

Abstract
Aims
To investigate the role of nitric oxide synthase and intracellular free zinc ion (Zn2+) in regulation of ultraviolet B light (UVB)-induced cell damage and apoptosis.
Main methods
Real time confocal microscopy measurement was used to determine the changes of intracellular free zinc concentration under different conditions. Cell apoptotic death was determined using fluorescein isothiocyanate (FITC) conjugated-annexin V (ANX5)/PI labeling followed by flow cytometry. Western analysis was used to determine cell apoptosis and eNOS uncoupling.
Key findings
UVB induced an elevation of Zn2+ within 2 min of exposure. The UVB-induced intracellular Zn2+ elevation was dependent on the increase of constitutive nitric oxide synthase (cNOS) activity and production of superoxide. Removal of Zn2+ with a lower concentration (<25 µM) of N,N,N',N'-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN), a Zn2+-specific chelator, did not induce cell death or prevent cells from UVB-induced apoptosis. However, a higher [TPEN] (> 50 µM) was cytotoxic to cells, but prevented cells from further UVB-induced apoptosis. The higher [TPEN] also induced cNOS uncoupling. Furthermore, treating the cells with a membrane permeable superoxide dismutase (PEG-SOD) inhibited Zn2+ release and reduced apoptotic cell death after UVB treatment. The results demonstrated a complex and dynamic regulation of UVB-induced cell damage.
Significance
Our findings not only advance our understanding of the correlations between cNOS activation and Zn elevation, but also elucidated the role of cNOS in regulation of oxidative stress and apoptosis upon UVB-irradiation.
Keywords: Ultraviolet B light (UVB), nitric oxide synthase (NOS), nitric oxide (NO•), peroxynitrite (ONOO−), oxidative stress, superoxide (O2•−), zinc (Zn2+) dyshomeostasis, apoptosis
Introduction
The elevation of nitric oxide (NO•) in skin cells after ultraviolet light B (UVB) irradiation plays an important role in regulation of apoptosis (Lee et al. 2000; Yamaoka et al. 2004). In keratinocytes, NO• is produced by constitutive nitric oxide synthase (cNOS), which is activated immediately by UVB-induced calcium flux (Deliconstantinos et al. 1996a; Lu et al. 2009). UVB also induces the production of superoxide (O2•−) (Aitken et al. 2007; Deliconstantinos et al. 1996b), which could rapidly react with NO• to form peroxynitrite (ONOO−) (Beckman and Koppenol 1996; Groves 1999). Our recent study indicated that UVB-induced imbalance of NO•/ONOO− promotes apoptotic cell death (Wu et al. 2010). A higher level of ONOO− could also induce cNOS uncoupling by oxidizing the Zn2+-thiolate-complex and releasing Zn2+ from the enzyme (Aravindakumar et al. 1999; Zou et al. 2002).
An elevation of intracellular Zn2+ ([Zn2+]i) has been shown to correlate to NO• production and oxidative-stress-induced apoptosis (Bernal et al. 2008; Suh et al. 2008). Zinc is the second most highly abundant transition metal found in the human body and is released from zinc-binding proteins under oxidative stress. Zn2+ is essential for DNA and protein synthesis and the maintenance of DNA and RNA (Vallee and Falchuk 1993). Skin contains twenty percent of total body zinc. Maintaining zinc homeostasis is important in regulation of skin health. A chelation of zinc promotes apoptosis of keratinocytes and supplement of zinc protects skin from UV-induced DNA fragmentation and apoptosis (Leccia et al. 1999; McGowan et al. 1994; Parat et al. 1997). However, the physiological relationships among NO• production, [Zn2+]i elevation and apoptosis upon UVB-irradiation are not clear. In the present study, we show that UVB-irradiation induced a quick elevation of [Zn2+]i, which was dependent on the increase of cNOS activity and production of O2•−. We also provide evidence that [Zn2+]i elevation was not a cause of apoptosis, but Zn2+ dyshomeostasis could lead to uncoupling of cNOS. The increased amount of uncoupled cNOS results in an imbalance of NO•/ONOO−, which promotes apoptosis of UVB-irradiated keratinocytes.
Materials and Methods
Cell culture
The immortalized human keratinocyte cell line HaCaT was kindly provided by Dr. Hongtao Yu (Jackson State University, MS) and cultured as a monolayer in DMEM (Cellgro) with 10% FBS (Cellgro) at 37°C with 5% CO2. Human umbilical vein endothelial (HUVEC) cells were cultured in MCDB-131 medium (VEC Technologies) with 10% FBS (Cellgro) at 37°C with 5% CO2.
Analysis of intracellular zinc concentration ([Zn2+]i)
For Real-time measurement, the cells were seeded in 35 mm plates one day before analysis. The cells were treated with NG-monomethyl-L-Arginine (LNMMA, 100 µM, Sigma) for 2 h, N-Acetyl-L-Cysteine (LNAC, 25 mM, Sigma) for 2 h, TPEN (50 µM, Molecular Probes) for 15 min or PEG-SOD (100 unit/mL, Sigma) for 1 h before stained with NG-DCF (10 µM, N7991, Invitrogen) in the dark for 30 min. The NG-DCF dye was removed and cells were washed twice with phosphate buffered saline (PBS). The cells were then covered with PBS (400 µL) and UVB (50 mJ/cm2) irradiated. The fluorescent images were acquired from 60 sec before to 600 sec after UVB-irradiation by a camera connected to a confocal microscope (LSM 510, Carl Zeiss) at the speed of 1 frame/5 sec at excitation and emission wavelengths of 506 nm and 531 nm, respectively.
