

Abstract
Transcriptional factors of the NF-κB family and STAT3 are ubiquitously expressed and control numerous physiological processes including development, differentiation, immunity, metabolism and cancer. Both NF-κB and STAT3 are rapidly activated in response to various stimuli including stresses and cytokines, although they are regulated by entirely different signaling mechanisms. Once activated, NF-κB and STAT3 control the expression of anti-apoptotic, pro-proliferative and immune response genes. Some of these genes overlap and require transcriptional cooperation between the two factors. The activation of and interaction between STAT3 and NF-κB plays a key role in controlling the dialog between the malignant cell and its microenvironment, especially with inflammatory/immune cells that infiltrate tumors. Quite often, cytokines whose expression is induced in response to NF-κB in immune cells of the tumor microenvironment lead to STAT3 activation in both malignant and immune cells. While within malignant and pre-malignant cells STAT3 exerts important oncogenic functions, within inflammatory cells it may also suppress tumor promotion through its anti-inflammatory effects. Other interactions and forms of crosstalk between NF-κB and STAT3 include physical interaction between the two, cooperation of these factors at gene promoters/enhancers, the NF-κB dependent expression of inhibitors of STAT3 activation and the participation of STAT3 in inflammatory cells in the negative regulation NF-κB. Despite these versatile and occasionally antagonistic interactions, NF-κB and STAT3 cooperate to promote the development and progression of colon, gastric and liver cancers. In addition to explaining the molecular pathogenesis of cancer, these interactions also offer opportunities for the design of new therapeutic interventions.
Introduction
The search for genes that control tumor growth and progression has resulted in the discovery of oncogenes, which are subject to mutational activation in cancer cells, as well as tumor suppressors (1). However, not all oncogenes are targets for mutational activation. Notable examples are the NF-κB and STAT3 transcriptional factors which were found to play pivotal roles in various aspects of the tumorigenic process in a number of malignancies (2, 3). Most often, NF-κB and STAT3 are constitutively activated in neoplastic cells due to upregulation of upstream signaling pathways in response to autocrine and paracrine factors that are produced within the tumor microenvironment (4). Although NF-κB and STAT3 do not match the classical oncogene definition, they are powerful activators of the malignant state and control expression of target genes important for cell proliferation, survival, angiogenesis and tissue repair (5–8). However, the functions of NF-κB and STAT3 extend far beyond the cancer cell and both transcriptional factors are important regulators of immune and inflammatory functions (9, 10). While being activated by cytokines and growth factors, both NF-κB and STAT3 control the expression of other cytokines and inflammatory/immune mediators, thus serving as a regulatory hub that coordinates immunie and inlfmamatory responses. Therefore, NF-κB and STAT3 also affect cancer cell physiology through their effects on immune and inflammatory cells in the tumor microenvironment (11–13).
NF-κB
The NF-κB term refers to a family of signal-responsive transcription factors that includes RelA/p65, c-Rel, RelB, NF-κB1/p50 and NF-κB2/p52 (14). A potential link between NF-κB and cancer first became obvious when the RelA gene, encoding its p65 subunit, was cloned and identified to be homologous to the viral oncogene v-Rel (15). While most solid and lymphoid tumors show constitutive NF-κB activity (7), mutations in genes encoding NF-κB family members are rare. In most cases NF-κB is maintained in an active state within cancer cells through mutational activation of upstream signaling molecules or in response to extracellular stimuli within the tumor microenvironment (6). In normal resting cells, NF-κB transcription factors are maintained in an inactive state within the cytoplasm through binding to inhibitory proteins called I-κB, which also include the unprocessed p105 and p100 forms of NF-κB1 and NF-κB2 (16). Dependent on the cell type, NF-κB is most potently activated upon engagement of pattern-recognition receptors (TLRs, NLRs and others), receptors for pro-inflammatory cytokines, such as TNF-α or IL-1, and antigen receptors (17–20). However, many other receptors as well as physical stimuli can also lead to NF-κB activation. Taken together, in various cell types NF-κB can be activated by many different stimuli and the actual response depends on the receptors expressed by the individual cell (Table 1).
Table 1.
Typical inducers of NF-κB and STAT3 include pattern recognition receptors such as TLRs and NLRs, antigen receptors of lymphocytes and various cytokines
| Cell type | STAT3 activators | NF-κB activators |
|---|---|---|
| CD4 and CD8 T cells | IL-6, IL-10, IL-27, IL-23, IL-21, IL-25 | TNF, IL-1, Ag Receptor, IL-17 |
| T reg cells | IL-6, IL-10, IL-23 | TNF, IL-1, Ag Receptor |
| Macrophages, DC | IL-10, VEGF, IL-23, IL-6, leptin, | TLR, NLR, TNF, M-CSF, IL-17, IL-1 |
| Neutrophils | IL-10, IL-6 | TLR, NLR |
| Epithelial and tumor cells | IL-6, IL-11, IL-22, EGF | TNF, IL-1, IL-17, TLR, NLR |
| Fibroblasts and myofibroblasts | IL-6, FGF, leptin | IL-17, TNF, TLR? |
Activation of NF-κB is most commonly mediated by the IκB kinase (IKK) complex, which is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit IKKγ/NEMO (21). IKK-mediated IκB phosphorylation leads to their ubiquitin-dependent degradation and the subsequent nuclear entry of the released NF-κB dimers (21, 22). Once in the nucleus, NF-κB dimers are further regulated by protein phosphorylation (16) and other port-translational modifications, such as protein acetylations (23)and activate genes whose products inhibit cell death (24–27), stimulate cell proliferation (28), and promote migratory and invasive phenotypes, as well as epithelial-mesenchymal transition (EMT) (29), that are associated with tumor progression (30, 31).
