

Abstract
Alzheimer disease (AD) is the most common neurodegenerative disorder worldwide and is at present, incurable. The accumulation of toxic amyloid-beta (Aβ) peptide aggregates in AD brain are thought to trigger the extensive synaptic loss and neurodegeneration linked to cognitive decline, an idea that underlies the ‘amyloid hypothesis’ of AD etiology in both the familal (FAD) and sporadic forms of the disease. Mutations causing FAD also result in the dysregulation of neuronal calcium (Ca2+) handling and may contribute to AD pathogenesis, an idea termed the ‘calcium hypothesis’ of AD. In particular, Ca2+ dysregulation by the endoplasmic reticulum (ER) in AD mouse models results in augmented cytosolic Ca2+ levels which can trigger signaling cascades that are detrimental to neuronal function and health. However, there is growing evidence to suggest that not all forms of Ca2+ dysregulation in AD neurons are harmful and some of them instead may be compensatory. These changes may help modulate neuronal excitability and slow AD pathology, especially in the early stages of the disease. Clearly, a better understanding of how dysregulation of neuronal Ca2+ handling contributes to neurodegeneration and neuroprotection in AD is needed as Ca2+ signaling modulators are targets of great interest as potential AD therapeutics.
Introduction
Alzheimer’s disease (AD) is characterised clinically by the progressive impairment of higher cognitive function, loss of memory and altered behaviour that follows a gradual progression. The pathological hallmarks of the disease are characterised at autopsy; the presence of senile plaques composed of extracellular amyloid-beta (Aβ) protein aggregates, intracellular neurofibrillary tangles (NFTs) composed of hyper-phosphorylated tau (τ) protein deposits, and the shrinkage of the cerebral cortex due to extensive neuronal loss [1]. The cause of AD is unknown but it is widely accepted that Aβ, particularly the highly fibrillogenic fragment 1–42 and its various assemblies, plays a central role in both familial, early-onset AD (FAD) and sporadic, late-onset AD (LOAD) neuropathology, termed “the amyloid hypothesis of AD” [2]. The study of Aβ-related mechanisms that occur prior to irreversible cognitive impairment and neurodegeneration in AD could reveal targets for therapeutic intervention and disease prevention.
Marked and sustained changes to intracellular calcium Ca2+ signalling occurs prior to cognitive decline and extensive neuronal death in AD [3]. The regulation of intracellular Ca2+ by the endoplasmic reticulum (ER) has been a focus of study since it was reported that fibroblasts from asymptomatic patients at risk for AD had enhanced cytosolic Ca2+ levels after application of bradykinin, a G-protein-coupled receptor agonist that increases intracellular Ca2+ by generating inositol-1,4,5-trisphosphate (IP3) and activation of IP3 receptors (IP3Rs) on the ER [4, 5]. Ryanodine receptors (RyanRs) receptors are the other major Ca2+ release channels found on the ER. Neuronal RyanRs activated via Ca2+-induced Ca2+ release (CICR) mechanism [6]. The sarco/endoplasmic reticulum ATPase (SERCA) pump refills depleted ER Ca2+ stores. The goal of this review is to discuss how changes in intracellular Ca2+ signalling by the ER may contribute to neurodegeneration in AD.
Calcium signalling in neurons
Calcium signalling is utilized by neurons to control a variety of functions, including membrane excitability, neurotransmitter release, gene expression, cellular growth, differentiation, free radical species formation and cell death [6]. Because of the ubiquitous nature of Ca2+ in second-messenger signalling, neurons have strict mechanisms to maintain low concentrations (50–300 nM) of cytosolic Ca2+ ([Ca2+]cyto) when neurons are at rest or have minimal activity [7]. Calcium-ATPases and the sodium/calcium (Na+/Ca2+) exchanger on the plasma membrane (PM) extrude Ca2+ into the extracellular space while the sarco/endoplasmic reticulum ATPase (SERCA) on the endoplasmic reticulum (ER) membrane pumps Ca2+ from the cytosol into intracellular stores. Thus, a large electrochemical gradient is created across the PM and ER membrane. Upon activation, Ca2+ can flux into the cytosol through channels on the PM that are either voltage-gated and/or ligand-gated (eg. N-methyl-D-aspartate (NMDA) receptors) or store-operated (eg. transient receptor potential channels, TRPC). Stimulation of G-protein-coupled receptors (eg, metabotrophic glutamate receptors, mGluR) can activate phospholipase C and increase the generation of inositol-1,4,5-trisphosphate (IP3), which can bind receptors IP3 receptors (IP3R) on the ER to release Ca2+ into the cytosol. The ER-resident ryanodine receptors (RyanR) are Ca2+ sensitive and serve to amplify the Ca2+ signals from IP3Rs or the extracellular pool, termed Ca2+-induced Ca2+ release (CICR) [6]. In response to stimulation neuronal [Ca2+]cyto can be elevated to 1–5 μM and trigger Ca2+-dependent cellular signalling cascades. The specific pathway activated will depend on the source of Ca2+, the spatio-temporal pattern of cytosolic Ca2+ accumulation and the resulting [Ca2+]cyto. Cytosolic Ca2+ can also flux into mitochondria and activate Ca2+-dependent mitochondrial matrix dehygrogenases and ATP production [8, 9]. The overall effect of these Ca2+ fluxes can range from the modulation of membrane excitability, enzyme/kinase activity, gene expression, mitochondrial function, reactive oxygen/nitrogen species (ROS/RNS) formation and apoptosis/necrosis.
