

Abstract
It has been proposed that hypermutability is necessary to account for the high frequency of mutations in cancer. However, historically, the mutation rate (μ) has been difficult to measure directly, and increased cell turnover or selection could provide an alternative explanation. We recently developed an assay for μ using PIG-A as a sentinel gene and estimated that its average value is 10.6 × 10-7 mutations per cell division in B-lymphoblastoid cell lines (BLCLs) from normal donors. Here we have measured μ in human malignancies and found that it was elevated in cell lines derived from T cell acute lymphoblastic leukemia, mantle cell lymphoma, follicular lymphoma in transformed phase, and 2 plasma cell neoplasms. In contrast, μ was much lower in a marginal zone lymphoma cell line and 5 other plasma cell neoplasms. The highest μ value that we measured, 3286 × 10-7, is 2 orders of magnitude above the range we have observed in non-malignant human cells. We conclude that the type of genomic instability detected in this assay is a common but not universal feature of hematologic malignancies.
1.0 Introduction
A model sentinel gene to measure spontaneous somatic mutations must be non-essential for growth or viability, and the mutant phenotype must be detectable among a vastly larger population of normal cells. Since a single mutation conferring loss of function could be complemented by the wild type allele on the homologous chromosome, most autosomal genes are not suitable for this purpose. However, due to hemizygosity in males and X-inactivation in females, a single mutation is sufficient to inactivate X-linked genes. For example, among lymphocytes in normal adults, resistance to 6-thioguanine occurs as a consequence of a spontaneous inactivating mutation in HPRT (Xq26-q27.2), at a frequency (f) of ∼ 2 - 10 × 10-6 [1].
While f represents the proportion of mutants within a population, the mutation rate (μ) represents the probability of a new mutation per cell division. If mutations are growth-neutral, f will increase over time according to the formula Δf = μ × Δd, where d represents number of cell divisions. By analysis of f values for HPRT in human population groups of different ages and estimates for Δd in human lymphocytes in vivo, it has been proposed that the normal μ value for this gene is ∼8.4 × 10-7 per cell division [2]. Similarly, based on population variances in f values for the GPA gene [3], μ has been estimated to be 2 - 4 × 10-7. Given such estimates, Loeb proposed that hypermutability would be essential for carcinogenesis, if the number of mutations required for malignancy (n) is >2 [4]. It was subsequently shown that n is likely > 5, and it may be much higher [5,6].
In support of this model, a great increase in the frequency of an intronic p53 mutation has been demonstrated in diverse tumor types [7]. While an increase in f suggests an elevation in μ, selection or an elevation in d could have the same effect. Measurements of μ in tumors by fluctuation analysis have suggested hypermutability [8], but this technique has not been universally accepted for human tissue culture [9]. Others have calculated μ by the formula Δf = μ × Δd, after depletion of pre-existing HPRT mutants using hypoxanthine-aminopterin-thymidine (HAT) [10]. Whereas colon cancers have exhibited an elevated μ [11], mutant frequency data has been variable for hematologic malignancies [12], which some have attributed to metabolic cooperation [13,14].
We have recently developed a technique for the direct measurement of μ, which uses the PIG-A gene (Xp22.1) as a sentinel [15]. PIG-A encodes a subunit of a protein complex in the endoplasmic reticulum, which catalyzes the transfer of N-acetylglucosamine to phosphatidylinositiol, the first reaction in the biosynthesis of glycosylphosphatidylinositol (GPI) [16]. PIG-A mutations disrupt the synthesis of GPI and the expression of proteins (e.g. CD48, CD52, CD55, CD59) that require GPI for their attachment to the cell surface [17,18]. Expanded populations of hematopoitetic stem cells with mutations in PIG-A mutations are the hallmark of Paroxysmal Nocturnal Hemoglobinuria (PNH). From this condition, it is known that a broad spectrum of mutations can disrupt the function of PIG-A [19]. Males are hemizygous for X-linked genes, and PIG-A is completely silenced on the inactivated X-chromosome in females [20]. Therefore only one mutation is required to produce the GPI (-) phenotype, and males and females are equally affected [19].
Apart from the peculiar expansion of the PIG-A mutant stem cell clone in PNH, there is evidence that under almost all other circumstances, PIG-A mutations are growth neutral in vivo in nucleated cells in humans and mice as well as in vitro [15,21,22]. As for HPRT, normal individuals harbor rare cells with PIG-A mutations [23], which can also arise in vitro in cell lines [15,24]. By flow cytometry, pre-existing mutants can be depleted, and a large number of cells can be analyzed to determine not only f, but also μ [15]. Here we have applied this approach to investigate the role of genomic instability in human hematologic cancers.
2.0 Materials and Methods
2.1 Cell Lines and culture media
HBL2 (derived from a human mantle cell lymphoma), PR1 (derived from a human transformed follicular lymphoma), SSK41 (derived from a human marginal zone lymphoma) [25,26], Jurkat (derived from a human T-cell acute lymphoblastic leukemia) and lines derived from human plasma cell neoplasms as well as an EBV transformed B-lymphoblastoid cell line from a normal individual were analyzed to determine the mutation rate. Plasma cell neoplasm lines ARH-77, RPMI-8226, KMS-11, ARP-1, U266, and SKMM2 were gifts from Dr. Hearn Cho, and NCI-H929 was obtained from the ATCC. Cells were grown in RPMI, 15% FCS, non-essential amino acids, L-glutamine, and penicillin-streptomycin. A BLCL from a patient with PNH that had both GPI (+) and GPI (-) populations served as a control. Preliminary studies demonstrated that HBL2 and PR1 had significant pre-existing GPI (-) populations. For PR1, GPI (+) cells were selected by sorting, from which clones were then isolated by limiting dilution in 96 well plates. A GPI (+) clone (PR1-C) was selected for further studies. For HBL2, GPI (+) clones designated HBL2-A, HBL2-B, and HBL2-C were also isolated in this manner. Limiting dilution cloning was also performed to establish 5 clones of Jurkat and 4 subclones of PR1-C.