STAT3
STAT3 belongs to the STAT (signal transducer and activator of transcription) family of signal responsive transcription factors, which like NF-κB are kept in an inactive form in the cytoplasm of non-stimulated cells (10, 32). However, STAT3 activation, like other members of its family, does not require inducible degradation of an inhibitor. Instead, it is mediated by phosphorylation of a critical tyrosine residue (Tyr 705) that induces STAT3 dimerization through phosphotyrosine-SH2 domain interaction (33). Once dimerized, STAT transcription factors enter the nucleus and activate a broad array of target genes. However, unphosphorylated STAT3 is still capable for dimerization and induction of transcription (34, 35). STAT activation is most commonly mediated by members of the JAK family of tyrosine-kinases and in the case of STAT3 the major activator is JAK1 (36). STAT3 transcriptional activity and DNA binding are further enhanced through serine (Ser 727) phoshorylation (37). Another mechanism that fine tunes of STAT3 activity, is its reversible acetylation (38), which also affects the activity of NF-κB family members (39).
Ablation of the STAT3 gene results in multiple developmental abnormalities and is lethal, underscoring a critical role in development and homeostasis (40). Normally, STAT3 activity is tightly regulated by multiple feedback mechanisms and its prolonged activation is associated with various malignancies (2, 5, 32, 41). Feedback regulation of STAT3 activation is a complex process and the two major players are the SHP2 phosphatase and the product of the STAT3-inducible gene SOCS3 (suppressor of cytokine signaling), which blocks STAT3 activation by gp130 receptor (42).
Activated nuclear STAT3 has been detected in many forms of cancer, including breast, colon, gastric, lung, head and neck, skin, prostate and others (2, 32, 43, 44). Conversely, conditional STAT3 ablation in enterocytes, keratinocytes and other epithelial cell types was found to inhibit tumor development and progression (41, 45–51). Importantly, both cancer cells and infiltrating immune and stromal cells harbor activated STAT3. Although constitutively active mutants of STAT3 (for example, STAT3C) were produced in the lab (2), naturally occurring STAT3 activating mutations are rare. It was observed that many tumor cells, which display constitutive STAT3 activation in vivo rapidly lose STAT3 phosphorylation once put into culture without neighboring immune or stromal cells (5). This implies that akin to NF-κB, STAT3 activation in cancer often is the result of chronic stimulation by extracellular signals (autocrine or paracrine) present in the tumor microenvironment. Indeed many cytokines and growth factors capable of activating STAT3 are found in the tumor microenvironment (Table 1). Such STAT3 activators include cytokines of the IL-6 family, which signal through the gp130 common signaling subunit (IL-6, IL-11, oncostatin M, LIF, CNTF, IL-27 and IL-35), cytokines of the IL-10 family (IL-10, IL-22, IL-19 and IL-20), epidermal growth factor (EGF) family members, HGF, VEGF, IL-23 and IL-21 (Table 1). Once activated in malignant or immune cells, STAT3 can induce the expression of a subset of genes whose products are important for further STAT3 activation, including IL-6, IL-22, EGF, IL-23 and IL-10 (52, 53) as well as STAT3-dependent induction of cell surface growth factor and cytokine receptors (EGFR, c-Met, IL-23R) or cytoplasmic proto-oncogenes such as K-Ras, Src and c-Abl, whose products are capable of inducing STAT3 phosphorylation (8, 54–56). It is therefore of no wonder that STAT3 is one of the most commonly activated transcription factors in cancer (32).
Interactions between STAT3 and NF-κB
Global chromatin binding surveys revealed that STAT3 binds at least 3,000 different gene promoters and the number of genes targeted by NF-κB family members is even larger. Importantly, NF-κB and STAT3 control both distinct and overlapping groups of genes during tumorigenesis. This can be explained in part by the distribution of NF-κB and STAT3 binding sites in the regulatory regions of such genes. For instance, a gene that contains only NF-κB sites may be NF-κB, but not STAT3, responsive, whereas a gene that contains both NF-κB and STAT3 binding sites may be regulated by both factors in a cooperative manner. Indeed, the Stark lab has shown that overexpression of a constitutively active form of STAT3 in HME cells induced 427 genes, whereas TNF-α (a major NF-κB activator) induced the expression of 1225 genes and only 123 of these genes were dependent on both NF-κB and STAT3 (35). Another global profiling of STAT3-dependent genes in mouse lung cells also revealed a large number of genes whose expression is controlled by STAT3, amongst which a number of typical NF-κB target genes, including chemokine genes, PAI-1, Bcl-3, Bcl-2, GADD45β and SOCS3, are also present (57). Therefore, it seems that the induction of a certain gene subset requires cooperation between STAT3 and NF-κB pathways.