Calcium regulation in neurons and aging
It is thought that as neurons age, their ability to maintain such tight regulation of Ca2+ gradients across the PM and ER membrane becomes compromised, likely due to inefficient energy metabolism and accumulating oxidative stress. Compared to young neurons, old neurons display Ca2+ dysregulation, or changes in Ca2+ regulation that lead to sustained increases in [Ca2+]cyto [10]. This observation has inspired the hypothesis that such changes in Ca2+ signalling could contribute to brain aging, neuronal dysfunction and neurodegeneration [11, 12]. Such age-dependent changes to Ca2+ handling in brain have been recently reviewed [10]. Ca2+ influx associated with action potentials induces larger Ca2+-dependent after hyperpolarizations (AHPs) [13–15] and impaired short-term synaptic plasticity [16] in aged neurons from rats and rabbits compared to young neurons. Pharmacologically isolated Ca2+ action potentials, whole-cell Ca2+ currents and Ca2+ transients during repetitive spike trains are larger in hippocampal neurons from aged animals [15–17]. Aging enhanced the activity of large (L-type) voltage-gated Ca2+ channels (L-VGCC) in partially dissociated hippocampal slices [18]. Functionally, antagonists of L-VGCC appear to improve learning and memory in aged animals [19] and in some patients with dementia [20]. Clearly, there are changes to different components of Ca2+ handling with aging and such alterations lead to the augmented susceptibility to induction of long-term depression (LTD) and an increase in the threshold frequency for induction of long-term potentiation (LTP) in aging neurons [21]. LTD and LTP refer to activity-dependent changes to synaptic strength and remodelling and are proported to be the basis for memory formation and storage [21].
Calcium and neurodegeneration in Alzheimer disease
Alzheimer disease (AD) is the most common form of progressive dementia in the elderly. Most cases of AD are sporadic but approximately 1–2% are genetically linked or familial (FAD) and are distinguished by the early onset of dementia (< 65 years old). Missense mutations in the amyloid precursor protein (APP), the presenilin-1 (PS1) and the presenilin-2 (PS2) genes [22] result in a shift in the proteolysis of APP by the γ-secretase complex such that the ratio of Aβ42/40 protein fragments in the brain is increased [23, 24] (Figure 1). This leads to the aggregation and formation of toxic Aβ42 oligomers that induce the loss of synapses and neuronal toxicity in AD [2]. Because FAD is pathologically identical to sporadic AD, it is thought that Aβ42 over-production is the causative factor in AD, dubbed the “amyloid cascade hypothesis”.
Figure 1. Calcium dysregulation in Alzheimer disease.

Sequential cleavages of β-amyloid precursor protein (APP) by β-secretase (β) and β-secretase (β) generate amyloid β peptide (Aβ). Aβ forms oligomers, which can insert into the plasma membrane and form Ca2+-permeable pores. The association of Aβ oligomers with the plasma membrane is facilitated by binding to surface phosphatidylserine (PtdS); age and Ca2+-related mitochondrial impairment leads to ATP depletion and might trigger flipping of PtdS from the inner portion of the plasma membrane to the cell surface. Reduction in ATP levels and loss of membrane integrity causes membrane depolarization, which leads to facilitation of Ca2+ influx through NMDAR and VGCC. Aβ oligomers can also affect activity of NMDAR, AMPAR and VGCC directly. Glutamate stimulates activation of mGluR1/5 receptors, production of InsP3 and InsP3R -mediated Ca2+ release from the ER. Presenilins (PS) function as an ER Ca2+-leak channels and many FAD mutations impair Ca2+-leak-channel function of PS, resulting in excessive accumulation of Ca2+ in the ER. Increased ER Ca2+ levels result in enhanced Ca2+ release through InsP3 -gated InsP3R 1 and Ca2+-gated RyanR(2/3). PS might also modulate activity of InsP3R, RyanR and SERCA pump directly. The activity of store-operated Ca2+ channels on the plasma membrane can be affected indirectly by PS mutations through the modulation of SERCA activity. Elevated cytosolic Ca2+ levels result in the activation of calcineurin (CaN) and calpains and lead to facilitation of LTD, inhibition of LTP, modification of neuronal cytoskeleton, synaptic loss and neuritic atrophy. Excessive Ca2+ is taken up by mitochondria through mitochondrial Ca2+ uniporter (MCU), eventually leading to opening of mitochondrial permeability-transition pore (mtPTP) and apoptosis.