2.2 Calculation of the mutation rate
Calculation of the mutation rate using the PIG-A gene was performed as described [15,27]. Cells were first stained with an anti-CD59 antibody, and the upper 50th percentile of the distribution curve was collected by sorting with a Dako-Cytomation Moflow instrument, to deplete pre-existing mutants. The collected GPI (+) cells were counted by trypan blue exclusion and then expanded in culture. To determine the mutant frequency after expansion in vitro, cells were first stained with a mixture of antibodies specific for multiple GPI-linked proteins (e.g. CD48, CD52, CD55, CD59, from Serotec), followed by rabbit anti-mouse immunoglobulin conjugated to-PE (DAKO-Cytomation), and then a FITC-conjugated antibody specific for a relevant transmembrane protein: HLA-DR (BD Pharmingen) for B cell neoplasms, CD45 (Serotec) for Jurkat, and HLA-class I (W6/32, Serotec) for plasma cell neoplasms. Antibodies were added to pelleted cells, which were resuspended, repelleted, and again resuspended, to ensure that all cells came in contact with the antibody. Cells were washed twice with cold media between incubations. Staining incubations were performed at a cell density of 100 million/ml, on ice, for at least 30 minutes. Propidium iodide was added at a final concentration of 0.1 to 0.15 μg/ml. Flow cytometric analysis was performed on a Becton Dickinson FACScan instrument, using Cellquest and FlowJo software. Live cells were identified by FSC/SSC, exclusion of propidium iodide, and expression of transmembrane proteins. By this approach GPI (+) cells register in the upper right quadrant and GPI (-) cells in the lower right quadrant. The mutant frequency, f, was calculated by taking the number of events in the lower right quadrant divided by total number of events analyzed. d, representing population doublings, was calculated by live cell counts before and after expansion [where d = LOG2 (number of cells after expansion ÷ number of cells before expansion)]. The mutation rate was calculated by the formula μ = f ÷ d.
2.3 FLAER reagent for detection of the loss of the GPI anchor
FLAER-Alexa 488 (Protox Biotech, Victoria, BC, Canada) binds to the GPI structure directly, conferring fluorescence to wild type cells but not to PIG-A mutant cells [28], with an emission spectra almost identical to FITC. In some experiments cells were stained first with the FLAER reagent at 37°C for 30 minutes at a concentration of 10-7M and a cell density of 100 million/ml. Cells were then stained on ice with a mixture of antibodies specific for multiple GPI-linked proteins (e.g. CD48, CD52, CD55, CD59), washed twice, followed by staining with PE-conjugated rabbit anti-mouse antibody.
2.4 Cloning Efficiency in Bulk Culture
To determine whether significant adjustments would have to be made to the estimate of d described above, we determined the cloning efficiency (CE) of the cell lines in bulk culture [15]. Briefly, 2 × 106 cells from liquid culture were seeded at a density of 106/ml and counted daily by trypan blue exclusion, with media added daily to keep the cells at this density. The cell counts were plotted on a log scale as a function of time in culture, and a regression line was estimated for the data points. CE was estimated as the y-intercept of this regression curve divided by the original number of cells seeded.
2.5 Cytogenetic Analysis
Chromosome preparations were made from cells harvested from the various cell line suspension cultures, with G-banding and metaphase analysis performed following standard procedures. FISH analysis was performed from the fixed cells prepared for chromosome studies using DNA probes specific for chromosomes X and Y (CEP X SpectrumGreen/ CEP Y SpectrumOrange DNA Probe Kit, Abbott Molecular, Des Plaines, IL) according to the manufacturer's protocols. Fifty interphase cells were scored for each cell line.
3.0 Results
3.1 Characteristics of control GPI (+) and GPI (-) cells and the purity of sorting
Flow cytometry readily distinguished distinct populations of GPI (+) cells (upper right quadrant) and GPI (-) cells (lower right quadrant) within a single BLCL from a patient with PNH (figure 1A). As expected, the GPI (-) cells from this patient did not bind to the FLAER reagent (data not shown). Artificial mixtures of control GPI (+) cells mixed with ∼1% GPI (-) cells from this patient were then stained with FITC-conjugated anti-CD59 antibody, and the upper 50th percentile of the distribution curve with respect to CD59 expression was collected. Post sort analysis demonstrated that the instrument was typically effective at eliminating > 98% of the GPI (-) population.
FIGURE 1. GPI (-) populations arise spontaneously in vitro in malignant cell lines.
