues (Center, second row). The dominant wavelet mode of transforming growth factor α (TGF-α), which binds to the EGF receptor, is WL ≅ 7.44 (data not shown), and demonstrates a well-marked density in the sequence location of about Ai, i = 83–120. EGF/TGF chimeric exchanges have shown this region to be required for the high affinity binding of TGF-α to the EGF receptor (66).
Figure 4.

(Left) Plots of the leading ligand and receptor eigenfunctions, ΨL, ΨR, of EGF and FGF. (Center) Corresponding graphs of their wavelet transformations, WL, WR. (Right) Maximum entropy, complex poles power spectra, h(ω) values of their ΨL and ΨR. The sequence positions along the abscissas of the WL and WR plots are n − 18 of the indicated amino acid sequence lengths where n of the EGF propeptide = 1,188; extracellular EGF REC. = 355; FGF = 154; extracellular FGF REC. = 700.
Fig. 4 Left (third row) portrays the leading eigenfunction of the Hi, i = 1–154 sequence of acid fibroblast growth factor, aFGF, its leading hydrophobic mode, h(ω) = 12.26 residues (Right, third row) and its discrete, trigonometric wavelet transformation (Center, third row) with WL ≅ 12.50 residues for the N-terminal densities. Studies have suggested that the wavelet mode density at the N-terminal end of aFGF is essential for cell nuclear localization (67) and, when given intracerebrally, the induction of consummatory behavior in rodents (68). The density at the N-terminal end of the WR of the aFGF receptor has also been found to be essential for the discriminative binding of basic FGF and aFGF (69). The ΨR of the predominantly extracellular domain of the aFGF receptor (Left, fourth row) manifests a globally dominant h(ω) = 13.10 (Right, fourth row) and a WR ≅ 12.61 residue band, on average (Center, fourth row). The multiplicity and distributed character of the isopotential densities in WR is consistent with chimerical studies that indicate multiple Ig-like binding regions distributed in the central portions of the extracellular aFGF receptor sequence (70). In a pilot study of the eigenfunctions of representative lambda, kappa, and gamma chains of the Ig superfamily, we have found ΨR values with h(ω) = 12.5–13.7.
Technical Comments and Limitations
The eigenfunction construction technique used here (34, 35) is derived from the singular value decomposition of autocovariance matrices, which has been successfully applied to many discrete, relatively short sample length biological data series including neuronal interspike intervals, digitized electromyographic, and brain wave potentials (71). Consistency is achieved across results, as in our ligand-receptor families of mode matches, rather than through the use of physiologically unrealistic sample lengths. These approaches to physiological data often involve the partition and representation of apparently continuous quantitative data by a small discrete alphabet composing a finite set of data values (72, 73), quite like our use of the finite set of discrete values for amino acid hydrophobic free energies used in these analyses. It should also be noted that with respect to both the ligands and receptors, we have found that hydrophobic free energy receptor eigenfunction modes from physiologically similar families vary around only 10 or so commonly found wavelengths, for example, h(ω) of ΨR ≈ 2.1, 3.0, 3.6, 4.0, 4.4, 5.6, 7.5, 8, 10, and 13.0 amino acid residues (16, 17).
In addition, we offer the caveat that we do not expect similarities in hydrophobic mode distributions among ligands and receptors to necessarily order relative binding affinities, discriminate between agonist and antagonist influences, or predict subsets of the range of biochemical and physiological actions differentially evoked by members of a family of peptides.
ABBREVIATIONS
- FGF
fibroblast growth factor
- EGF
epidermal growth factor
Footnotes
Swiss-Prot (1996) Protein Sequence Data Bank (Med. Biochem. Department, University of Geneva, Switzerland), Release 32.5.