In addition to changes in Aβ42/40 ratio, a number of studies point to dysfunctional endoplasmic reticulum (ER) Ca2+ signalling in AD, particularly those involving PS mutations [3]. In 1989, Khachaturian proposed that sustained changes in Ca2+ homeostasis could provide the common pathway for aging and the neuropathological changes associated with AD, termed the “calcium hypothesis of brain aging and Alzheimer’s disease” [25] and the evidence to support the hypothesis came soon after. Fibroblasts from asymptomatic patients at risk for AD displayed enhanced IP3R-mediated Ca2+ signalling [4]. Expression of Ca2+-handling genes are significantly altered in brain tissue from AD patients [26]. Recently, memantine has been approved by the FDA for use in the treatment of patients with moderate to severe AD. Because memantine antagonizes the Ca2+-permeable NMDA receptor by blocking open, over-activated channels [27], its efficacy illustrates the potential involvement of altered Ca2+ signalling in the clinical manifestation of AD. Nimodipine, a dihydropyridine derivative and L-VGCC antagonist, has beneficial effects in AD patients and slows the progression of the disease [28]. A nimodipine derivative, MEM-1003, has completed Phase II clinical trials [29]. Finally, a single nucleotide polymorphism in a newly identified plasma membrane Ca2+ channel, CALHM1, interferes with Ca2+ permeability and slightly increases susceptibility to sporadic, late-onset AD [30, 31]. It should be noted, however, that the role of CALHM1 in AD is controversial, with several studies showing no association between the two [32–34]. Further study of CALHM1 function in neuronal Ca2+ signaling is clearly required. In any case, there is a universal agreement that Ca2+ dysregulation in neurons appears to be a genuine consequence AD pathology (Figure 1).
Do changes in Ca2+ homeostasis affect Aβ production? If so, changes in Ca2+ handling could be the trigger of AD pathology. Past studies have addressed this question but there are conflicting conclusions. The earliest study reported that cultured human embryonic kidney cells expressing APP had increased Aβ production after treatment with the Ca2+ ionophore, A23187 [35]. The same result was obtained after treating the cells with caffeine, a RyR agonist, suggesting that both Ca2+ influx and release of Ca2+ from intracellular stores promoted Aβ production [36]. However, after treatment with thapsigargin, which irreversibly blocks SERCA activity and increases cytosolic Ca2+ levels, cultured cells showed dose-dependent decreases in their production of Aβ [37]. More recently it was demonstrated that influx of Ca2+ from L-VGCC and elevated [Ca2+]cyto increased intraneuronal Aβ42 production, while release of ER Ca2+ was inadequate for Aβ production [38]. Conversely, it has been shown that loss of Ca2+influx through plasma membrane channels due to polymorphisms increases Aβ42 formation and Aβ42/40 ratio in CHO cells expressing APP [30]. The processing of APP is complex and is clearly affected by altered cytosolic Ca2+ signalling but further study, particularly in neuronal systems, is required to determine which Ca2+ sources are important for Aβ production.
The effects of APP and its metabolites on cytosolic Ca2+ signalling are well established and have been reviewed recently [39]. Aβ40 and Aβ42 can form Ca2+-permeable pores on the plasma membrane, generally leading to an increase in [Ca2+]cyto [40]. Pore formation is enhanced by exposure of phosphatidylserine on the cell surface; an indication that a cell will undergo apoptosis [41, 42]. Because destabilization of cytosolic Ca2+ levels can trigger free radical formation [43], lipid peroxidation [44] and apoptosis [45], such mechanisms could be involved in Aβ neurotoxicity. Aβ oligomers can modulate NMDA receptor activity [46–48] and sensitivity to NMDA-mediated excitotoxicity [47, 49]. Aβ oligomers can also suppress activity of presynaptic P/Q-type (neuronal) VGCC [50] and direct modulation of L-type Ca2+ VGCC activity by APP was recently reported [51]. Secreted APPs have neuroprotective qualities because they attenutate the elevated [Ca2+]cyto evoked by Aβ [52] and moderate glutamate-induced cytosolic Ca2+ levels in hippocampal neurons by increasing cyclic GMP [53]. Changes in Ca2+ dynamics are thought to contribute to the altered synaptic transmission observed in PDAPP mice [54]. More recently, in vivo Ca2+ imaging experiments with Tg2567 mice displayed elevated [Ca2+]cyto, or Ca2+ overload, in neurites and spines that were in close proximity to Aβ plaques [55] and induction of Ca2+ waves in astrocytes [56]. Ca2+ disturbances observed in both cases were most likely caused by direct effects of soluble Aβ oligomers on Ca2+ signaling in neurons and astrocytes [57].
Downstream effects of sustained dysregulated cytosolic Ca2+ is activation of Ca2+-dependent phosphatase calcineurin and neuritic atrophy [55]. Activation of calcineurin also has profound effects on synaptic plasticity [58]. Excessive Ca2+ signals also activate Ca2+-dependent proteases calpains which degrade signaling enzymes involved in learning and memory [59, 60]. Prolonged Ca2+ dysregulation causes accumulation of ROS, mitochondrial dysfunction and neuronal death. Increases in [Ca2+]cyto can cause excessive Ca2+ flux into mitochondria and increase ROS production [43, 45, 61]. Evidence of ROS, reduced energy metabolism and decreased cytochrome c oxidase activity have been described in the brains of AD patients [62, 63]. These processes could ultimately lead to the extensive cortical and hippocampal atrophy and neurodegeneration characteristic of AD [64] (Figure 1).