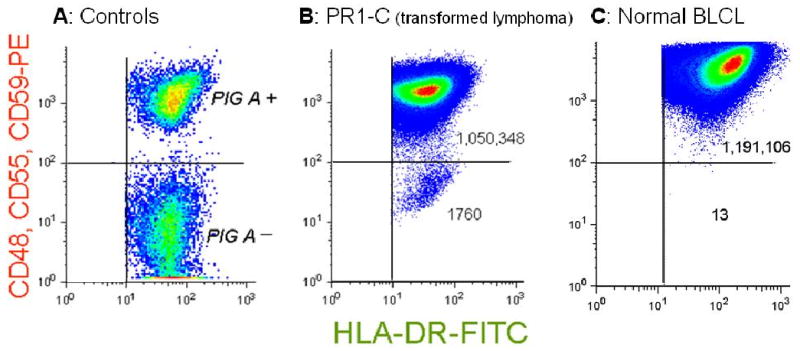
Flow cytometry pseudo-density dot plots. (A) PIG-A (+) cells and PIG-A (-) cells within a single BLCL from a patient with PNH. The PIG-A (-) population does not express GPI-linked proteins but does express the transmembrane protein HLA-DR. (B) PR1-C, a transformed follicular lymphoma cell line, analyzed after expansion after sorting to eliminate pre-existing mutants. The normal population expresses GPI-linked proteins and transmembrane proteins, registering in the upper right quadrant. There is a large population of spontaneously arising mutants registering in the lower right quadrant that appear similar to the PIG-A (-) cells from the patient with PNH. The mutant frequency, f, is calculated as the number of events in the lower right quadrant divided by the total number events analyzed (see table). (C) A BLCL from a normal donor, demonstrating a much smaller number of spontaneously arising GPI (-) cells.
3.2 Mutation rate in cell lines from lymphoid malignancies
Cell line PR1-C was separated by sorting, yielding 7.3 × 105 GPI (+) cells, which were expanded in vitro over 21 days, undergoing 6.45 cell divisions. Re-analysis using antibodies specific for 3 GPI-linked antigens demonstrated a spontaneously arising population of GPI (-) cells in the lower right quadrant (figure 1B). The mutant frequency was calculated to be 1673 × 10-6, and μ was calculated to be 2593 × 10-7 mutations per cell division (table 1). This value is two orders of magnitude above previous estimates for μ for PIG-A in BLCLs from normal donors [15,27] and similarly above the estimated in vivo mutation rate for the HPRT gene in the general population [2]. In contrast, analysis of a BLCL from a healthy donor demonstrated much fewer spontaneously arising mutants (figure 1C), with a mutation rate of 17 × 10-7 (table 1), which is within our previous estimates. The cloning efficiency of the malignant cell lines was high (table 1 and table 2) and was comparable to our previous estimates for B-lymphoblastoid cell lines [15], suggesting that major differences in the kinetics of cell turnover would not account for the elevation in μ that we observed.
Table 1. Lymphoid neoplasms: determination of f and μ.
| Cell Type | Designation | * Starting # cells (× 10-6) | CE** | # days in culture¶ | # cell divisions, d¶¶ | # GPI(−) cells | Total # cells analyzed§ | Mutant frequency f (× 106)‡ | ‡‡ Mutation rate μ (× 107) |
|---|---|---|---|---|---|---|---|---|---|
| BLCL | Normal Donor | 1.0 | 20 | 6.42 | 13 | 1,191,119 | 11 | 17 | |
| Transformed Lymphoma | 0.57 | ||||||||
| PR1-C parental | .73 | 21 | 6.45 | 1760 | 1,052,108 | 1673 | 2593 | ||
| PR1-C-sub clone 1 | 2.08 | 21 | 5.44 | 2893 | 1,618,270 | 1787 | 3286 | ||
| PR1-C-sub clone 2 | 2.68 | 21 | 5.14 | 288 | 486,909 | 591 | 1151 | ||
| PR1-C-sub clone 3 | 2.49 | 21 | 5.82 | 1017 | 1,710,695 | 595 | 1022 | ||
| PR1-C-sub clone 4 | 2.06 | 21 | 5.27 | 2368 | 2,006,108 | 1180 | 2240 | ||
| Mantle Cell Lymphoma | 0.79 | ||||||||
| Experiment 1 | |||||||||
| HBL2- A | 1.4 | 21 | 6.23 | 1324 | 2,433,723 | 544 | 874 | ||
| Experiment 2 | |||||||||
| HBL2-A | 1.0 | 20 | 8.09 | 2236 | 2,274,028 | 983 | 1215 | ||
| Experiment 3 | |||||||||
| HBL2- A | 3.3 | 21 | 5.41 | 829 | 2,086,003 | 397 | 734 | ||
| HBL2- B | 3.8 | 26 | 6.17 | 952 | 2,913,268 | 326 | 529 | ||
| HBL2- C | 4.8 | 21 | 5.10 | 506 | 1,278,381 | 395 | 776 | ||
| T cell ALL | 0.93 | ||||||||
| Experiment 1 | |||||||||
| Jurkat Parental | 1.14 | 41 | 10.15 | 108 | 624,573 | 173 | 170 | ||
| Experiment 2 | |||||||||
| Jurkat Parental | 1.38 | 20 | 7.95 | 276 | 2,348,927 | 118 | 148 | ||
| Jurkat Clone 1 | 2.67 | 20 | 7.30 | 192 | 1,694,854 | 113 | 155 | ||
| Jurkat Clone 2 | 1.72 | 20 | 7.99 | 648 | 1,136,989 | 570 | 713 | ||
| Jurkat Clone 3 | 1.11 | 21 | 8.78 | 346 | 1,160,399 | 298 | 340 | ||
| Jurkat Clone 4 | 1.11 | 21 | 6.47 | 985 | 793,749 | 1240 | 1918 | ||
| Jurkat Clone 5 | 1.85 | 21 | 8.15 | 506 | 713,762 | 709 | 870 | ||
| Marginal Zone Lymphoma | SSK41 | 1.6 | 0.65 | 25 | 8.27 | 21 | 2,346,400 | 9 | 11 |
Number of GPI (+) cells collected by sorting, in millions
Cloning Efficiency in Bulk Culture
Number of days in culture between sorting and analysis
Number of cell divisions occurring in culture between sorting and analysis
Number of cells in the upper right quadrant plus number of cells in the lower right quadrant
f = number of GPI (-) cells divided by total number of cells analyzed. Results expressed as mutants per 106 cells.