References
- 1.Rose G D. Nature (London) 1978;272:586–590. doi: 10.1038/272586a0. [DOI] [PubMed] [Google Scholar]
- 2.Lesk A M, Chothia C. J Mol Biol. 1980;136:225–270. doi: 10.1016/0022-2836(80)90373-3. [DOI] [PubMed] [Google Scholar]
- 3.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 4.Chothia C. Annu Rev Biochem. 1984;53:537–572. doi: 10.1146/annurev.bi.53.070184.002541. [DOI] [PubMed] [Google Scholar]
- 5.Nozaki Y, Tanford C. J Biol Chem. 1971;246:2211–2217. [PubMed] [Google Scholar]
- 6.Manavalen P, Ponnuswamy P K. Nature (London) 1978;275:673–674. doi: 10.1038/275673a0. [DOI] [PubMed] [Google Scholar]
- 7.Israelachvili J, Wennerstrom H. Nature (London) 1996;379:219–225. doi: 10.1038/379219a0. [DOI] [PubMed] [Google Scholar]
- 8.Pashley R M, McGuiggan P M, Ninham B W, Evans D F. Science. 1985;229:1088–1098. doi: 10.1126/science.4035349. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser E T, Kèzdy F J. Proc Natl Acad Sci USA. 1983;80:1137–1143. doi: 10.1073/pnas.80.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaiser E T, Kèzdy F J. Science. 1984;223:249–255. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- 11.Kamtekar S, Hecht M H. FASEB J. 1995;9:1013–1017. doi: 10.1096/fasebj.9.11.7649401. [DOI] [PubMed] [Google Scholar]
- 12.Gronenbron A M, Clore G M. Science. 1994;263:536–538. doi: 10.1126/science.8290964. [DOI] [PubMed] [Google Scholar]
- 13.Dill K A, Fiebig K M, Chan H S. Proc Natl Acad Sci USA. 1993;90:1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lumry R. Methods Enzymol. 1995;259:1–103. doi: 10.1016/0076-6879(95)59065-x. [DOI] [PubMed] [Google Scholar]
- 15.Beattie J, Shand J H, Flint D J. Eur J Biochem. 1996;239:479–485. doi: 10.1111/j.1432-1033.1996.0479u.x. [DOI] [PubMed] [Google Scholar]
- 16.Mandell A J. Annu Rev Pharmacol Toxicol. 1984;24:237–274. doi: 10.1146/annurev.pa.24.040184.001321. [DOI] [PubMed] [Google Scholar]
- 17.Mandell A J, Russo P V, Blomgren B. Ann NY Acad Sci. 1987;504:88–117. doi: 10.1111/j.1749-6632.1987.tb48727.x. [DOI] [PubMed] [Google Scholar]
- 18.Ramachandran G N, Sassiekharan V. Adv Protein Chem. 1968;28:283–437. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 19.Gabor D. J Inst Electron Eng. 1946;93:429–457. [Google Scholar]
- 20.Zetin F N, Rusk S F, Raymond V, Mandell A J. Endocrinology. 1988;122:1121–1128. doi: 10.1210/endo-122-3-1121. [DOI] [PubMed] [Google Scholar]
- 21.Mandell A J. In: Dynamic Patterns in Complex Systems. Kelso J A S, Mandell A J, Shlesinger M F, editors. Singapore: World Scientific; 1988. pp. 219–235. [Google Scholar]
- 22.Araga S, Leboef R D, Blalock J E. Proc Natl Acad Sci USA. 1993;90:8747–8751. doi: 10.1073/pnas.90.18.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Dowd B F, Lefkowitz R J, Caron M G. Annu Rev Neurosci. 1989;12:67–83. doi: 10.1146/annurev.ne.12.030189.000435. [DOI] [PubMed] [Google Scholar]
- 24.Ostrowski J, Kjelsberg M A, Caron M G, Lefkowitz R J. Annu Rev Pharmacol Toxicol. 1992;32:167–183. doi: 10.1146/annurev.pa.32.040192.001123. [DOI] [PubMed] [Google Scholar]