ER Ca2+ and neurodegeneration in AD
The role of the ER in the dysregulation of cytosolic Ca2+ in AD has been a major focus of research because mutations that cause AD also affect ER Ca2+ signalling. Skin fibroblasts from human patients that harbour a mutation in presenilin 1 (PS1)-A246E, a transmembrane protein that is the catalytic component of the γ-secretase complex, showed exaggerated Ca2+ release from IP3-gated stores compared to controls after treatment with bombesin and bradykinin [4]. These alterations in Ca2+ signalling were detected before the development of overt clinical symptoms and such changes were not present in cells from subjects that failed to develop AD [5]. Cells expressing mutant PS1 [65] and primary cortical neurons from mice expressing mutant PS1 displayed similar alterations in signalling [66, 67]. Clinical mutations of the PS2 gene also enhanced Ca2+ release from IP3R-gated ER stores [68]. Much data have been generated since these early studies to suggest that in addition to their γ-secretase function, PS mutations have a significant impact on Ca2+ signalling in AD models. It has been reported that mutations in PS can modulate capacitative Ca2+ entry, a refilling mechanism for depleted Ca2+ stores [66, 69, 70]. Explanation to these results has been provided by a recent discovery that PS also functions as ER Ca2+ leak channels and that FAD mutations in PS1 disrupts this function [71, 72], resulting in overloaded ER Ca2+ stores and exaggerated ER Ca2+ release in PS double knockout fibroblasts and in fibroblasts transfected with mutant PS1 and PS2 constructs. The PS1-M146V mutation augmented Ca2+ release from IP3- and caffeine- gated stores in hippocampal and cortical neurons in 3XTg-AD mice [14, 73]. The gating of IP3R was reported to be directly modulated by PS1-M146L in overexpression system [74], providing another potential mechanism for connection between mutations in presenilins and ER Ca2+ signaling. Xenopus laevis oocytes expressing PS1-M146V have increased SERCA activity compared to those with wild-type PS1 [75], a mechanism that could additionally contribute to the overfilling of ER Ca2+ store. It is clear that familial AD mutation in presenilins affect the activity and/or expression of many proteins involved in ER Ca2+ signalling and predominately results in enhanced release of Ca2+ from ER stores.
Several mouse models of AD demonstrate that RyanRs are up-regulated in expression and function in cultured neurons and in brain. For example, the exaggerated IP3- and caffeine-evoked Ca2+ responses in 3XTg-AD hippocampal and cortical neurons were attributed to increased RyanR expression and recruitment [14, 67, 73]. RyanRs are responsible for the amplification of intracellular Ca2+ signals from IP3R stores or Ca2+ influx from the plasma membrane by CICR. Given the involvement of RyanRs in CICR, modulation of membrane excitability [76, 77], neuronal function [78, 79] and hippocampal learning and memory [80, 81], it is rational to hypothesize that the up-regulation of the RyanRs could contribute to AD pathology. In human post-mortem tissue, ryanodine binding is elevated in hippocampal regions (subiculum, CA2 and CA1) of AD brain in the early stages of the disease prior to extensive neurodegeneration and overt Aβ plaque deposition [82]. Furthermore, it has been reported that RyanR levels are increased in 3 different mouse models of AD; PS1-M146V, PS2-N141I, 3XTg-AD and TgCRND8 [83–86]. Finally, increased RyR levels enhanced Ca2+ release from the ER and sensitized cortical neurons from PS1-M146V and PS2-N141I mice to neurotoxic insults, such as treatment with high glutamate concentration [83, 84] (Figure 1).
ER Ca2+ and neuroprotection in AD
Are all changes to neuronal ER Ca2+ signaling in AD detrimental? Recent evidence has revealed new insight into potential importance of enhanced Ca2+ release from neuronal ER in the context of AD. Chakroborty, et al. 2009, demonstrated that while pre-symptomatic 3XTg-AD mice had aberrant ryanodine-evoked Ca2+ responses in CA1 pyramidal neurons compared to non-Tg, due to increased RyanR type 2 expression, they displayed seemingly normal synaptic transmission [73]. These results suggest a mechanism by which neurons might maintain Ca2+ homeostasis and neuronal function in the early stages of the disease. Furthermore, TgCRND8 mice displayed increased RyanR type 3 expression and function and increased Ca2+ release from ryanodine-gated stores in primary cortical neurons [86] but showed no changes in global Ca2+ handling [87]. It followed that Tg neurons were no more susceptible to neurotoxicants, such as glutamate, compared to non-Tg and suppression of RyanR3 up-regulation sensitized neurons to death in culture [87].