For quantitative analysis of [Zn2+]i, the cells were seeded in 12-well tissue culture plates, treated with LNMMA, LNAC or TPEN and stained with NG-DCF as described above. Immediately after irradiation, the buffer was replaced with 1 mL of fresh medium and TPEN was added to the designated wells. Fluorescence intensities were recorded immediately and 30 min after UVB-irradiation at wavelengths of 506/531 nm, respectively, using a Spectramax M2 plate reader (Molecular Devices).
Determination of cell apoptosis by flow cytometry
The cells were treated with SOD for 1 h before or TPEN for 15 min before and after UVB irradiation. At 24 h post-irradiation, the cells were harvested by 0.01% trypsin digestion and combined with the cells floating in the medium. The apoptotic cell death was analyzed by determination of the loss of membrane phospholipid symmetry and membrane integrity (Schindl et al. 1998; Vermes et al. 1995) using a fluorescein isothiocyanate (FITC) conjugated-annexin V (ANX5)/PI apoptosis detection kit (BD Biosciences) following the manufacturer’s protocol. The ANX5/PI double-stained cells were analyzed by using a FACSort Flow Cytometer (Becton Dickinson) equipped with CellQuest software (Becton Dickinson). The parameters of the measurement were set at SSC 350; FL1 700; FL2 700, and a total 10,000 cells were counted.
Western blot analysis of PARP cleavage and eNOS uncoupling
The cells were treated with different concentration of TPEN for 15 min before and after UVB irradiation. The cells were lysed at 4°C in NP-40 lysis buffer (2% NP-40, 80 mM NaCl, 100 mM Tris-HCl, 0.1% SDS) containing a Proteinase Inhibitor Cocktail (Complete™, Roche Molecular Biochemicals) 24 h after UVB. The protein samples were then added to five-fold Laemmli buffer and boiled. These samples were separated on SDS-PAGE gel and then transferred onto a nitrocellulose membrane. The membrane was blocked with 5% (w/v) skim milk in Tris-Buffered Saline Tween-20 (TBST) for 1 h and then incubated with anti-poly (ADP-ribose) polymerase (anti-PARP) antibodies (Cell Signaling) at 4°C overnight. After washed with TBST, the membrane was incubated with HRP–conjugated anti-rabbit antibody for 1 h at room temperature. Membrane was then washed three times in TBST and two times in TBS, and developed in West Pico Supersignal Chemiluminescent substrate (Pierce).
To determine the SDS-resistant eNOS dimers and monomers, low-temperature SDS-PAGE under reducing conditions were performed as described before (Zou et al. 2002). The cells were treated with TPEN and harvested at the time points as indicated. The proteins were mixed with five-fold Laemmli buffer without boiling, electrophorezed on a reducing SDS-PAGE (with 2.5% 2-mercaptoethanol) at 4°C and then transferred onto a nitrocellulose membrane. The eNOS was detected by western blot analysis as described above. The intensities of the bands of coupled eNOS (eNOSc) and uncoupled eNOS (eNOSu) were quantitated using ImageJ (v1.42k, NIH).
UV Irradiation
UVB was generated from a 15 W UVB lamp (UVP), which has an emission spectra from 280–370 nm with a peak at 310 nm. The intensity of UVB was standardized by a UVB meter (UVP) and set at 3 mW/cm2. The medium was replaced with PBS during the irradiation. After UV irradiation, fresh medium was added to each plate.
Statistical Analysis
Student’s t-testwas used to analyze the significance of data. p<0.05 was considered significant.
Results
UVB triggers [Zn2+]i elevation in keratinocytes
Our recent studies indicate that UVB induced rapid increase of cNOS activity, which generated an imbalance of NO•/ONOO− and promoted apoptotic and skin damage (Wu et al. 2010). Since previous studies suggested that an alternation of Zn2+]i was associated with NO•-induced neuronal cell death (Bossy-Wetzel et al. 2004; Cuajungco and Lees 1997), we determined whether [Zn2+]i also plays a role in UVB-induced and NO•-mediated skin cell apoptosis. We first performed real-time measurement of [Zn2+]i in HaCaT cells using an inverted laser-scanning confocal microscope. Intracellular free zinc was labeled with a zinc specific fluorescence dye - Newport Green-DCF (NG-DCF) and the fluorescence intensity was recorded by a camera connected to the confocal microscope at the speed of 1 frame/5 sec. Our data shows that while [Zn2+]i remained the same in control cells (Fig. 1C), [Zn2+]i started to increase in 2 min and peaked in 3 min after UVB-irradiation (Fig. 1A, 1B and 1D). The UVB-induced fluorescence was inhibited by a zinc specific chelator N,N,N',N'-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN, Fig. 1E), which suggests that the increase of fluorescence is [Zn2+]i-dependent. Since NO• peaked in 40 s post-UVB (Wu et al. 2010), our results indicate that Zn2+ homeostasis is alternated after NO• elevation in skin cells upon UVB-irradiation.
Fig. 1.