In addition to binding to adjacent sites in the control regions of shared target genes, several NF-κB family members, in particular RelA/p65 and p50, were found to physically interact with STAT3 (39, 58–60). This interaction may result in either specific transcriptional synergy or repression of NF-κB/STAT3 regulated genes (35). There are several different scenarios for the STAT3:NF-κB interaction. First, STAT3, presumably in its unphosphorylated form can bind to NF-κB complexed with IκB, displace IκB from NF-κB and thereby facilitate NF-κB activation and nuclear entry even in the absence of conventional IKK signaling (35). Second, certain NF-κB induced inhibitory proteins, such as IκBζ, can bind to STAT3 and inhibit its binding to DNA (61, 62). Third, STAT3 may interact with p65 RelA/p65 in the nucleus and recruits the p300 histone acetylase (HAT) complex to the complex. In addition to histones, p300 can also acetylate p65 and increase its nuclear retention and thereby prolong its transcriptional activity (39). The interaction of STAT3 with p300 or other HATs is also required for STAT3-dependent transcription (63) and reversible acetylation of RelA regulates the duration of nuclear NF-κB activity (23). Acetylated RelA interacts only weakly with IκBα, and its deacetylation by histone deacetylases (HDACs) markedly increases IκBα binding, and therefore results in its nuclear export and deactivation (64). This means that STAT3 can prolong the presence of active NF-κB in the nucleus but would be unable to do so in the absence of upstream NF-κB activating signals. Since NF-κB activation is tightly regulated (65), the latter scenario may represent an important mechanism by which activated STAT3 in cancer cells can ensure constitutive NF-κB activation, even when the IKK complex is only temporarily activated (39). Furthermore, the tumor microenvironment also contains anti-inflammatory signals, such as TGFβ and IL-10, which downregulate the production of pro-inflammatory cytokines as well as some of the intracellular signaling pathways that lead to conventional NF-κB activation. It is therefore tempting to speculate that STAT3-mediated cell-autonomous nuclear NF-κB retention may be an important mechanism for STAT3-depedendent NF-κB activation in cancer. Fourth, while in the nucleus, the NF-κB:STAT3 complex can bind to unique DNA target sequences to which neither factor can bind on its own. For example, simultaneous activation of STAT3 and NF-κB results in their interaction, nuclear retention and STAT3 binding to non-classical sequence in SAA gene promoter (66). Nonetheless, NF-κB is not the only transcription factor, which cooperates with STAT3. Other STAT3 partners include androgen receptor, C/EBP, STAT6, STAT1, c-Jun, and β-catenin (63). These factors may further modulate the STAT3:NF-κB interaction.
Functional interaction or antagonism between NF-κB and STAT3 can also occur at the level of signal transduction regulators, such as SOCS proteins whose expression is controlled by both NF-κB and STAT3 (42, 67, 68). Particularly, SOCS3, whose expression is induced by both NF-κB and STAT3, binds to the gp130 receptor subunit and prevents further cytokine-dependent activation of STAT3 (33). Likewise, SOCS1 and SOCS3 can attenuate NF-κB activation driven by several cytokines and TLR agonists, although the exact mechanism underlying this inhibitory effect is not entirely clear (33). Given that STAT3 prolongs nuclear retention of NF-κB, SOCS3-mediated STAT3 inactivation may be also responsible for decreased NF-κB activity.
NF-κB and STAT3 control a protumorigenic gene expression
Amongst the many genes controlled by NF-κB and STAT3, either synergistically or individually, one can identify groups whose products play important roles in tumor development. One of the key hallmarks of cancer is the ability of malignant cells to execute an anti-apoptotic prosurvival program that prevents intrinsically-programmed or exogenously-induced cell death (69). Anti-apoptotic genes are prominent targets for NF-κB and STAT3, and genes such as Bcl-xL, Bcl-2, c-IAP2 are activated by both factors, whereas A1 and c-FLIP are mostly NF-κB-dependent and Mcl-1 and Survivin are STAT3-dependent (4, 7, 32, 70, 71). STAT3 also functions as a repressor of p53 expression, since blocking STAT3 in cancer cells results in upregulation of p53 and its target p21, leading to p53-mediated apoptosis (72). Remarkably, inhibition of NF-κB also augments p53-induced apoptosis (73, 74). It remains to be determined whether STAT3 or NF-κB activated in the context of chronic inflammation can suppress p53-mediated cell cycle arrest and apoptosis in premalignant cells harboring oncogenic mutations, thereby giving these cells an opportunity to overcome p53-dependent genome surveillance and progress into fully blown cancer. If this is indeed the case, the role of inflammation in tumor initiation may be even greater than its proposed role in generation of reactive oxygen and nitrogen species, which may induce oncogenic mutations. Taken together, it seems plausible that enhanced expression of anti-apoptotic genes makes a vital contribution to the oncogenic functions of NF-κB and STAT3, as it ensures the survival of pre-malignant cells harboring oncogenic mutations and makes these cells resistant to challenges such as genotoxic insults caused by accumulating mutations, various forms of immune surveillance and others.
Additional STAT3 and NF-κB targets are several cell cycle control and proliferation genes; Prominent amongst this group are cyclins D and B, as well as c-Myc (47, 63, 75). These genes mediate cell autonomous effects of STAT3 (and NF-κB) on tumor growth, as seen in several models where inactivation of STAT3 in tumor cells resulted in decreased tumor size (45, 50). Interestingly, in mammary epithelial cells, cyclinD1 is also activated by the alternative NF-κB signaling pathway in response to a member of the TNF cytokine family, RANK ligand (RANKL) (76).
Tissue resistance and repair genes are somewhat similar in function to the anti-apoptotic genes because they enhance the survival of transformed cells in the hostile microenvironment present during early stages of tumor development. These genes include STAT3 targets such as RegIIIβ and RegIIIγ, Tff3 and inducible form of Hsp70 (Hsp72) (36, 47, 77, 78). Anti-oxidant genes such as Mn-SOD, ferritin, catalase also belongs to this group and their expression is controlled by NF-κB as well as by STAT3 (75). These genes are important to deal with intracellular insults by reactive oxygen species generated during chronic inflammation, metabolic stress and hypoxia, thereby increasing the survival potential of pre-malignant tumor precursors.
Angiogenesis and hypoxia genes
STAT3 was shown to control the expression of vascular endothelial growth factor (VEGF), hypoxia inducible factor 1 alpha (HIF1α) and bFGF (basic fibroblast growth factor), all of which are important mediators of angiogenesis (79, 80). Conditional STAT3 inactivation in hematopoietic cells revealed that the effect of STAT3 on tumor angiogenesis originates from its function in myeloid suppressor dendritic cells (MDSC) and tumor associated macrophages (TAM) (79, 81). NF-κB was also shown to be an important regulator of HIF1α gene transcription and a regulator of VEGF gene expression (82), although it remains to be proven experimentally whether NF-κB directly activates VEGF gene expression or it does so via induction of HIF1α, which is a direct activation of VEGF transcription.