Mutation rate calculated as μ= f divided by d. Results expressed as mutations per 107 cell divisions
Table 2. Plasma cell neoplasms: determination of f and μ.
| Cell line | Starting # cells (× 10-6)* | # days in culture¶ | CE** | # cell divisions, d¶¶ | # GPI(−) cells | Total # cells analyzed§ | Mutant frequency f (× 106)‡ | Mutation rate μ (× 107)‡‡ |
|---|---|---|---|---|---|---|---|---|
| KMS-11 | .38 | 22 | 0.56 | 6.74 | 5 | 1082484 | 4.62 | 6.9 |
| ARP-1 | 2.0 | 33 | 0.66 | 5.35 | 3 | 539077 | 5.57 | 10.4 |
| U266 | 6.3 | 33 | 0.69 | 5.00 | 62 | 1619500 | 38.3 | 76.6 |
| SKMM2 | 1.6 | 22 | 1.0 | 4.51 | 2 | 2027237 | 0.99 | 2.2 |
| NCI-H929 | 2.1 | 21 | 0.55 | 3.48 | 16 | 1235227 | 12.95 | 37.2 |
| ARH-77 | 7.3 | 19 | 0.82 | 3.21 | 82 | 1736463 | 47.2 | 147 |
| RMPI-8226 | 0.88 | 22 | 0.73 | 5.24 | 16 | 1487327 | 10.8 | 20.5 |
Number of GPI (+) cells collected by sorting, in millions
Cloning Efficiency in Bulk Culture
Number of days in culture between sorting and analysis
Number of cell divisions occurring in culture between sorting and analysis
Number of cells in the upper right quadrant plus number of cells in the lower right quadrant
f = number of GPI (-) cells divided by total number of cells analyzed. Results expressed as mutants per 106 cells.
Mutation rate calculated by the formula μ = f divided by d. Results expressed as mutations per 107 cell divisions
PIG-A mutant cells demonstrate loss of GPI-linked membrane proteins and they also do not have any detectable GPI on their surface. To demonstrate that spontaneously arising CD59(-) CD48(-) CD55(-) cells did not produce any detectable GPI, we analyzed PR1-C cells by two methods in parallel: using FITC conjugated antibodies specific for transmembrane proteins as shown in figure 1, or pre-incubation with FLAER-Alexa-488 and omitting final the FITC-conjugated antibody step. With the FLAER reagent, GPI (+) cells were expected to appear in the upper right quadrant as in figure 1, but here GPI (-) cells were expected to appear in the lower left quadrant, because GPI is required for the cell to bind to FLAER. We confirmed that this was a case for a subclone of PR-1C (figure 2A), in an experiment where the mutation rate was shown to be 3286 × 10-7 (table 1). Similarly, for Jurkat and HBL2-A (figure 2B and 2C) we observed spontaneously arising GPI (-) populations that did not express any of the representative GPI-linked proteins on the surface, and which did not bind to the FLAER reagent. In these experiments the calculated μ values were 170 × 10-7 for Jurkat and 874 × 10-7 for HBL-2A. Of note, the calculated values for f and μ were similar using the two approaches.
FIGURE 2. Parallel analysis of cultures of malignant cell lines using antibodies specific for transmembrane proteins or the FLAER reagent.