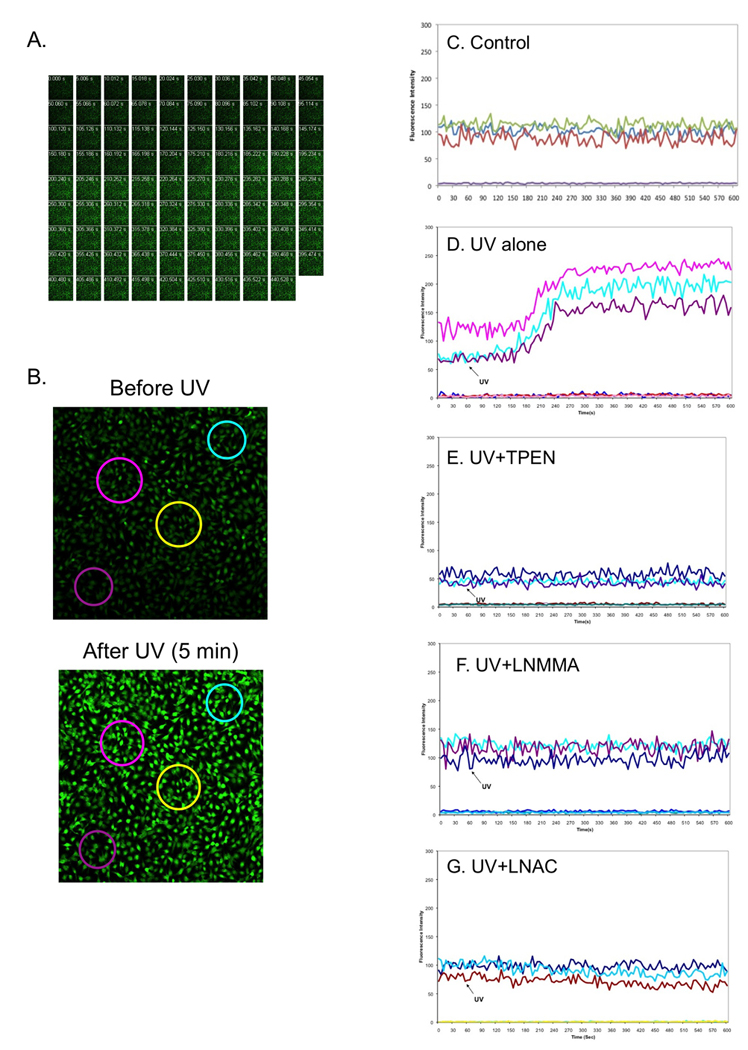
NOS activation and oxidative-stress mediate UVB-induced [Zn2+]i elevation in HaCaT cells. The cells were loaded with NG (10 µM) for 30 min and then the fluorescence of the cells were monitored using a Carl Zeiss confocal microscope. When the background signals of fluorescence were stabilized, the cells were irradiated with UVB (50 mJ/cm2) and fluorescence intensities at three different areas were continuously recorded. Meanwhile the fluorescence images were captured before and after UVB. Panel A: a series of [Zn2+]i fluorescence images captured in 5 sec intervals. Panel B: High-resolution pictures of cells captured before and 5 min after UVB-irradiation. Large circles represent individual regions of interests (ROIs) selected for analysis. Panels C–G: Fluorescence intensities were continuously recorded from the cells that were a control (non-UVB, C), treated by UVB (D), pretreated with 50 µM TPEN (E), 100 µM LNMMA (F) or 25 mM LNAC (G). Individual ROIs were graphed as a function of time. The draw for the first 60 sec was the baseline before cells were irradiated by UVB. Flat lines represent control ROIs drawn in regions free of cells.
A combination of NOS activation and oxidative stress leads to [Zn2+]i elevation upon UVB
Our recent study indicates that UVB induced an immediate release of O2•−, which rapidly reacts with NO• to form ONOO− (Wu et al. 2010). To determine the contribution of NO• and O2•− to UVB-induced elevation of [Zn2+]i, we assessed the extent of the effect of NOS inhibitor and antioxidant on [Zn2+]i upon UVB-irradiation. An N-substituted L-Arg analog LNMMA was used to inhibit NOS activity and the glutathione (GSH) synthesis precursor LNAC was used to reduce oxidative stress. Our data shows that pre-treating the cells with LNMMA or LNAC inhibited UVB-induced elevation of [Zn2+]i (Fig. 1F and 1G). These results suggest that [Zn2+]i elevation is caused by a combination effect of NOS activation and oxidative stress upon UVB-irradiation. As an important signaling molecule to regulate diverse physiological events, NO• is also produced by eNOS in endothelia HUVEC cells and regulates vascular remodeling and cell survival. The amount of NO• release from HUVECs depends on various stimuli, such as UV irradiation and shear stress (Di Pietro et al. 2006). To determine whether UVB-induced and NOS/oxidative stress-mediated elevation of [Zn2+]i is cell type independent, we analyzed the [Zn2+]i elevation in HUVEC cells in the presence of LNMMA and LNAC under the same conditions as above. The [Zn2+]i was quantitatively measured by a fluorescence plate reader at the indicated time points. The data shows that the [Zn2+]i was rapidly increased 2.33±0.12 folds in HUVEC cells after exposure to UVB-irradiation (Fig. 2). The increased [Zn2+]i was slowly decreased to 1.88±0.39 folds at 30 min post-irradiation. The UVB-irradiated cells that received TPEN (50 µM) did not fluoresce above the level of control cells or cells that had been treated with TPEN alone (Fig. 2). The UVB-induced increase of [Zn2+]i was again inhibited by pre-treating the cells with LNMMA or LNAC (Fig. 2). The similar results were also observed using MEF and MCF-7 cells (data not shown). These results indicate that rapid elevation of [Zn2+]i induced by UVB-irradiation is not cell-type specific.
Fig. 2.