Oncogenes and transcription factors
Additional NF-κB and STAT3 target genes include transcription factors that can activate the expression of additional constellation of target genes involved in tumorigenesis. For example, STAT3 induces the members of AP-1 family, such c-Jun, and c-Fos; and also upregulates its own expression (35, 36, 56). As mentioned above, NF-κB and STAT3 also control expression of the HIF1α genes (79, 82).
Chemoattractants and chemokines
Prominent amongst STAT3 and NF-κB targets are genes encoding chemokines, chemoattractants that play pivotal roles in the recruitment and renewal of different cell subsets in the tumor microenvironment. CCL2, CXCL2, and IL-1β in particular regulate the recruitment of myeloid cells, which give rise to TAM’s or MDSC; while the CCL20/CCR6 axis is important for attraction of pro-inflammatory Th17 cells (83). STAT3 also induces expression of calcium binding S100A9 protein, which is specifically required for tumorigenesis and attraction of MSDC (84). Immunosuppressive T regulatory cells are attracted in CCL22/CCL17/CCR4 and CCL19/CCL21/CCR7-dependent manner (83, 85). Hence, by regulating chemokine synthesis, STAT3 and NF-κB determine the selective presence of various subsets of immune cells in the tumor microenvironment.
Tumor promoting and immuno-suppressive cytokines
This cohort of genes expressed predominantly in immune cells includes TNF-α, IL-1α, IL-1β, IL-6, IL-22, IL-23, IL-10, IL-12p35 and p40, IL-17A, IFNγ and several others. These cytokines control and mediate the cross-talk between cancer cells and tumor infiltrating immune and inflammatory cells and are responsible for inflammation-promoted tumor growth and inhibition of tumor immunosurveillance (86). Quite a few of these cytokines are controlled by NF-κB, STAT3 or both (Table 2). Genetic or pharmacological inhibition or intervention with these cytokines in various models of cancer either increases or decreases tumor growth and also affects tumor multiplicity (29, 45, 87–91). For the sake of simplicity, the NF-κB and STAT3 regulated cytokines can be further classified according to their function in cancer as either tumor-promoting (e.g. TNF, IL-23, IL-6) or tumor-inhibiting (TRAIL, IFNγ and IFNα). Since cytokines regulate systemic and tumor associated inflammation, it should be noted that the involvement of different cytokines in cancer (with or without an obvious inflammatory component) may be tumor and model dependent. For example, in inflammation associated cancer IFNγ can promote tumorigenesis (92), while under non-inflammatory conditions it can activate potent anti-tumor responses (93, 94). The same applies for IL-10, whose ablation causes or enhances spontaneous gastrointestinal tumorigenesis due to chronic inflammation (89) but IL-10 is also believed to have a protumorigenic function due to its ability to suppress anti-tumor immune and inflammatory responses (95). It is important to point out that although cytokines in the tumor microenvironment are produced mostly by hematopoietic cells (3, 45, 46), some of the cytokines such as IL-6 and TNF-α can be also produced by the malignant cells themselves to establish an autocrine tumor-promoting signaling loop that further enhances NF-κB and STAT3 activation.
Table 2.
NF-κB and STAT3 control the production of cytokines, which have multiple effects on immune and cancer cells in the tumor microenvironment
| Cytokine | Effects on malignant/neoplastic cells | Effects on immune cells | Inducibility by STAT3 | Inducibility by NF-κB |
|---|---|---|---|---|
| TNF-α | survival, growth | survival, activation, recruitment | − | +++ |
| IL-6 | survival, growth | Th17 differentiation T cell survival Myeloid cell recruitment | +++ | +++ |
| IFNγ | no direct effect? | Activation of Th1, NK and CD8 cells | − | + |
| IL-23 | no direct effect? | Th17 differentiation, Treg suppression IL-17 and IL-22 production | +++ | +++ |
| IL-22 | survival? | +++ | + | |
| IL-17 | chemokine induction, survival | T cell regulation, monocyte and neutrophil recruitment | +++ | + |
| IL-27 | ? | Suppression of Th1/Th17 differentiation and immune cell activation, survival? | ++ | ++ |
| IL-1 | Survival and growth? | Immune activation | − | +++ |
NF-κB and STAT3 in immune and inflammatory cells
STAT3 and NF-κB are often activated in tumor associated immune and inflammatory cells, including myeloid cells and T lymphocytes (9, 79, 96, 97). Importantly, many of the protumorigenic signals generated by STAT3 and NF-κB are exerted within immune and inflammatory cells (4, 5). In some cells, NF-κB and STAT3 are critical for cell survival, for instance NF-κB in developing B cells or thymocytes (16), and STAT3 in activated T cells, subjected to activation-induced cell death (5). As mentioned above, NF-κB is a major downstream component of antigen receptor (TCR, BCR) patterns recognition receptor and inflammatory cytokine signaling pathways, resulting in the control of key immune and inflammatory functions (16). The cytokine and growth factor mileu with the tumor microenvironment controls both the function and the behavior of tumor-infiltrating immune and inflammatory cells as well as the growth and survival of malignant cells and NF-κB, as well as STAT3 are instrumental in this process. In other words, although NF-κB and STAT3 action within premalignant and fully transformed cells are critical for tumorigenesis, NF-κB and STAT3 in immune cells are also important in the control of tumor promotion and progression. Although the basic mechanisms by which STAT3 and NF-κB operate in immune cells are similar to those by which they act in normal and transformed epithelial cells, there are situations whereby STAT3 or NF-κB exert one effect in epithelial cells (normal and transformed) and a very different effect in immune/inflammatory cells. For instance, whereas ablation of STAT3 in enterocytes inhibits the development and growth of CAC (45, 47), its inactivation in myeloid cells actually enhances chronic inflammation and CAC development presumably due to defects in IL-10 signaling (98) (S.G. and M.K., unpublished observations). By contrast, the ablation of IKKβ, the critical activator for NF-κB in either cell type inhibits CAC development and growth (99). However, the inactivation of IKKβ or the IKKγ/NEMO regulatory subunit in liver epithelial cells (hepatocytes), enhances and accelerates the development of either chemically induced or spontaneous hepatocellular carcinoma (HCC) (100, 101). Enhanced HCC development in these mice is due to increased hepatocyte death in the absence of NF-κB resulting in compensatory proliferation of differentiated hepatocytes harboring oncogenic mutations. By contrast, ablation of IKKβ in Kupffer cells prevents HCC development because it inhibits the production of IL-6, which stimulates compensatory hepatocyte proliferation (101, 102).