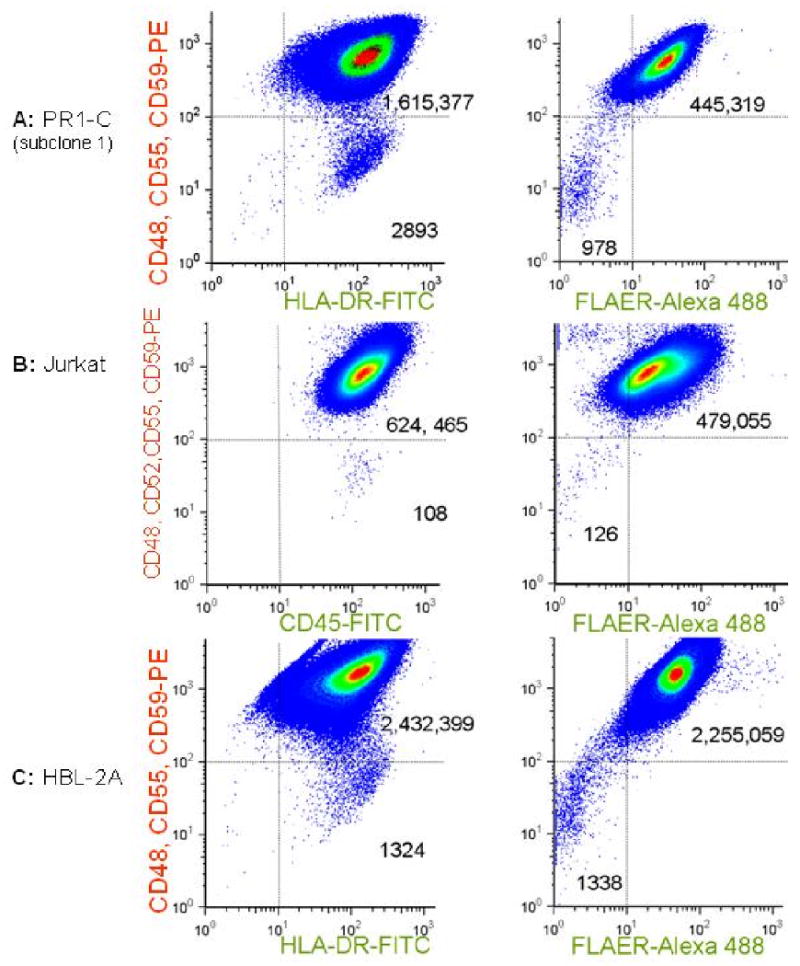
Samples were analyzed after expansion in vitro after sorting to eliminate preexisting mutants. Pseudo-density dot plots are shown for analyses using FITC-conjugated antibodies specific for transmembrane proteins (left panels) or the FLAER Alexa-488 reagent, which binds to the GPI structure directly (right panels). In all 3 examples, there are distinct populations of spontaneously arising cells that neither express GPI-linked proteins nor take up the FLAER reagent, but which do express transmembrane proteins. (A) A subclone of PR1-C (transformed lymphoma); (B) Jurkat, a T cell ALL cell line; (C) HBL2-A, derived from a Mantle Cell Lymphoma.
Although we always obtained a high degree of purity when sorting artificial mixtures of GPI (+) and GPI (-) cells, we wanted to confirm that these very high μ values in malignant cell lines were not somehow related to residual GPI (-) cells that had escaped sorting. We therefore stained HBL2-A cells with PE-conjugated anti-CD59 antibody, collected the upper 50th percentile of the population, and then stained a portion of the collected cells immediately after sorting using mouse anti-CD48, CD55, and CD59 antibodies, rabbit anti-mouse PE secondary antibody, and HLA-DR-FITC as described above. The sorted population was compared with an aliquot of cells that was stained with the same antibodies but which was not sorted. The unsorted aliquot demonstrated a mutant frequency of 3316 × 10-6. The sorted aliquot demonstrated a frequency of residual GPI (-) cells of 54 × 10-6, representing a 98.4% depletion of the pre-existing mutants. 106 cells from the sorted GPI (+) fraction were then returned to culture and expanded over 20 days, undergoing 8.09 cell divisions. f was then determined to be 983 × 10-6 and μ was calculated to be 1215 × 10-7. We then corrected for residual GPI-negative cells that had escaped sorting by subtracting 54 × 10-6, the post-sorting f value on day zero, from 983 × 10-6, the f value measured after 20 days in culture. This yielded an estimated μ value of 1148 × 10-7, which differs from the uncorrected value by < 6%. We therefore concluded that the purity of the sorting is sufficient for an accurate estimate of μ, even for cell cultures that have a very high mutation rate.
3.3 Mutation rate in clones
We next compared mutation rate analyses in clones of HBL2 and Jurkat and subclones of PR1-C. Among the four subclones of PR1-C, the mutation rate ranged from 1022 × 10-7 to 3286 × 10-7 (table 1). In a parallel analysis of 3 clones of HBL2, the mutation rate ranged from 529 × 10-7 to 776 × 10-7 (table 1). Analyses of parental cultures of Jurkat yielded estimates of μ of 170 × 10-7 and 148 × 10-7 in 2 separate experiments. Among 5 clones of Jurkat, the mutation rate ranged from 155 × 10-7 to 1918 × 10-7. From this data we concluded that hypermutability in these malignant cell lines is a consistent phenotype, but that bulk cultures might harbor sub-populations that differ substantially with respect to this parameter.
3.4 Mutation rate in cell lines derived from plasma cell neoplasms
Although these three malignant cell lines demonstrated a consistent elevation in μ, we found that a marginal zone lymphoma cell line, SSK41, demonstrated a mutation rate in the normal range (table 1). However, this was not unexpected, because marginal zone lymphoma represents an indolent neoplasm. To investigate whether aggressive malignancies might demonstrate a normal mutation rate, we analyzed plasma cell neoplasms, because it is believed that it is only the most advanced forms of this malignancy that can give rise to cell lines. Among 7 such cell lines, there was a wide range in the mutation rate, from 2.2 to 147 × 10-7 (figure 3, table 2). In two of these plasma cell neoplasm lines, there was a substantial population of spontaneously arising GPI (-) cells, and μ was elevated. We confirmed that this population did not have GPI anchors on the surface by staining with FLAER-Alexa 488, as in figure 2 (data not shown). In the other 5 plasma cell neoplasm lines, μ was similar to results that we have reported for BLCLs [15,27]. Overall, among cell lines derived from 11 different malignancies in this study, μ varied over 3 orders of magnitude and was elevated above the range for non-neoplastic human cells in almost half of the cases.
Figure 3. Representative cell lines derived from plasma cell neoplasms.