- 25.Wagner R L, Apriletti J W, McGrant M E, West B L, Baxter J D, Gletterick R J. Nature (London) 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 26.de Gennes P G. Scaling Concepts in Polymer Physics. Ithaca, NY: Cornell Univ. Press; 1985. [Google Scholar]
- 27.Di Marzio E A, Guttman C M. J Chem Phys. 1985;95:1189–1197. [Google Scholar]
- 28.White S H, Jacobs R E. Biophys J. 1990;57:911–921. doi: 10.1016/S0006-3495(90)82611-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degli Esposti M, Crimi M, Venturoli G. Eur J Biochem. 1990;190:207–219. doi: 10.1111/j.1432-1033.1990.tb15566.x. [DOI] [PubMed] [Google Scholar]
- 30.Zamyatnin A A. Prog Biophys Mol Biol. 1972;24:107–123. doi: 10.1016/0079-6107(72)90005-3. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds J A, Gilbert D V, Tanford C. Proc Natl Acad Sci USA. 1974;71:2925–2927. doi: 10.1073/pnas.71.8.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kyte J, Doolittle R F. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 33.Golub G H, Van Loan C F. Matrix Computations. Baltimore: Johns Hopkins Univ. Press; 1993. pp. 409–474. [Google Scholar]
- 34.Broomhead D S, King G P. Physica D. 1986;20:217–236. [Google Scholar]
- 35.Broomhead D S, Jones R, King G P. J Phys A. 1987;20:L563–L569. [Google Scholar]
- 36.Wickerhauser MV. Adapted Wavelet Analysis from Theory to Software. Wellesley, MA: Peters; 1994. pp. 103–150. [Google Scholar]
- 37.Daubechies I, Jaffard S, Jean-Lin J. SIAM J Math Anal. 1991;22:554–573. [Google Scholar]
- 38.Madan R N. In: Maximum Entropy and Bayesian Methods. Mohammad-Djafari A, Demoments G, editors. Dordrecht, The Netherlands: Kluwer; 1993. pp. 49–54. [Google Scholar]
- 39.Burg J P. Geophysics. 1972;37:375–376. [Google Scholar]
- 40.Priestly M D. Spectral Analysis and Time Series. San Diego: Academic; 1981. p. 600. [Google Scholar]
- 41.Henderson R, Baldwin J M, Ceska T A. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 42.Strader C D, Fong T M, Tota M R, Underwood D, Dixon R A. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 43.Dohlman H G, Thoner J, Caron M G, Lefkowitz R J. Annu Rev Biochem. 1991;60:653–667. doi: 10.1146/annurev.bi.60.070191.003253. [DOI] [PubMed] [Google Scholar]
- 44.Metzger T G, Ferguson D M. FEBS Lett. 1995;375:1–4. doi: 10.1016/0014-5793(95)01185-h. [DOI] [PubMed] [Google Scholar]
- 45.Meng F, Hoversten M, Thompson R C, Taylor L, Watson S J, Akil H. J Biol Chem. 1995;270:12730–12736. doi: 10.1074/jbc.270.21.12730. [DOI] [PubMed] [Google Scholar]
- 46.Wang W W, Shahrestanifar M, Jin J, Howells R D. Proc Natl Acad Sci USA. 1995;92:12436–12440. doi: 10.1073/pnas.92.26.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue J C, Chen C-G, Zhu J M, Kunapuli S, DeRiel J D, Lei Y, Liu-chen L-Y. J Biol Chem. 1994;269:30195–30199. [PubMed] [Google Scholar]
- 48.Gether U, Johansen T E, Snider R M, Lowe J A, Nakanishi S, Schwartz T W. Nature (London) 1993;362:345–348. doi: 10.1038/362345a0. [DOI] [PubMed] [Google Scholar]