Given the importance of Ca2+ signalling to synaptic plasticity [88] and that mouse models of AD display neuronal hyperexcitability, epileptiform activity and functional disruption of neuronal networks [55, 89–92], perhaps alterations in ER Ca2+ handling are utilized to depress membrane excitability and prevent excitotoxicity. RyanRs are appropriately situated in the dendritic spines of CA1 hippocampal neurons to contribute to the CICR required to induce synaptic changes [73]. Recently, it has been shown that RyanR3 plays a substantial role in mediating the slow AHP current in hippocampal CA1 pyramidal neurons, important for the depression of membrane excitability [79]. Up-regulation of RyR3 could prove important for the modulation of excitability in cortical neurons of TgCRND8 mice, due to accumulating Aβ in vitro, and neurons that are unable to up-regulate RyR3 due to siRNA treatment could be more vulnerable to the excitotoxicity, oxidative stress and death related to Aβ exposure [93]. Such a role for RyanR3 could be relevant in brain as well. Adult (4–4.5 month old) TgCRND8 mice have increased expression of RyanR3 in the cortex and hippocampus compared to controls [86]. It is interesting to note that pre-plaque TgCRND8 mice display presynaptic depression of basal synaptic transmission in hippocampal CA1 mediated by large current Ca2+-activated K+ (BK) channels, which participate in AHP and are activated in epilepsy [94]. Altered neuronal excitability and epileptic activity in AD are thought to be a manifestation of neuronal circuit remodelling that can occur very early in the disease. In AD patients, seizures may accompany the onset of mild cognitive impairment and can occur at the time of diagnosis prior to extensive neurodegeneration [95]. This is an emerging area of study in AD and changes to RyanR-mediated Ca2+ signalling could be an important factor in these phenomena.
Furthermore, when AD-related mutations negate the ability of PS to “leak” Ca2+ from the ER [71], the results are damaging to neuronal function and health. The up-regulation of RyanR function in hippocampal neurons from the 3X-TgAD mice expressing PS1-M146V knockin mutation appear to partially compensate for the loss of ER Ca2+ leak function in these neurons [96]. It was demonstrated that when up-regulated RyanRs were blocked by RNA interference or dantrolene in hippocampal neurons from 3X-TgAD mice, the results were significantly overloaded ER Ca2+ pools [96]. Moreover, when APPPS1 transgenic mice harbouring the FAD PS1-L166P mutation were chronically fed dantrolene, Aβ plaque load was increased by 3-fold and PSD95 expression, a marker for excitatory synapse formation, was decreased in brain from 8 month old mice compared to control [96]. Such data indicate that the loss of ER Ca2+ leak function of presenilins can be partially compensated by an increased RyanR-mediated Ca2+ flux from the ER.
These findings prompt us to suggest that perhaps some forms of dysregulated ER Ca2+ signalling are a response to the adverse conditions of AD, such as for example the accumulation of Aβ peptides or increased neuronal excitability, and are an effort to maintain intracellular Ca2+ and cellular homeostasis (Figure 2). If such compensatory mechanisms exist, they may slow down the progression of disease but can not stop it completely, as TgCRND8, APPPS1 and 3X-TgAD mice go on to develop cognitive dysfunction and plaque formation [97–100]. Possibly such subtle alterations to neuronal Ca2+ regulation are only effective in the early stages of the disease. Another possibility is that these small changes to ER Ca2+ signalling are intended for the short-term and when they are sustained over the duration of AD progression, they may actually contribute to neuronal dysfunction and aberrant neuronal network formation leading to cognitive deficits [90]. The mechanisms by which changes to intracellular Ca2+ could be protective in AD are still not clear and further studies are required to determine how they may affect neuronal function and viability (Figure 2).
Figure 2. Pathways to neurodegeneration and neuroprotection in Alzheimer disease.
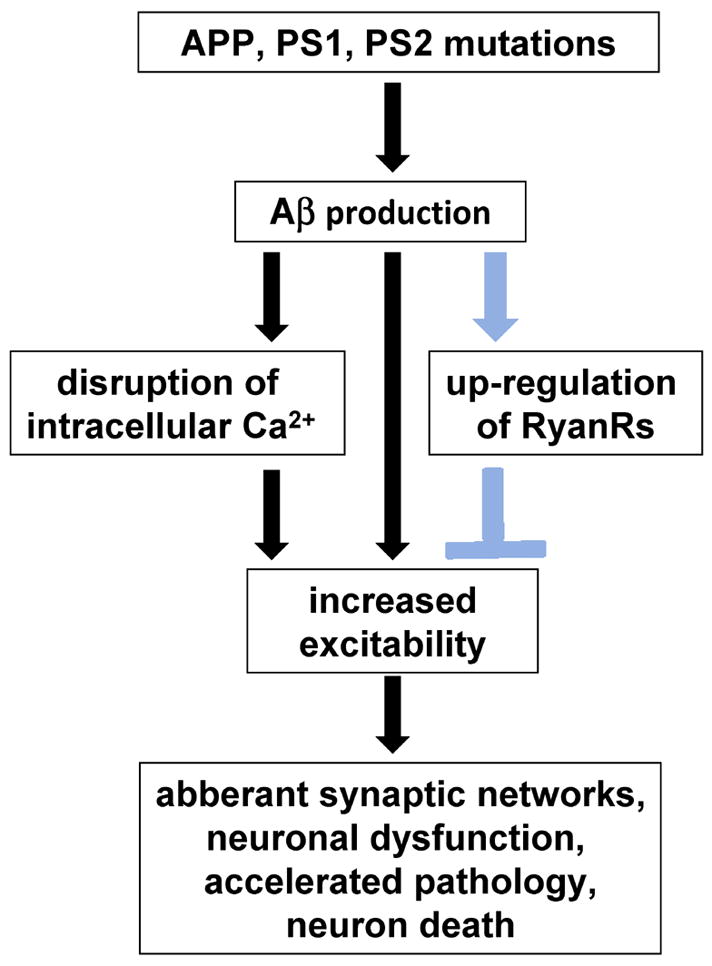
Illustration of how changes to intracellular Ca2+ handling could lead to neurodegeneration (black arrows) or potentially neuroprotection (blue arrows). It is generally accepted that Aβ oligomers and sustained Ca2+ dysregulation contributes to neurodegeneration in AD. Recent studies have demonstrated that both contribute to increased neuronal excitability, which can trigger the aberrant remodelling of neuronal networks, neuronal dysfunction, Aβ production and cell death. New data suggests that particular types of Ca2+ dysregulation in AD could be compensatory, eg. the up-regulation of RyanRs, and are triggered in parallel to attenuate or delay neurodegenerative mechanisms.