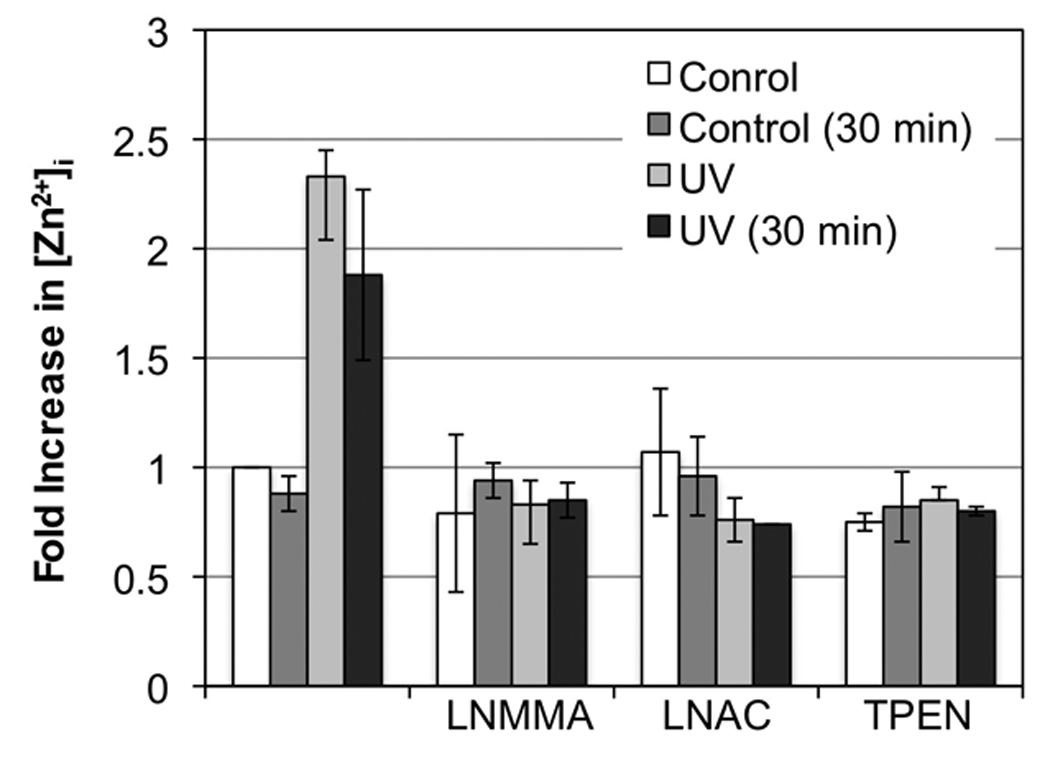
NOS activation and oxidative-stress mediate UVB-induced [Zn2+]i elevation in HUVEC cells. The cells were not treated or treated with LNMMA (100 µM), LNAC (25 mM) and TPEN (50 µM) before loaded with NG for 30 min. The fluorescence intensity was determined using a Spectramax plate reader. Fluorescence intensities were measured immediately and 30 min after UVB irradiated (50 mJ/cm2) and then normalized to the control group to obtain a ratio for relative [Zn2+]i. The error bars represent the standard deviation of two independent experiments.
Complex roles of Zn2+ in regulation of apoptosis without or with UVB-irradiation
An increase of [Zn2+]i has been shown to be associated with apoptotic death of cells under various stimuli (Aizenman et al. 2000; Canzoniero et al. 1999; Lobner et al. 2000). However, free Zn2+ could also directly or indirectly protect cells from oxidative stress-induced apoptosis (Ha et al. 2006; Powell 2000; Taylor et al. 1997). To determine the role of released intracellular Zn2+ in UVB-induced apoptosis of keratinocytes, we determined apoptosis of HaCaT cells, which were treated with various concentrations of TPEN before and after UVB-irradiation. Our data show that UVB-irradiation induced apoptosis was not affected by TPEN at a concentration below 25 µM (Fig. 3A). However, while higher concentrations of TPEN (50 µM and 75 µM) induced apoptosis by themselves, they also inhibited further apoptotic death induced by UVB (Fig. 3A). The results were further confirmed by determination of the extent of effects of high/low [TPEN] on UVB-induced poly (ADP-ribose) polymerase (PARP) cleavage, evidence widely used for detecting cells undergoing apoptosis. Treated cells by UVB caused cleavage of 116-kDa PARP to an 89-kDa fragment which is the marker for cell apoptosis. Neither low concentration (12.5 µM) nor high concentration (75 µM) of TPEN protects keratinocytes from PARP cleavage upon UVB irradiation (Fig. 3B).
Fig. 3.
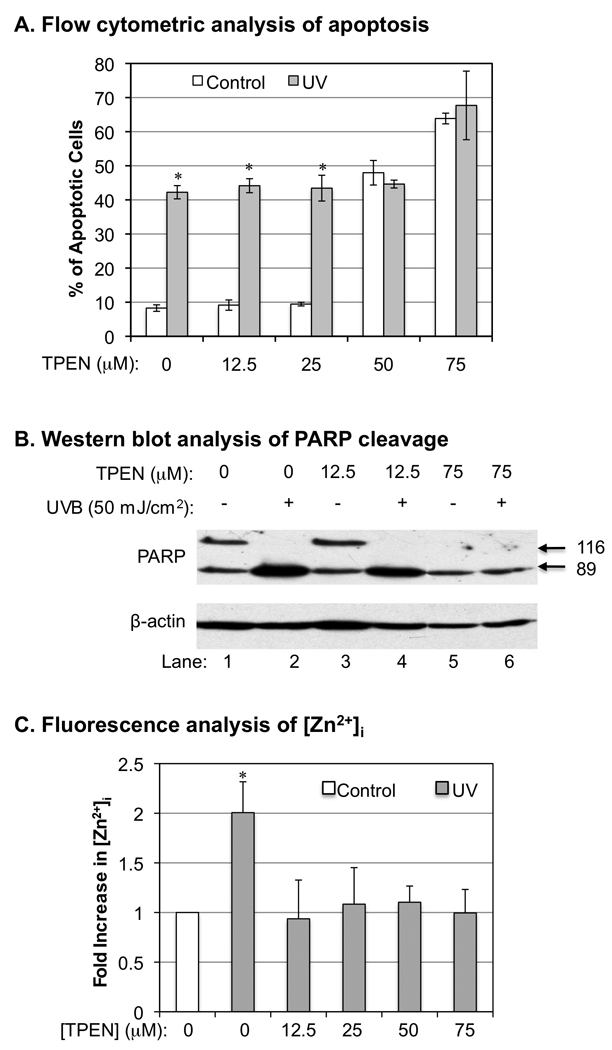
The correlations between UVB-induced [Zn2+]i elevation and apoptosis. HaCat cells were treated with different concentration of TPEN as indicated 15 min before and after UVB-irradiation (50 mJ/cm2). The amount of Annexin V-FITC and PI double-stained cells were quantitatively analyzed by flow cytometry at 24 h post-irradiation. Panel A: the percentage of Annexin V-FITC/PI positive cells, which represent apoptotic dead cells. The error bars represent the standard deviation of three independent experiments. * p < 0.05 vs. UVB alone. Panel B: Western blot analysis of PARP cleavage. Panel C: Relative fluorescence intensities of [Zn2+]i in UVB or TPEN treated HaCaT cells. The error bars represent the standard deviation of three independent experiments. * p < 0.05 vs. UVB alone.