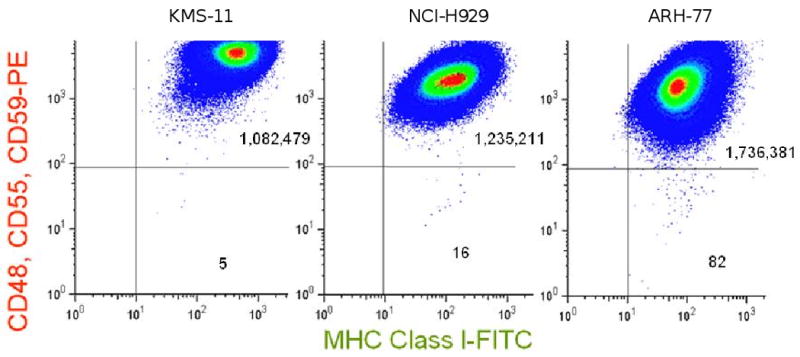
Cells were analyzed after expansion in vitro after sorting to eliminate pre-existing mutants. KMS-11 demonstrates a low number of spontaneously arising GPI (-) cells and a low mutation rate. Within NCI-H929, there is a larger population of spontaneously arising GPI (-) cells, and μ is close to the upper limit of the values we have previously reported for BLCLs from normal individuals [15,27]. In contrast, ARH-77 demonstrates a higher frequency of spontaneously arising GPI (-) cells and a higher μ value.
3.5 Cytogenetic Analysis
We wanted to rule out the possibility that loss of GPI-anchored proteins on the surface might in some cases be due to loss of an entire X chromosome, particularly in cell lines that exhibited chromosomal instability. We first analyzed PR1-C and HBL2-A by standard karyotyping. For PR1-C, G-banding confirmed the t(14;18) translocation and additional complex abnormalities. For HBL2-A, G-banding confirmed the t(11;14) translocation and additional complex abnormalities. For these cell lines as well as Jurkat, we then collected GPI-negative cells by gating on cells that had lost CD59 expression. After expansion in bulk culture without cloning, we treated these sorted cells with proaerolysin and verified that at least 95% of the viable cells were GPI-negative. These GPI-negative cells were then prepared for metaphase and FISH analysis using probes specific for the X and Y centromeres. For the GPI-negative Jurkat culture, the X and Y chromosomes were examined on 100 G-banded metaphases, 2 of which demonstrated an XY karyotype, and 98 of which demonstrated an X, -Y karyotype. [An abnormality of 18p was also identified, and the X chromosome appeared to be normal by G-banding.] Interphase FISH demonstrated 12% XY, and 88% X,-Y (figure 4). A separately derived GPI-negative clone of Jurkat demonstrated 40% X,-Y, 38% XY, and 20% XXY by FISH. For the GPI-negative HBL2-A culture, 20 G-banded metaphases demonstrated the t(11;14) translocation, the presence of an X chromosome, loss of the Y, and complex additional abnormalities. Interphase FISH demonstrated 100% X,-Y (figure 4). For the GPI-negative PR1-C cultures, adequate metaphases were not obtained, but interphase FISH demonstrated 82% XY, 12% X,-Y, and 4% XXYY (figure 4). Overall, from this data we concluded that nullizygosity for the X chromosome is probably not likely to be a common cause of the spontaneous appearance of the GPI-negative phenotype in these cell lines.
Figure 4. FISH analysis for the X and Y chromosomes.

Analyses were performed on spontaneously arising GPI-negative sub-populations of Jurkat, HBL2, and PR1-C, isolated by sorting and proaerolysin selection. X and Y chromosome centromeric probes generate green and orange fluorescence respectively. Representative interphase and metaphase cells are shown. Spontaneous loss of the Y chromosome was observed, but not the spontaneous loss of the X chromosome in any of the cells analysed by FISH or G-banded karyotyping.
4.0 Discussion
This study supports the model that an elevation of the mutation rate is a common feature of lymphoid and plasma cell malignancies. We believe that a μ value of almost 3300 × 10-7 is the highest mutation rate ever reported in a sentinel gene in human cells, apart perhaps from the special cases of VDJ recombination and microsatellite instability [13,29]. We can not rule out the possibility that mutations in the other genes involved in GPI-anchor biosynthesis could result in the GPI (-) phenotype. For example, it is possible that in some cell lines with an elevated μ, a somatic mutation has, at first, inactivated one copy of the more than 20 autosomal genes involved in the synthesis or trafficking pathway [30] for GPI. If this were to be the case, then some of the cells with spontaneous GPI-anchor loss could actually reflect a second loss of heterozygosity event rather than a mutation in PIG-A. We believe, though, that genomic instability is the most likely explanation for why this mutation would occur in the first place.
We also can not completely exclude the possibility that the GPI-negative phenotype could arise due to large deletions involving PIG-A or perhaps even loss of the entire X chromosome. However, our cytogenetic analysis has not revealed this in selected GPI-negative subcultures for 3 cell lines, 2 of which demonstrate gross instability of the autosomes. Indeed, nullizygosity for the X chromosome is not generally seen in malignancies, probably because it is not compatible with viability. We suspect that any mitoses that result in nullizygosity for the X chromosome would yield cells that would be excluded from the live cell gate before their GPI-anchors would be lost from the surface.