Conclusion
There is much evidence to suggest that dysregulated Ca2+ signalling has a significant role to play in the pathology of AD. Evidence suggests that the various Ca2+ handling channels and pumps in the ER are prominent contributors to the alterations of intracellular Ca2+ signalling in AD. Some of the Ca2+ signaling changes observed in AD appear to be detrimental to neuronal health, but some may be compensatory. A more comprehensive understanding of the role of dysregulated Ca2+ handling in neurodegeneration and neuroprotection in AD will be imperative for the future design of effective disease-modifying therapeutics.
Acknowledgments
IB is a holder of Carla Cocke Francis Professorship in Alzheimer’s Research and supported by the McKnight Neuroscience of Brain Disorders Award, Alzheimer’s Disease Drug Discovery Foundation, and NIH grant R01AG030746.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Giannakopoulos P, Kovari E, Gold G, von Gunten A, Hof PR, Bouras C. Pathological substrates of cognitive decline in Alzheimer’s disease. Front Neurol Neurosci. 2009;24:20–9. doi: 10.1159/000197881. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–63. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:534–8. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Etcheberrigaray R, Hirashima N, Nee L, Prince J, Govoni S, Racchi M, Tanzi RE, Alkon DL. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol Dis. 1998;5:37–45. doi: 10.1006/nbdi.1998.0176. [DOI] [PubMed] [Google Scholar]
- 6.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 7.Verkhratsky A, Mattson MP, Toescu EC. Aging in the mind. Trends Neurosci. 2004;27:577–8. doi: 10.1016/j.tins.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 8.Hajnoczky G, Csordas G, Yi M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium. 2002;32:363–77. doi: 10.1016/s0143416002001872. [DOI] [PubMed] [Google Scholar]
- 9.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toescu EC, Verkhratsky A. The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell. 2007;6:267–73. doi: 10.1111/j.1474-9726.2007.00296.x. [DOI] [PubMed] [Google Scholar]
- 11.Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8:345–6. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- 12.Landfield PW. ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging. 1987;8:346–7. doi: 10.1016/0197-4580(87)90074-1. [DOI] [PubMed] [Google Scholar]
- 13.Landfield PW. Increased hippocampal Ca2+ channel activity in brain aging and dementia. Hormonal and pharmacologic modulation. Ann N Y Acad Sci. 1994;747:351–64. doi: 10.1111/j.1749-6632.1994.tb44422.x. [DOI] [PubMed] [Google Scholar]
- 14.Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci. 2006;26:3482–90. doi: 10.1523/JNEUROSCI.4171-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21:9744–56. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemond P, Jaffe DB. Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow after hyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience. 2005;135:413–20. doi: 10.1016/j.neuroscience.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 18.Thibault O, Landfield PW. Increase in single L-type calcium channels in hippocampal neurons during aging. Science. 1996;272:1017–20. doi: 10.1126/science.272.5264.1017. [DOI] [PubMed] [Google Scholar]
- 19.Disterhoft JF, Oh MM. Pharmacological and molecular enhancement of learning in aging and Alzheimer’s disease. J Physiol Paris. 2006;99:180–92. doi: 10.1016/j.jphysparis.2005.12.079. [DOI] [PubMed] [Google Scholar]
- 20.Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S, Bossini A, Fagard R, Gil-Extremera B, Laks T, Kobalava Z, Sarti C, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Birkenhager WH. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Intern Med. 2002;162:2046–52. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- 21.Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–25. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 22.Williamson J, Goldman J, Marder KS. Genetic aspects of Alzheimer disease. Neurologist. 2009;15:80–6. doi: 10.1097/NRL.0b013e318187e76b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–90. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 24.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–7. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 25.Khachaturian ZS. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- 26.Emilsson L, Saetre P, Jazin E. Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis. 2006;21:618–25. doi: 10.1016/j.nbd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Lipton SA. Pathologically activated therapeutics for neuroprotection. Nat Rev Neurosci. 2007;8:803–8. doi: 10.1038/nrn2229. [DOI] [PubMed] [Google Scholar]
- 28.Lopez-Arrieta JM, Birks J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst Rev. 2002:CD000147. doi: 10.1002/14651858.CD000147. [DOI] [PubMed] [Google Scholar]
- 29.Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2007;29:625–55. [PubMed] [Google Scholar]
- 30.Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao HT, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, Pasquier F, Galimberti D, Scarpini E, Mann D, Lendon C, Campion D, Amouyel P, Davies P, Foskett JK, Campagne F, Marambaud P. A polymorphism in CALHM1 influences Ca2+ homeostasis, A beta levels, and Alzheimer’s disease risk. Cell. 2008;133:1149–1161. doi: 10.1016/j.cell.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui PJ, Zheng L, Cao L, Wang Y, Deng YL, Wang G, Xu W, Tang HD, Ma JF, Zhang T, Ding JQ, Cheng Q, Chen SD. CALHM1 P86L Polymorphism is a Risk Factor for Alzheimer’s Disease in the Chinese Population. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1207. [DOI] [PubMed] [Google Scholar]