To determine whether the concentration-dependent effect of TPEN on UVB-induced apoptosis correlates with the inhibition of cytosolic Zn2+ release, we analyzed [Zn2+]i after treating the cells with different concentration of TPEN before UVB-irradiation. Our data shows that TPEN prevented UVB-induced [Zn2+]i elevation even at 12.5 µM (Fig. 3C), a concentration of TPEN that had no effect on UVB-induced apoptosis (Fig. 3A). These results indicate that cytosolic free Zn2+ release did not cause apoptosis directly after UVB-irradiation. However, since high [TPEN] induced apoptosis and meanwhile protected cells from further apoptosis induced by UVB (Fig. 3A and 3B), our results also suggest that intracellular Zn2+ homeostasis plays a critical role in regulation of apoptosis without or with UVB-irradiation.
UVB induced production of ONOO− mediates [Zn2+]i elevation and promotes apoptosis
Since inhibition of NOS activation eliminated UVB-induced free Zn2+ release (Fig. 1F), we investigated the mechanism of NO•-mediated alternation of intracellular Zn2+ homeostasis upon UVB-irradiation. Previous studies indicated that NO• reacts quickly with O2•− to form ONOO− (Beckman and Koppenol 1996; Groves 1999), which has been shown to oxidize the zinc-thiolate complex and zinc-fingers of zinc-binding proteins including cNOS and result in the release of zinc from the complex (Aravindakumar et al. 1999; Pacher et al. 2007; Zou et al. 2002). Our recent study demonstrated that ONOO− was released in HaCaT cells almost immediately after UVB-irradiation (Wu et al. 2010). To determine whether UVB-induced ONOO− production leads to [Zn2+]i elevation, we treated the cells with a membrane permeable polyethylene glycol modified superoxide dismutase (PEG-SOD) to decrease cellular O2•− and therefore reduce the oxidative stress generated by ONOO−. The extent of the effects of SOD on UVB-induced [Zn2+]i elevation and apoptosis were determined by the real-time fluorescence measurement and flow cytometry methods as described previously. Our data shows that UVB-induced [Zn2+]i elevation was totally inhibited by treating the cells with SOD (Fig. 4A). These results suggest that oxidative stress generated from ONOO− mediates UVB-induced [Zn2+]i elevation.
Fig. 4.
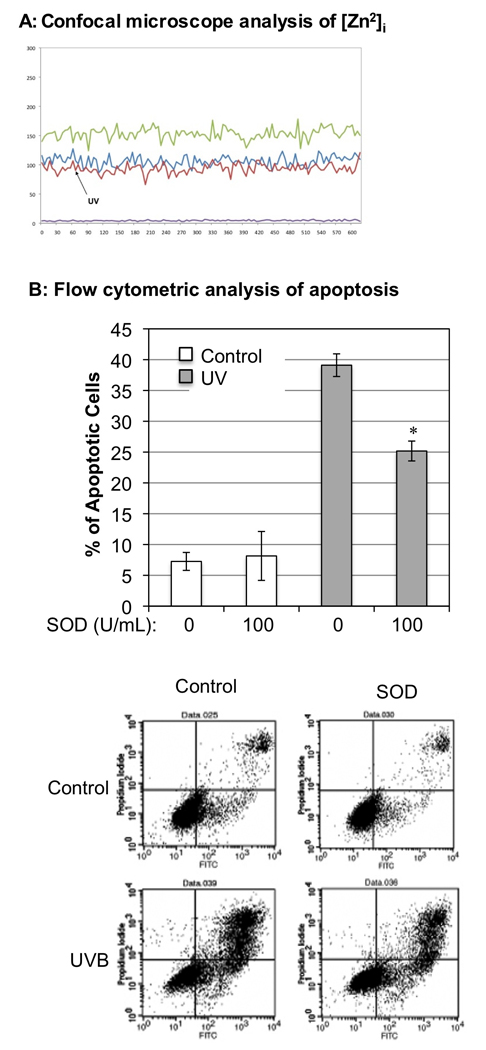
Removal of O2•− prevents [Zn2+]i elevation and reduces apoptotic cell death after UVB-irradiation. Panel A: NG-DCF fluorescence intensity was recorded from the cells that were treated with PEG-SOD from individual ROIs graphed as a function of time. The background lines represent control ROIs drawn in regions free of the cells. Panel B: The relative percentage of apoptotic cells with or without being pretreated with PEG-SOD after UVB-irradiation (50 mJ/cm2). The apoptotic cell death was analyzed at 24 h post-irradiation using the Annexin-V/PI double staining and flow cytometry method. The error bars represent the standard deviation of three independent experiments. * p < 0.05 vs. UVB alone. The lower panel shows the dot-plot graphs of flow cytometry.