The Yu and Pardoll lab’s have investigated the basis for the pro-tumorigenic function of STAT3 in immune/inflammatory cells and have found that inhibition of STAT3 within immune cells significantly enhances anti-tumor immune responses (96, 97). They used several transplanted tumors and found that Mx1-Cre driven “floxed” STAT3 gene inactivation (which primarily affects bone marrow, myeloid cells, lymphocytes and endothelial cells) dampens tumor growth (96, 103). Several mechanisms, which are not mutually exclusive, were proposed, including a role for STAT3 in the immunosuppressive action of IL-10 and inhibition of DC maturation as well as its effects on MDSC differentiation and recruitment (5, 97), STAT3- dependent production of angiogenic factors (79) as well as the role of STAT3 in differentiation and cytokine production by tumor-promoting T cells (46) and its role in reciprocal regulation of IL-23p19 versus IL-12p35 gene transcription (52). The latter possibility suggests the involvement of the NF-κB factors p65 RelA and c-Rel, which work in close collaboration with STAT3 to ensure IL-23p19 gene activation and IL-12p35 gene repression (52). However, IL-10 ablation as well as myeloid cell-specific STAT3 ablation enhance chronic inflammation and augment tumorigenicity in colon cancer (89) (S.G. and M.K., unpublished observations). NF-κB inhibition in myeloid cells in response to Mx1-Cre or LysM-Cre mediated deletion of IKKβ, however, always results in reduced tumor loads in CAC or HCC models and defective production of cytokines including IL-6, IL-1, IL-12/IL-23 and TNF-α (99, 101). Therefore, STAT3 in immune cells can play either a pro- or anti-tumorigenic role depending on the cell type in which it is activated, while most of the functions of NF-κB in immune cells are clearly pro-tumorigenic (3).
What are the effects of cytokines produced by immune cells in the tumor microenvironment? This is a complex question and in many cases cytokines act on more then one cell type and target both cancer cells and immune inflammatory cells and can exert both synergistic or antagonistic function (Table 2). NF-κB induced IL-6, IL-1 and TNF-α have been implicated in tumor development and growth in several models, primarily by their ability to increase the survival and growth of cancer cells, but also through their effects on the immune system, which sustain tumor-associated inflammation (45, 86, 104, 105). In recent years, an important protumorigenic role for IL-23 has also emerged (94). This cytokine is produced by myeloid cells in the tumor microenvironment in a STAT3- and NF- κB-dependent manner (52) and is upregulated in many cancers (94). However, the mechanism of IL-23 action in cancer is poorly understood. IL-23 regulates the stability of the Th17 lineage and ensures IL-22 and IL-17 production which may be important in cancer (46, 94). IL-23 is also involved in the regulation of FoxP3 expression and IL-10 production in inflammation and cancer, although there are conflicting reports on the effects of IL-23 on FoxP3 gene expression and protein amounts (52, 106–108). Although IL-23R is rarely expressed by malignant cells, two IL-23 targets, IL-17 and IL-22, are capable of directly activating NF-κB and STAT3 in neoplastic cells. In addition, at least in chronic inflammatory settings, IL-23 controls Treg function and sustains inflammation and may have a broad impact on cytokine production, including other activators of NF- κB and STAT3, such as IL-6 and TNF-α (109).
Below we describe several examples in which cytokine-mediated cross talk between NF-κB and STAT3 in immune and cancer cells facilitates tumorigenesis in a mouse model of CAC.
The interplay between NF- κB and STAT3 in colitis associated cancer: malignant cooperation between immune and cancer cells matter
CAC was the first cancer model in which a role of NF- κ or any defined molecular entity in providing a critical link between inflammatory cells and premalignant epithelial cells in tumor development was demonstrated (99). Using a conditional disruption of the IKKβ gene in mice, we found that NF-κ B activation in enterocytes is essential for the development of colonic adenomas (99). The oncogenic role of NF-κ B in neoplastic epithelial cells is mediated through induction of anti-apoptotic proteins, particularly Bcl-XL (99), whose expression is also controlled by STAT3 (45, 47). Indeed, we and others found that inactivation of STAT3 in epithelial cells not only reduces tumorigenicity in a mouse model of CAC, but more specifically, attenuates the expression of the anti-apoptotic protein Bcl-XL, as well as other pro-survival and tissue protective proteins and cell cycle regulators, which eventually mediate STAT3-dependent tissue regeneration and tumor growth (45, 47, 78). NF-κB activation in myeloid cells also enhances tumor multiplicity and growth (99). Early studies implicated IL-6 in tumor growth in CAC (104) and we and others have confirmed that IL-6 produced by myeloid cells, T cells and other cell types is important for tumor initiation and growth, and that IL-6 production relies on IKKβ-dependent activation of NF-κB in myeloid cells and on STAT3 and NFATc2 in T cells (45, 110).