- 32.Bertram L, Schjeide BM, Hooli B, Mullin K, Hiltunen M, Soininen H, Ingelsson M, Lannfelt L, Blacker D, Tanzi RE. No association between CALHM1 and Alzheimer’s disease risk. Cell. 2008;135:993–4. doi: 10.1016/j.cell.2008.11.030. author reply 994–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minster RL, Demirci FY, DeKosky ST, Kamboh MI. No association between CALHM1 variation and risk of Alzheimer disease. Hum Mutat. 2009;30:E566–9. doi: 10.1002/humu.20989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beecham GW, Schnetz-Boutaud N, Haines JL, Pericak-Vance MA. CALHM1 polymorphism is not associated with late-onset Alzheimer disease. Ann Hum Genet. 2009;73:379–81. doi: 10.1111/j.1469-1809.2009.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Querfurth HW, Selkoe DJ. Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry. 1994;33:4550–61. doi: 10.1021/bi00181a016. [DOI] [PubMed] [Google Scholar]
- 36.Querfurth HW, Jiang J, Geiger JD, Selkoe DJ. Caffeine stimulates amyloid beta-peptide release from beta-amyloid precursor protein-transfected HEK293 cells. J Neurochem. 1997;69:1580–91. doi: 10.1046/j.1471-4159.1997.69041580.x. [DOI] [PubMed] [Google Scholar]
- 37.Buxbaum JD, Ruefli AA, Parker CA, Cypess AM, Greengard P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc Natl Acad Sci U S A. 1994;91:4489–93. doi: 10.1073/pnas.91.10.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierrot N, Ghisdal P, Caumont AS, Octave JN. Intraneuronal amyloid-beta1–42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J Neurochem. 2004;88:1140–50. doi: 10.1046/j.1471-4159.2003.02227.x. [DOI] [PubMed] [Google Scholar]
- 39.Kawahara M, Negishi-Kato M, Sadakane Y. Calcium dyshomeostasis and neurotoxicity of Alzheimer’s beta-amyloid protein. Expert Rev Neurother. 2009;9:681–93. doi: 10.1586/ern.09.28. [DOI] [PubMed] [Google Scholar]
- 40.Arispe N, Rojas E, Pollard HB. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A. 1993;90:567–71. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-beta-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23:1249–63. doi: 10.1016/s0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- 42.Simakova O, Arispe NJ. The cell-selective neurotoxicity of the Alzheimer’s Abeta peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for Abeta toxicity. J Neurosci. 2007;27:13719–29. doi: 10.1523/JNEUROSCI.3006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s Disease. J Neurochem. 2005;93:1361–70. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- 44.Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–94. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 45.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–99. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 46.Ye C, Walsh DM, Selkoe DJ, Hartley DM. Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett. 2004;366:320–5. doi: 10.1016/j.neulet.2004.05.060. [DOI] [PubMed] [Google Scholar]
- 47.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 48.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–75. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 50.Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid beta oligomers (A beta(1–42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–97. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Wang Z, Wang B, Justice NJ, Zheng H. Amyloid Precursor Protein Regulates Cav1.2 L-type Calcium Channel Levels and Function to Influence GABAergic Short-Term Plasticity. J Neurosci. 2009;29:15660–8. doi: 10.1523/JNEUROSCI.4104-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 53.Barger SW, Mattson MP. The secreted form of the Alzheimer’s beta-amyloid precursor protein stimulates a membrane-associated guanylate cyclase. Biochem J. 1995;311(Pt 1):45–7. doi: 10.1042/bj3110045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larson J, Lynch G, Games D, Seubert P. Alterations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mice. Brain Res. 1999;840:23–35. doi: 10.1016/s0006-8993(99)01698-4. [DOI] [PubMed] [Google Scholar]
- 55.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous Hyperactivity and Intercellular Calcium Waves in Astrocytes in Alzheimer Mice. Science. 2009 doi: 10.1126/science.1169096. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bezprozvanny I. Amyloid goes global. Sci Signal. 2009;2:pe16. doi: 10.1126/scisignal.263pe16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol. 2004;91:2437–44. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- 59.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 62.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 63.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–62. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 64.de Leon MJ, Mosconi L, Blennow K, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Tsui W, Saint Louis LA, Sobanska L, Brys M, Li Y, Rich K, Rinne J, Rusinek H. Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:114–45. doi: 10.1196/annals.1379.012. [DOI] [PubMed] [Google Scholar]
- 65.Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999;72:1061–8. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- 66.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol. 2000;149:793–8. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–13. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leissring MA, Parker I, LaFerla FM. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1, 4,5-trisphosphate-mediated calcium signals. J Biol Chem. 1999;274:32535–8. doi: 10.1074/jbc.274.46.32535. [DOI] [PubMed] [Google Scholar]