Since ONOO− is considered a highly reactive oxidant and potent inducer of cell death (Szabo et al. 2007), we examined whether treating cells with SOD also reduces UVB-induced apoptosis of the cells. Our data showed that removal of O2•− reduced apoptotic death of the irradiated cell from 39.1±1.8% to 25.2±1.6% (Fig. 4B). Removal of O2•− by SOD reduced the apoptotic death of UVB-treated cells by approximately 47% after correcting with the 7−8% of apoptotic cells observed in untreated or SOD alone treated cells. In our previous study, LNAC, the ONOO− scavenger, was also used to evaluate the effect of peroxynitrite on apoptotic death induced by UVB. Upon LNAC treatment, the UVB-induced apoptotic cell death was reduced 38.6% at 24 h post-irradiation (Wu et al. 2010). These results also indicate that UVB-induced formation of ONOO− contributes to apoptotic death of UVB-irradiated cells, which agrees with our recent report (Wu et al. 2010).
TPEN uncouples eNOS in a concentration-dependent manner
In disease condition or extracellular stimuli, the increased monomerization of eNOS alters the enzyme function and generates superoxide instead of NO•. This monomerization of eNOS is referred to eNOS uncoupling (Yang et al. 2009). Our data showed that while a higher concentration (> 50 µM) of TPEN induced apoptotic death of HaCaT cells, it also prevented further UVB-induced apoptosis of the cells (Fig. 3A). This result suggests that high [TPEN] may share the upstream signaling pathway with UVB in induction of apoptosis (Fig. 6). Previous report indicated that TPEN could uncouple eNOS by removing Zn2+ from the enzyme in a cell free system and E. Coli. (Li et al. 1999; Zou et al. 2002). Our recent studies suggested that UVB could also uncouple eNOS and increase production of ONOO− (Liu and Wu 2009; Wu et al. 2010). Since uncoupled eNOS produces O2•− (Huk et al. 1997) that was critical for UVB-induced apoptosis (Fig. 4B), we analyzed the effect of [TPEN] on eNOS uncoupling in HaCaT cells. Our data showed that eNOS dimers were significantly reduced immediately after the treatment of 75 µM TPEN (Fig. 5, Lane 4 vs. 1) and were partially recovered at 2 h post-treatment (Fig. 5A, Lane 5 vs. 4). The eNOS dimers were only slightly reduced after the treatment of 12.5 µM TPEN (Fig. 5A, Lane 2 vs. 1) and were totally recovered at 2 h post-treatment (Fig. 5A, Lane 3 vs. 2). These results indicated that TPEN uncouples eNOS in a concentration-dependent manner. These results suggest that eNOS uncoupling might be the pathway that shared by high [TPEN] or UVB induced apoptosis of keratinocytes.
Fig. 6.
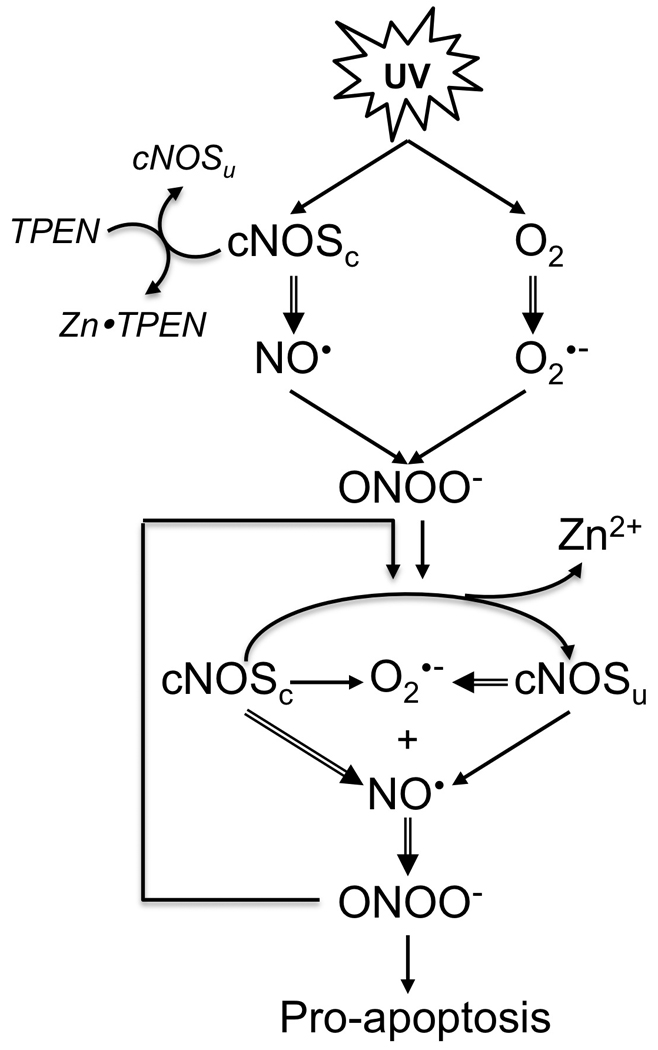
Proposed signaling circuits for UVB-induced cNOS uncoupling, Zn2+ release and apoptosis. cNOSc: coupled cNOS. cNOSu: uncoupled cNOS.
Fig. 5.