Ablation of IL-6 reduced both the multiplicity and size of colonic adenomas in mouse CAC (45). Both the proliferative and survival effects of IL-6 are in part mediated through activation of STAT3 in enterocytes. Yet, the effect of IL-6 may be additive to the effects of IL-11, IL-22 and EGF, which also act on enterocytes and can also activate STAT3 (45, 78, 111). Therefore, STAT3 as well as NF-κB activators in cancer may act redundantly or at least have overlapping effects. However, both NF-κB and STAT3 have essential effunctions in CAC development, as ablation of STAT3 in enterocytes also compromised cell survival and greatly reduced CAC growth (45). NF-κB and STAT3 in myeloid cells drive the expression of not only IL-6, IL-11 and IL-22, but also of IL-23, which has direct effects on T cells and some types of monocytes (Figure 3). IL-23 then may induce expression of IL-17 and IL-22 and may have indirect effects on production of other cytokines (Figure 3). Overall, such findings suggest that many of the pro-tumorigenic effects of NF-κB (and STAT3) activation in immune cells are mediated through paracrine signaling by a network of cytokines that further control STAT3 and NF-κB activity in epithelial cells and their malignant derivatives (Figure 3).
Figure 3.
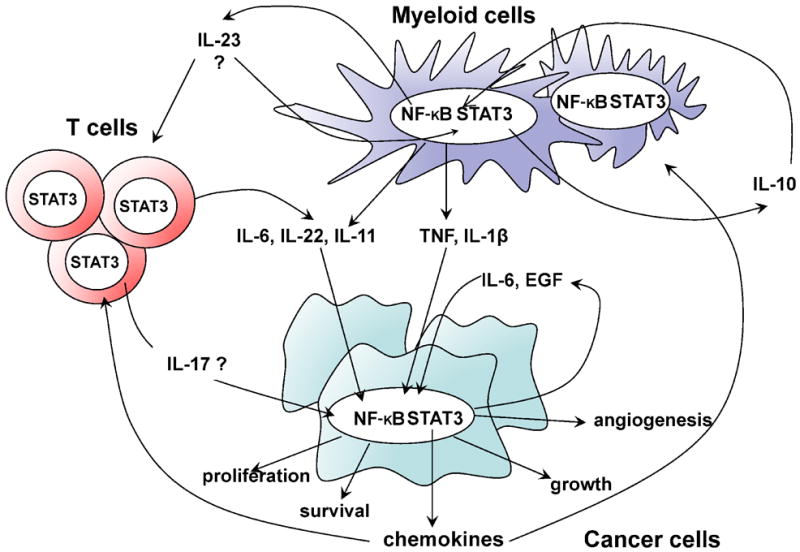
Functional interactions between NF-κB and STAT3 in immune cells control the production of pro-inflammatory cytokines that maintain inflammation and stimulate tumor growth by activating NF-κB and STAT3 in cancer cells
Conclusions and future directions
It has become clear that NF-κB and STAT3, a highly interactive duo are key regulators of epithelial tumorigenesis. Pro-inflammatory cytokines that are produced by immune and inflammatory cells and signal in on cancer cells functionally link NF-κB and STAT3 within the two cell types. NF-κB and STAT3 control the expression of proliferative and survival genes in premalignant cells and their neoplastic progeny. In addition, STAT3 in immune/inflammatory cells modulates the effect of NF-κB on perpetuation of inflammation and production of tumor-promoting cytokines.
These findings suggest that regardless of the effect of NF-κB on survival or proliferation of neoplastic cells, IKKβ inhibitors can be used to slow down tumor growth and enhance susceptibility to therapeutics capable of inducing malignant cell death because they should prevent the expression of key tumor promoting cytokines. The same may apply for STAT3, which has been proposed as a very good target for anti-cancer therapy (32). However, it is important that the inhibition of STAT3 in epithelial cells will be dominant over its inhibition in myeloid cells, which may produce pro-inflammatory and pro-tumorogenic effects. Given the recently observed complications associated with inhibition of IKKβ-driven NF-κB activation (112), the inhibition of JAK-driven STAT3 activation in cancer may be a preferred strategy in some cases. Nevertheless, it should be considered that the mere disruption of either NF-κB or STAT3 signaling does not lead to cell death and therefore their inhibitors should be combined with cancer-specific cytotoxic drugs. It should also be recognized that unless the cytocidal agent being used in conjunction with NF-κB or STAT3, inhibitors are highly cancer cell specific, the systemic inhibition of NF-κB or STAT3 function may result in enhanced damage to normal cells and tissues, as the prosurvival effects of these transcription factors are widespread. Another potential complication of IKKβ or NF-κB inhibition is long lasting immune suppression and unpredictable effects on the inflammatory response. The same may apply to STAT3 inhibition, which has prominent immune functions. Despite their potential side effects, NF-κB and STAT3 inhibition may represent a good approach to combat cancer, given that their inhibition not only affects growth and survival of malignant cells that rapidly accumulate mutation that confer drug resistant but also acts within normal immune/inflammatory cells that do not accumulate mutations.
To avoid the potential toxicity of global NF-κB and STAT3 inhibition, it is worthwhile considering a more specific strategies that target NF-κB- and STAT3-dependent tumor promoting cytokines, such as TNF-α, IL-6 and IL-23. This should alleviate the systemic toxicity associated with total NF-κB and STAT3 inhibition and retain most of their immune functions but may effect tumor growth NF-κB and STAT3 themselves. Also of interest are inhibitors of NF-κB:STAT3 interaction, as such drugs may spare some of the essential homeostatic functions executed by NF-κB and STAT3 on their own, while inhibiting their malicious cooperation in cancer cells. Taken together, the dangerous liason formed by NF-κB and STAT3 may also serve as the Achilles Hill of many cancers, providing new opportunities for therapeutic intervention.
Figure 1.