- 49.Fukuda K, Terasako K, Kato S, Mori K. FEBS Lett. 1995;373:177–181. doi: 10.1016/0014-5793(95)01034-c. [DOI] [PubMed] [Google Scholar]
- 50.Watson B, Meng F, Akil H. Neurobiol Dis. 1996;3:87–96. doi: 10.1006/nbdi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 51.Zadina J E, Hackler L, Ge L-J, Kastin A J. Nature (London) 1997;386:499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, Fau Y, Liu J. FEBS Lett. 1994;347:279–283. doi: 10.1016/0014-5793(94)00560-5. [DOI] [PubMed] [Google Scholar]
- 53.Meunier J C, Mollereau C, Toll L, Suaudeau C. Nature (London) 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 54.Wang J B, Johnson P S, Wu J M, Wang W F, Uhl G R. J Biol Chem. 1994;269:25966–25969. [PubMed] [Google Scholar]
- 55.Walker P, Munoz M, Martinez R, Peitsch M C. J Biol Chem. 1994;269:2863–2869. [PubMed] [Google Scholar]
- 56.Sautel M, Rudolf K, Wittneben H, Herzog H. Mol Pharmacol. 1996;50:285–292. [PubMed] [Google Scholar]
- 57.Ross P C, Kostas C M, Ramabhadran T V. Biochem Biophys Res Commun. 1994;205:1836–1842. doi: 10.1006/bbrc.1994.2884. [DOI] [PubMed] [Google Scholar]
- 58.Strnad J, Hadcock J R. Biochem Biophys Res Commun. 1995;216:913–921. doi: 10.1006/bbrc.1995.2708. [DOI] [PubMed] [Google Scholar]
- 59.Nehring R B, Meyerhoff W, Richter D. DNA Cell Biol. 1995;14:939–944. doi: 10.1089/dna.1995.14.939. [DOI] [PubMed] [Google Scholar]
- 60.Ozenberger B A, Hadcock J R. Mol Pharmacol. 1995;47:82–87. [PubMed] [Google Scholar]
- 61.Liapakis G, Fitzpatrick D, Hoeger C, Rivier J. J Biol Chem. 1996;271:20331–20339. doi: 10.1074/jbc.271.34.20331. [DOI] [PubMed] [Google Scholar]
- 62.Bell G I, Reisine T. Trends Neurosci. 1993;16:34–38. doi: 10.1016/0166-2236(93)90050-v. [DOI] [PubMed] [Google Scholar]
- 63.Tseng M J, Coon S, Stuenkel E, Struk V, Logsdon C D. J Biol Chem. 1995;270:17884–17891. doi: 10.1074/jbc.270.30.17884. [DOI] [PubMed] [Google Scholar]
- 64.Tseng M J, Detjen K, Struk V, Logsdon C D. J Biol Chem. 1995;270:18858–18864. doi: 10.1074/jbc.270.32.18858. [DOI] [PubMed] [Google Scholar]
- 65.Benya R V, Akeson M, Mrozinski J, Jensen R T, Battey J F. Mol Pharmacol. 1994;46:495–501. [PubMed] [Google Scholar]
- 66.Kramer R H, Lengerink A E, van Bueren-Koornneef I L, van der Meer A. J Biol Chem. 1994;269:8708–8711. [PubMed] [Google Scholar]
- 67.Fantal W J, Johnson D E, Williams L T. Annu Rev Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- 68.Lin Y Z, Yao S Y, Hawiger J. J Biol Chem. 1996;271:5305–5308. doi: 10.1074/jbc.271.10.5305. [DOI] [PubMed] [Google Scholar]
- 69.Reich-Slotkey R, Shaoul E, Berman B, Graziani G, Ron D. J Biol Chem. 1995;270:29813–29818. doi: 10.1074/jbc.270.50.29813. [DOI] [PubMed] [Google Scholar]
- 70.Zimmer Y, Givol D, Yayon A. J Biol Chem. 1995;268:7899–7903. [PubMed] [Google Scholar]
- 71.Hamer R N. Brain Topography. 1990;3:43–47. doi: 10.1007/BF01128860. [DOI] [PubMed] [Google Scholar]
- 72.Mandell A J, Selz K A. Chaos. 1997;7:67–81. doi: 10.1063/1.166241. [DOI] [PubMed] [Google Scholar]
- 73.Walters P. Symbolic Dynamics and Its Applications. Providence, RI: Am. Math. Soc.; 1991. [Google Scholar]