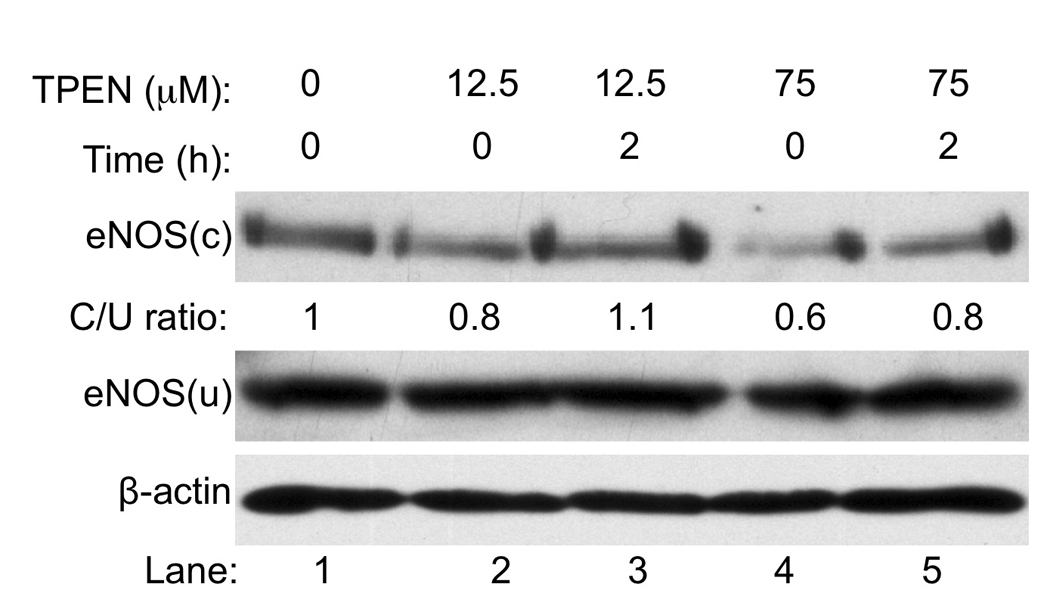
The eNOS uncoupling is TPEN concentration-dependent. HaCaT cells were not treated or treated with 12.5 or 75 µM [TPEN] as indicated. Immediately or 2 h after treatment, total lysates were subjected to a low-temperature SDS-PAGE under reducing conditions. The monomer and dimer of eNOS were monitored by Western blot analysis. The intensities of the bands of eNOS were quantitated using ImageJ (v1.42k, NIH) and normalized to the control.
Discussion
UVB induces a rapid release of NO• and O2•− in cultured keratinocytes (Aitken et al. 2007). Our recent studies demonstrated that UVB induces an immediate increase of cNOS activity and an immediate elevation of NO• and ONOO− in both cultured cells and skin tissue (Wu et al. 2010). An elevation of intracellular level of NO• has been shown to alter intracellular Zn2+ homeostasis (Bernal et al. 2008; Frederickson et al. 2002; St Croix et al. 2002), which plays a role in regulation of UV-induced apoptosis (Leccia et al. 1999; McGowan et al. 1994; Parat et al. 1997). However, it has never been reported that UV-irradiation increases intracellular free Zn2+ concentration. Our data shows that UVB-irradiation induced a quick elevation of [Zn2+]i in both HaCaT and HUVEC cells (Fig. 1 and Fig. 2). However, the [Zn2+]i elevation was behind an increase NO• and ONOO−, which were elevated within 45 sec after UVB (Wu et al. 2010). Our data also showed that UVB-induced [Zn2+]i elevation was dependent on the increase of cNOS activity (Fig. 1F), which has been shown to be responsible for early production of NO• after UVB-irradiation (Wu et al. 2010). In addition to NO•, the generation of O2•− was also critical for UVB-induced [Zn2+]i elevation since treating the cells with either LNAC or SOD completely eliminated free zinc release after UVB-irradiation (Fig. 1G and Fig. 4A). Since NO• can rapidly react with O2•− to form ONOO− (Beckman and Koppenol 1996; Groves 1999), our results suggested that UVB-induced [Zn2+]i elevation is mediated by ONOO−.
Zinc homeostasis has been shown to play a role in regulation of apoptosis of cultured skin cells. While chelation of zinc promotes apoptotic cell death, supplement of zinc protects skin cells from UV-induced DNA-damage and apoptosis (Leccia et al. 1999; McGowan et al. 1994; Parat et al. 1997). Our data showed that while higher concentration of TPEN (>50 µM) induced apoptosis, which agreed with a previous report (Parat et al. 1997), it also prevented further apoptosis induced by UVB (Fig. 3). The protective effect was not dependent on the removal of released intracellular Zn2+ from UVB-irradiation since lower concentration of TPEN (<25 µM) could also chelate the Zn2+, but had no effect on cell apoptosis with or without UVB-irradiation (Fig. 3). Interestingly, while PARP cleavage is not affected by 12.5 µM TPEN (Fig. 3B, Lane 3 vs. 1), all PARP is cleaved to 89-kD fragment under 75 µM TPEN treatment (Fig. 3B, Lane 5). However, the total amount of PARP is less in the cells treated with 75 µM TPEN than the amount in the cells treated with 12.5 µM TPEN or without treatment (Fig. 3B Lanes 5,6 vs. 1–4). This could be due to that TPEN activates a caspase-3 independent apoptotic pathway by reduction of PARP expression as previously reported (Parker et al. 2006; Yun et al. 2003). The preventative effect of TPEN on the further UVB-induced apoptosis is likely due to the uncoupling of eNOS (Fig. 5), which leads to a reduction of NO• production upon UVB-irradiation (Fig. 6). Without an elevation of NO•, there would be reduced production of ONOO−, which is the cause of UVB-induced apoptosis as we recently reported (Wu et al. 2010). However, it was surprising to observe that TPEN could only partially reduced the dimerized cNOS, even at 75 µM (Fig. 5). With Kd for Zn2+ at 2.58 × 10–16 M, 75 µM TPEN could reduce cytosolic Zn2+ to a subfemtomole level as previously reported (Colvin et al. 2008). The results suggest that zinc may not be essential for the dimerization of cNOS. However we could not rule out that some Zn-cNOS complexes exist in a conformation in which TPEN could not reach the bound ion.