- 69.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–72. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 70.Giacomello M, Barbiero L, Zatti G, Squitti R, Binetti G, Pozzan T, Fasolato C, Ghidoni R, Pizzo P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol Dis. 2005;18:638–48. doi: 10.1016/j.nbd.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 71.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER calcium leak channels, a function disrupted by mutations linked to familial Alzheimer’s disease. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson O, Tu H, Lei T, Bentahir M, de Strooper B, Bezprozvanny I. Familial Alzheimer disease-linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J Clin Invest. 2007;117:1230–9. doi: 10.1172/JCI30447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–70. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP(3) receptor channel gating. Neuron. 2008;58:871–83. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J Cell Biol. 2008;181:1107–16. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Savic N, Sciancalepore M. Intracellular calcium stores modulate miniature GABA-mediated synaptic currents in neonatal rat hippocampal neurons. Eur J Neurosci. 1998;10:3379–86. doi: 10.1046/j.1460-9568.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- 77.Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22:21–8. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–60. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van de Vrede Y, Fossier P, Baux G, Joels M, Chameau P. Control of IsAHP in mouse hippocampus CA1 pyramidal neurons by RyR3-mediated calcium-induced calcium release. Pflugers Arch. 2007;455:297–308. doi: 10.1007/s00424-007-0277-4. [DOI] [PubMed] [Google Scholar]
- 80.Futatsugi A, Kato K, Ogura H, Li ST, Nagata E, Kuwajima G, Tanaka K, Itohara S, Mikoshiba K. Facilitation of NMDAR-independent LTP and spatial learning in mutant mice lacking ryanodine receptor type 3. Neuron. 1999;24:701–13. doi: 10.1016/s0896-6273(00)81123-x. [DOI] [PubMed] [Google Scholar]
- 81.Galeotti N, Quattrone A, Vivoli E, Norcini M, Bartolini A, Ghelardini C. Different involvement of type 1, 2, and 3 ryanodine receptors in memory processes. Learn Mem. 2008;15:315–23. doi: 10.1101/lm.929008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O’Neill C. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer’s disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- 83.Chan SL, Mayne M, Holden CP, Geiger JD, Mattson MP. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J Biol Chem. 2000;275:18195–200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 84.Lee SY, Hwang DY, Kim YK, Lee JW, Shin IC, Oh KW, Lee MK, Lim JS, Yoon DY, Hwang SJ, Hong JT. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006;20:151–3. doi: 10.1096/fj.05-4017fje;1. [DOI] [PubMed] [Google Scholar]
- 85.Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J Neurochem. 2005;94:1711–8. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- 86.Supnet C, Grant J, Kong H, Westaway D, Mayne M. Amyloid-beta-(1–42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J Biol Chem. 2006;281:38440–7. doi: 10.1074/jbc.M606736200. [DOI] [PubMed] [Google Scholar]
- 87.Supnet C, Noonan C, Richard K, Bradley J, Mayne M. Upregulation Of The Type 3 Ryanodine Receptor Is Neuroprotective In The TgCRND8 Mouse Model Of Alzheimer’s Disease. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06487.x. in press. [DOI] [PubMed] [Google Scholar]
- 88.Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 89.Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett. 2004;367:164–7. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- 90.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–9. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 92.Wang Y, Zhang G, Zhou H, Barakat A, Querfurth H. Opposite effects of low and high doses of abeta42 on electrical network and neuronal excitability in the rat prefrontal cortex. PLoS One. 2009;4:e8366. doi: 10.1371/journal.pone.0008366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res. 2002;36:1307–13. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 94.Ye H, Jalini S, Mylvaganam S, Carlen P. Activation of large-conductance Ca(2+)-activated K(+) channels depresses basal synaptic transmission in the hippocampal CA1 area in APP (swe/ind) TgCRND8 mice. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.05.012. [DOI] [PubMed] [Google Scholar]
- 95.Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;22:121–4. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- 96.Zhang H, Sun S, Ozkan E, Herreman A, DeStrooper B, Bezprozvanny I. Neuroscience. Chicago, Il: 2009. Role of presenilins in neuronal calcium homeostasis. (ed. Sf Neuroscience) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–70. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- 98.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 99.Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM, Jucker M. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 2006;7:940–6. doi: 10.1038/sj.embor.7400784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horre K, Van Houtvin T, Esselmann H, Paul S, Schafer MK, Berezovska O, Hyman BT, Sprangers B, Sciot R, Moons L, Jucker M, Yang Z, May PC, Karran E, Wiltfang J, D’Hooge R, De Strooper B. gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science. 2009;324:639–42. doi: 10.1126/science.1171176. [DOI] [PMC free article] [PubMed] [Google Scholar]