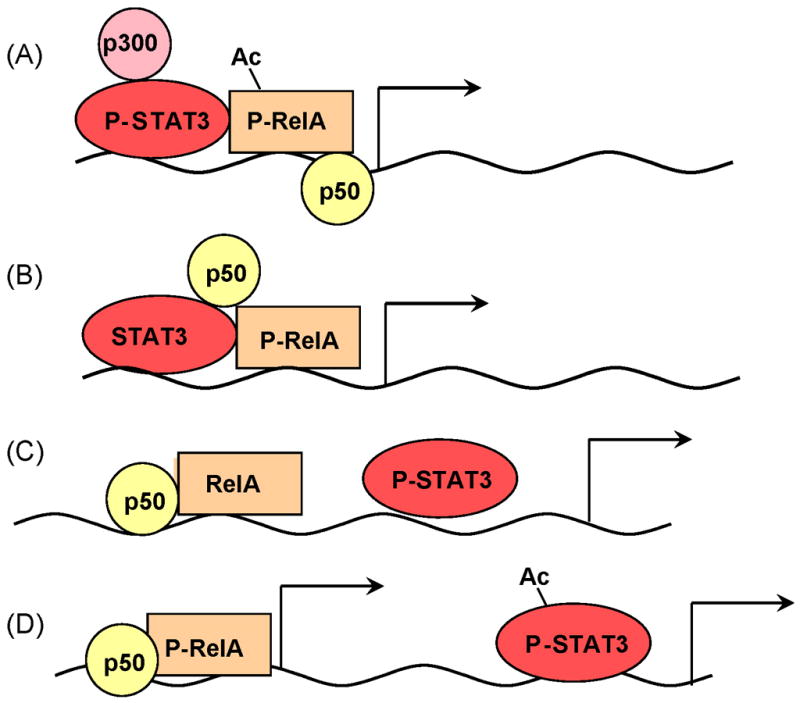
Different modes of NF-κB and STAT3 interaction in transcriptional control. A) Activated STAT3 interacts with p65 RelA to recruit the p300 HAT complex to cause RelA, acetylation that prolongs its nuclear retention. This mechanism may mediate STAT3-NF-κB dependent gene transcription. B) STAT3 (either phosphorylated or non-phosphorylated) interacts with p50/NF-κB and/or RelA and together they induce gene transcription through binding to composite sites. C,D) STAT3 and NF-κB do not interact physically, but bind to the same (C) or different (D) promoters, thereby synergistically activating gene expression (C) or acting individually on genes that are either NF-κB- or STAT3-dependent (D).
Figure 2.

Signaling pathways that activate NF-κB and STAT3. Different receptors can lead to NF-κB and STAT3. Different receptors can lead to NF-κB activation via IKK and STAT3 activating via JAK1.
Acknowledgments
This work was supported in part by Research Fellowship Award from Crohn’s and Colitis Foundation of America (CCFA #1762) to S.G. and grants from the NIH and a Jeannik M. Littlefield-AACR grant in metastatic colon cancer to M.K., who is an American Cancer Society Research Professor.
Footnotes
Authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bishop JM. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- 2.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 3.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 4.Karin M. NF-kB and cancer: mechanisms and targets. Mol Carcinog. 2006;45:355–361. doi: 10.1002/mc.20217. [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 6.Karin M, Cao Y, Greten FR, Li ZW. NF-kB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 7.Karin M, Lin A. NF-kB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 8.Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–324. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- 9.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 11.Greten FR, Karin M. NF-kB: Linking Inflammation and Immunity to Cancer Development and Progression. Nature Reviews Immunology. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 12.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 13.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh S, Karin M. Missing pieces in the NF-kB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- 15.Gilmore TD. The Re1/NF-kappa B/I kappa B signal transduction pathway and cancer. Cancer Treat Res. 2003;115:241–265. [PubMed] [Google Scholar]
- 16.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 17.Bezbradica JS, Medzhitov R. Integration of cytokine and heterologous receptor signaling pathways. Nat Immunol. 2009;10:333–339. doi: 10.1038/ni.1713. [DOI] [PubMed] [Google Scholar]
- 18.Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- 19.Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- 20.Kruglov AA, Kuchmiy A, Grivennikov SI, Tumanov AV, Kuprash DV, Nedospasov SA. Physiological functions of tumor necrosis factor and the consequences of its pathologic overexpression or blockade: Mouse models. Cytokine Growth Factor Rev. 2008;19:231–244. doi: 10.1016/j.cytogfr.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Karin M. The IkappaB kinase - a bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- 22.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkB kinase that activates the transcription factor NF-kB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 24.Beg AA, Baltimore D. An essential role for NF-kB in preventing TNF-a induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z-G, Hu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis, while NF-kB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 26.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNFa-induced apoptosis by NF-kB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 27.Wang C-Y, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 28.Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001;12:73–90. doi: 10.1016/s1359-6101(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 29.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 30.Huang TT, Miyamoto S. Postrepression activation of NF-kappaB requires the amino-terminal nuclear export signal specific to IkappaBalpha. Mol Cell Biol. 2001;21:4737–4747. doi: 10.1128/MCB.21.14.4737-4747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 34.Braunstein J, Brutsaert S, Olson R, Schindler C. STATs dimerize in the absence of phosphorylation. J Biol Chem. 2003;278:34133–34140. doi: 10.1074/jbc.M304531200. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007;21:1396–1408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 37.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 38.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Herrmann A, Deng JH, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeda K, Noguchi K, Shi W, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 42.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 43.Sansone P, Storci G, Tavolari S, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin W, Cheepala S, Roberts JN, Syson-Chan K, DiGiovanni J, Clifford JL. Active Stat3 is required for survival of human squamous cell carcinoma cells in serum-free conditions. Mol Cancer. 2006;5:15. doi: 10.1186/1476-4598-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009 doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 48.Jenkins BJ, Roberts AW, Najdovska M, Grail D, Ernst M. The threshold of gp130-dependent STAT3 signaling is critical for normal regulation of hematopoiesis. Blood. 2005;105:3512–3520. doi: 10.1182/blood-2004-09-3751. [DOI] [PubMed] [Google Scholar]
- 49.Judd LM, Alderman BM, Howlett M, et al. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 50.Chan KS, Sano S, Kiguchi K, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kataoka K, Kim DJ, Carbajal S, Clifford JL, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis. 2008;29:1108–1114. doi: 10.1093/carcin/bgn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15:114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu XY. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006;16:214–219. doi: 10.1038/sj.cr.7310029. [DOI] [PubMed] [Google Scholar]