This analysis of mutation rate in a culture will represent a population mean, and indeed, we have provided some evidence that there may be differences between subclones derived from the same culture. If cells with the highest mutation rate are at a disadvantage in bulk culture, then we might expect that further analysis of subclones will elucidate the maximal spontaneous mutation rate that is compatible with survival of human cells. It should be noted that the mutant frequency that we measure may be influenced by the point during the cell cycle in which the mutation occurs; DNA damage occurring before the PIG-A gene is replicated may result in 0, 1 or 2 daughter cells with the mutation, depending on how the lesion is repaired. However, mutations that occur after replication can result in only one mutant daughter cell during that division cycle. Any differences in timing of spontaneous mutations between cell lines could then influence the calculated μ values by up to two-fold, which is considerably less than the magnitude of the differences reported here.
This study also suggests that at an elevation of the mutation rate—as detected by this assay-- is a not a universal feature of hematologic malignancies. It remains a possibility that genomic instability is still essential for the development of malignancy but may be transient in some cases, perhaps due to mutagen exposure. While PIG-A can be inactivated by most types of mutations [15,19,23,31-33] as well as, hypothetically, by epigenetic silencing [34], some forms of hypermutability would escape detection by this assay. Examples include whole gene translocations, inversions, and duplications, and it is possible that some malignancies could exhibit only these forms of genomic instability. Alternatively, n might be small for some cancers, as may be the case for acute lymphocytic leukemias with MLL rearrangements [35].
The rate of mutation in PIG-A would likely correlate with the types of mutation that alter cellular targets of chemotherapy and result in drug resistance. Indeed, PIG-A mutations themselves would probably confer resistance to Campath—a therapeutic anti-CD52 antibody [32]. Conversely, malignancies with a spontaneous mutation rate that is sufficiently high as to compromise viability might be more susceptible to the mutagenic effects of some chemotherapy agents. We predict that with this approach, it may be possible to investigate the relationship between hypermutability and clinical outcome using primary leukemia and lymphoma cells from patients.
Acknowledgments
Support: NIH-RO1-CA109258, The Spanish Ministry of Science and Innovation, and the UTE-CIMA project.
Footnotes
There are no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albertini RJ, Nicklas JA, O'Neill JP, Robison SH. In vivo somatic mutations in humans: measurement and analysis. Ann Rev Genet. 1990;24:305–326. doi: 10.1146/annurev.ge.24.120190.001513. [DOI] [PubMed] [Google Scholar]
- 2.Green MHL, O'Neill JP, Cole J. Suggestions concerning the relationship between mutant frequency and mutation rate at the hprt locus in human peripheral T-lymphocytes. Mutation Research. 1995;334:323–339. doi: 10.1016/0165-1161(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 3.Vickers MA, Hoy T, Lake H, Kyoizumi S, Boyse J, Hewitt M. Estimation of Mutation Rate at Human Glycophorin A Locus In Hematopoietic Stem Cell Progenitors. Env Mol Mut. 2002;39:333–341. doi: 10.1002/em.10076. [DOI] [PubMed] [Google Scholar]
- 4.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 5.Hahn WC, Weinberg RA. Rules for Making Human Tumor Cells. N Engl J Med. 2002;347:1593–1603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- 6.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber T, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JKV, Gazdar AF, Hartigan J, Wu L, Changsheng L, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 7.Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci, USA. 2006;103:18238–18242. doi: 10.1073/pnas.0607057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seshadri R, Kutlaca RJ, Trainor K, Matthews C, Morley AA. Mutation rate of normal and malignant human lymphocytes. Cancer Res. 1987;47:407–409. [PubMed] [Google Scholar]
- 9.Kendal WS, Frost P. Pitfalls and practice of Luria-Delbruck fluctuation analysis: a review. Cancer Res. 1988;48:1060–1065. [PubMed] [Google Scholar]
- 10.Rossman TG, Goncharova EI, Nadas A. Modeling and measurement of the spontaneous mutation rate in mammalian cells. Mutat Res. 1995;328:21–30. doi: 10.1016/0027-5107(94)00190-g. [DOI] [PubMed] [Google Scholar]
- 11.Glaab WE, Tindall KR. Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects. Carcinogenesis. 1997;18:1–8. doi: 10.1093/carcin/18.1.1. [DOI] [PubMed] [Google Scholar]
- 12.Lin YW, Perkins JJ, Zhang Z, Aplan PD. Distinct Mechanisms Lead to HPRT Gene Mutations in Leukemic Cells. Genes, Chromosomes & Cancer. 2004;39:311–323. doi: 10.1002/gcc.20005. [DOI] [PubMed] [Google Scholar]