- 54.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 55.Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang J, Chatterjee-Kishore M, Staugaitis SM, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 57.Dauer DJ, Ferraro B, Song L, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24:3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 58.Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida Y, Kumar A, Koyama Y, et al. Interleukin 1 activates STAT3/nuclear factor-kappaB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004;279:1768–1776. doi: 10.1074/jbc.M311498200. [DOI] [PubMed] [Google Scholar]
- 60.Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–392. doi: 10.2174/1566524023362456. [DOI] [PubMed] [Google Scholar]
- 61.Wu Z, Zhang X, Yang J, et al. Nuclear protein IkappaB-zeta inhibits the activity of STAT3. Biochem Biophys Res Commun. 2009 doi: 10.1016/j.bbrc.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 62.Totzke G, Essmann F, Pohlmann S, Lindenblatt C, Janicke RU, Schulze-Osthoff K. A novel member of the IkappaB family, human IkappaB-zeta, inhibits transactivation of p65 and its DNA binding. J Biol Chem. 2006;281:12645–12654. doi: 10.1074/jbc.M511956200. [DOI] [PubMed] [Google Scholar]
- 63.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 64.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. Embo J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hagihara K, Nishikawa T, Sugamata Y, et al. Essential role of STAT3 in cytokine-driven NF-kappaB-mediated serum amyloid A gene expression. Genes Cells. 2005;10:1051–1063. doi: 10.1111/j.1365-2443.2005.00900.x. [DOI] [PubMed] [Google Scholar]
- 67.Yoshimura A, Mori H, Ohishi M, Aki D, Hanada T. Negative regulation of cytokine signaling influences inflammation. Curr Opin Immunol. 2003;15:704–708. doi: 10.1016/j.coi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 68.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 70.Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFa-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 71.Rebouissou S, Amessou M, Couchy G, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457:200–204. doi: 10.1038/nature07475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niu G, Wright KL, Ma Y, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25:7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Egan LJ, Eckmann L, Greten FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 75.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cao Y, Bonizzi G, Seagroves TN, et al. IKKa provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 77.Tebbutt NC, Giraud AS, Inglese M, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 78.Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118:3367–3377. doi: 10.1172/JCI35213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim DJ, Chan KS, Sano S, Digiovanni J. Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol Carcinog. 2007;46:725–731. doi: 10.1002/mc.20342. [DOI] [PubMed] [Google Scholar]
- 81.Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 82.Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci. 2009;14:540–551. doi: 10.2741/3261. [DOI] [PubMed] [Google Scholar]
- 84.Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mailloux AW, Young MR. NK-dependent increases in CCL22 secretion selectively recruits regulatory T cells to the tumor microenvironment. J Immunol. 2009;182:2753–2765. doi: 10.4049/jimmunol.0801124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Langowski JL, Kastelein RA, Oft M. Swords into plowshares: IL-23 repurposes tumor immune surveillance. Trends Immunol. 2007;28:207–212. doi: 10.1016/j.it.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 88.Popivanova BK, Kitamura K, Wu Y, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. Journal of Clinical Investigation. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 91.Swann JB, Vesely MD, Silva A, et al. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A. 2008;105:652–656. doi: 10.1073/pnas.0708594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hanada T, Kobayashi T, Chinen T, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J Exp Med. 2006;203:1391–1397. doi: 10.1084/jem.20060436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 94.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 95.Dercamp C, Chemin K, Caux C, Trinchieri G, Vicari AP. Distinct and overlapping roles of interleukin-10 and CD25+ regulatory T cells in the inhibition of antitumor CD8 T-cell responses. Cancer Res. 2005;65:8479–8486. doi: 10.1158/0008-5472.CAN-05-1319. [DOI] [PubMed] [Google Scholar]
- 96.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11:1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 97.Wang T, Niu G, Kortylewski M, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 98.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 99.Greten FR, Eckmann L, Greten TF, et al. IKKb links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 100.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 101.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKb couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 102.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 103.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24:315–327. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 104.Becker C, Fantini MC, Schramm C, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 105.Balkwill F. Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 2002;13:135–141. doi: 10.1016/s1359-6101(01)00020-x. [DOI] [PubMed] [Google Scholar]
- 106.Izcue A, Hue S, Buonocore S, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhou L, Ivanov II, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 108.Stewart CA, Trinchieri G. Reinforcing suppression using regulators: a new link between STAT3, IL-23, and Tregs in tumor immunosuppression. Cancer Cell. 2009;15:81–83. doi: 10.1016/j.ccr.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 109.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weigmann B, Lehr HA, Yancopoulos G, et al. The transcription factor NFATc2 controls IL-6-dependent T cell activation in experimental colitis. J Exp Med. 2008;205:2099–2110. doi: 10.1084/jem.20072484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rose-John S, Mitsuyama K, Matsumoto S, Thaiss WM, Scheller J. Interleukin-6 trans-signaling and colonic cancer associated with inflammatory bowel disease. Curr Pharm Des. 2009;15:2095–2103. doi: 10.2174/138161209788489140. [DOI] [PubMed] [Google Scholar]
- 112.Greten FR, Arkan MC, Bollrath J, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]