- 13.Bachl J, Dessing M, Olsson C, vonBorstel RC, Steinberg C. An experimental solution for the Luria-Delbruck flucutation problem in measuring hypermutation rates. Proc Natl Acad Sci, USA. 1999;96:6847–6849. doi: 10.1073/pnas.96.12.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicolas JF, Jakob H, Jacob F. Metabolic cooperation between mouse embryonal carcinoma cells and their differentiated derivatives. Proc Natl Acad Sci, USA. 1978;75:3292–3296. doi: 10.1073/pnas.75.7.3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Araten DJ, Golde DW, Zhang RH, Thaler HT, Gargiulo L, Notaro R, Luzzatto L. A Quantitative Measurement of the Human Somatic Mutation Rate. Cancer Res. 2005;65:8111–8117. doi: 10.1158/0008-5472.CAN-04-1198. [DOI] [PubMed] [Google Scholar]
- 16.Hillmen P, Bessler M, Mason PJ, Watkins WM, Luzzatto L. Specific defect in N-acetylglucosamine incorporation in the biosynthesis of the glycosylphosphatidylinositol anchor in cloned cell lines from patients with paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci, USA. 1993;90:5272–5276. doi: 10.1073/pnas.90.11.5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, Maeda K, Kitani T, Kinoshita T. The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesis. Science. 1993;259:1318–1320. doi: 10.1126/science.7680492. [DOI] [PubMed] [Google Scholar]
- 18.Rosse WF, Ware RE. The Molecular Basis of Paroxysmal Nocturnal Hemoglobinuria. Blood. 1995;86:3277–3286. [PubMed] [Google Scholar]
- 19.Luzzatto L, Nafa K, Genetics of PNH . In: PNH and the GPI-linked Proteins. Young N, Moss J, editors. Academic Press; San Diego: 2000. pp. 21–47. [Google Scholar]
- 20.Carrel L, Willard H. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 21.Araten DJ, Bessler M, McKenzie S, Castro-Malaspina H, Childs BH, Boulad F, Karadimitris A, Notaro R, Luzzatto L. Dynamics of Hematopoiesis in Paroxysmal Nocturnal Hemoglobinuria (PNH): No evidence for intrinsic growth advantage of PNH clones. Leukemia. 2002;16:2243–2248. doi: 10.1038/sj.leu.2402694. [DOI] [PubMed] [Google Scholar]
- 22.Keller P, Payne JL, Tremml G, Greer PA, Gaboli M, Pandolfi PP, Bessler M. FES-Cre Targets Phosphatidylinositol Glycan Class A (PIGA) Inactivation to Hematopoietic Stem Cells in the Bone Marrow. J Exp Med. 2001;194:581–590. doi: 10.1084/jem.194.5.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araten D, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci, USA. 1999;96:5209–5214. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Eshleman JR, Brodsky RA, Medof ME. Glycosylphosphatidylinositol-anchored protein deficiency as a marker of mutator phenotypes in cancer. Cancer Res. 2001;61:654–658. [PubMed] [Google Scholar]
- 25.Mestre-Escorihuela C, Rubio-Moscardo F, Richter J, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marín M, Rosenwald A, Sugimoto K, Wheat L, Karran E, García J, Sanchez L, Prosper F, Staudt L, Pinkel D, Dyer M, Martinez-Climent J. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109 doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 26.Rubio-Moscardo F, Blesa D, Mestre C, Siebert R, Balasas T, Benito A, Rosenwald A, Climent J, Martinez JI, Schilhabel M, Karran EL, Gesk S, Esteller M, deLeeuw R, Staudt LM, Fernandez-Luna JL, Pinkel D, Dyer M, Martinez-Climent JA. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood. 2005;106:3214–3222. doi: 10.1182/blood-2005-05-2013. [DOI] [PubMed] [Google Scholar]
- 27.Araten DJ, Luzzatto L. The mutation rate in PIG-A is normal in patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2006;108:734–736. doi: 10.1182/blood-2006-01-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brodsky R, Mukhina G, Nelson K, Lawrence T, Jones R, Buckley J. Resistance of Paroxysmal Nocturnal Hemoglobinuria Cells to the Glycosylphosphatidylinositol-Binding Toxin Aerolysin. Blood. 1999;93:1749–1756. [PubMed] [Google Scholar]
- 29.Bhattacharyya N, Skandalis A, Ganesh A, Groden J, Meuth M. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci, USA. 1994;91:6319–6323. doi: 10.1073/pnas.91.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tashima Y, Taguchi R, Murata C, Ashida H, Kinoshita T, Maeda Y. PGAP2 Is Essential for Correct Processing and Stable Expression of GPI-anchored Proteins. Molecular Biology of the Cell. 2006;17:1410–1420. doi: 10.1091/mbc.E05-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA. PIG-A mutations in normal hematopoiesis. Blood. 2005;105:3848–3854. doi: 10.1182/blood-2004-04-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rawstron AC, Rollinson SJ, Richards S, Short MA, English A, Morgan GJ, Hale G, Hillmen P. The PNH phenotype cells that emerge in most patients after CAMPATH-1H therapy are present prior to treatment. Br J Haematol. 1999;107:148–153. doi: 10.1046/j.1365-2141.1999.01676.x. [DOI] [PubMed] [Google Scholar]
- 33.Ware RE, Pickens CV, DeCastro CM, Howard TA. Circulating PIG-A mutant T lymphocytes in healthy adults and patients with bone marrow failure syndromes. Exp Hematol. 2001;29:1403–1409. doi: 10.1016/s0301-472x(01)00746-9. [DOI] [PubMed] [Google Scholar]
- 34.Hu R, Mukhina G, Lee S, Jones R, Englund P, Brown P, Sharkis S, Buckley J, Brodsky R. Silencing of genes required for glycosylphosphatidylinositol anchor biosynthesis in Burkitt lymphoma. Experimental Hematology. 2009;37:423–434. doi: 10.1016/j.exphem.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullighan C, Goorha S, Radtke I, Miller C, Coustan-Smith E, Dalton J, Girtman K, Mathew S, JM J, Pounds S, Su X, Pui C, Relling M, Evans W, Shurtleff S, Downing J. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]