Conclusion
Our results indicate that UVB-induced accumulation of ONOO− and oxidative stress lead to the eNOS uncoupling, intracellular zinc dyshomeostasis, and cell damage via a complex and dynamic regulation. Based on our findings, we proposed a signaling circuit for UVB-induced cNOS uncoupling, Zn2+ release and cell death (Fig. 6). UVB activates cNOS, which generates NO•. Meanwhile, UVB also generates O2•−, which reacts rapidly with NO• to from ONOO−. The elevated ONOO− oxidizes the Zn2+-thiolate center in cNOS and induces cNOS uncoupling and Zn2+ release. The uncoupled cNOS produces more O2•− and leads to further elevation of ONOO−, cNOS uncoupling and [Zn2+]i elevation. The increased ONOO− also leads to an NO•/ONOO− imbalance and promotes apoptosis.
Supplementary Material
Acknowledgments
We thank Drs. Tadeusz Malinski (Ohio University) and Robert A. Colvin (Ohio University) for their insightful discussions. We thank Mr. Josh K. Ketterman for technical assistance in using the confocal microscope. We thank Mr. Oliver L. Carpenter and Ms. Kimberly S. George for editorial assistance. This work is partially supported by 2R56CA086928 (to S. W.) and 2RO1CA086928 (to S. W.).
Abbreviations
- ANX5
annexin V
- CAG-AM
Calcium Green-1 acetoxymethyl ester
- cNOS
constitutive nitric oxide synthase
- FITC
fluorescein isothiocyanate
- HUVEC
human umbilical vein endothelial
- LNAC
N-Acetyl-L-Cysteine
- LNMMA
NG-monomethyl-L-Arginine
- MEF
murine embryonic fibroblast
- NG-DCF
Newport Green-DCF
- NO•
nitric oxide
- O2•−
superoxide
- ONOO−
peroxynitrite
- PBS
phosphate buffered saline
- PEG-SOD
polyethylene glycol-conjugated superoxide dismutase
- PI
propidium iodide
- ROS
reactive oxygen species
- TPEN N
N,N',N'-tetrakis (2-pyridylmethyl) ethylenediamine
- UVB
ultraviolet B light
- [Zn2+]i
intracellular Zn2+ concentration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aitken GR, Henderson JR, Chang SC, McNeil CJ, Birch-Machin MA. Direct monitoring of UV-induced free radical generation in HaCaT keratinocytes. Clinical and experimental dermatology. 2007;32:722–727. doi: 10.1111/j.1365-2230.2007.02474.x. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. Journal of Neurochemistry. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- Aravindakumar CT, Ceulemans J, De Ley M. Nitric oxide induces Zn2+ release from metallothionein by destroying zinc-sulphur clusters without concomitant formation of S-nitrosothiol. The Biochemical Journal. 1999;344 Pt 1:253–258. doi: 10.1042/0264-6021:3440253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. The American Journal Of Physiology. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bernal PJ, Leelavanichkul K, Bauer E, Cao R, Wilson A, Wasserloos KJ, Watkins SC, Pitt BR, St Croix CM. Nitric-oxide-mediated zinc release contributes to hypoxic regulation of pulmonary vascular tone. Circulation Research. 2008;102:1575–1583. doi: 10.1161/CIRCRESAHA.108.171264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Canzoniero LM, Turetsky DM, Choi DW. Measurement of intracellular free zinc concentrations accompanying zinc-induced neuronal death. The Journal Of Neuroscience. 1999;19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, Holmes WR. Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. American journal of physiology Cell physiology. 2008;294:C726–C742. doi: 10.1152/ajpcell.00541.2007. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Lees GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiology Of Disease. 1997;4:137–169. doi: 10.1006/nbdi.1997.0163. [DOI] [PubMed] [Google Scholar]
- Deliconstantinos G, Villiotou V, Stavrides JC. Alterations of nitric oxide synthase and xanthine oxidase activities of human keratinocytes by ultraviolet B radiation. Potential role for peroxynitrite in skin inflammation. Biochemical pharmacology. 1996a;51:1727–1738. doi: 10.1016/0006-2952(96)00110-4. [DOI] [PubMed] [Google Scholar]
- Deliconstantinos G, Villiotou V, Stavrides JC. Increase of particulate nitric oxide synthase activity and peroxynitrite synthesis in UVB-irradiated keratinocyte membranes. The Biochemical Journal. 1996b;320 ( Pt 3):997–1003. doi: 10.1042/bj3200997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro R, Mariggio MA, Guarnieri S, Sancilio S, Giardinelli A, Di Silvestre S, Consoli A, Zauli G, Pandolfi A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) regulates endothelial nitric oxide synthase (eNOS) activity and its localization within the human vein endothelial cells (HUVEC) in culture. Journal Of Cellular Biochemistry. 2006;97:782–794. doi: 10.1002/jcb.20686. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Cuajungco MP, LaBuda CJ, Suh SW. Nitric oxide causes apparent release of zinc from presynaptic boutons. Neuroscience. 2002;115:471–474. doi: 10.1016/s0306-4522(02)00399-8. [DOI] [PubMed] [Google Scholar]
- Groves JT. Peroxynitrite: reactive, invasive and enigmatic. Current Opinion In Chemical Biology. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- Ha KN, Chen Y, Cai J, Sternberg P., Jr Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Investigative Ophthalmology & Visual Science. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- Huk I, Nanobashvili J, Neumayer C, Punz A, Mueller M, Afkhampour K, Mittlboeck M, Losert U, Polterauer P, Roth E, Patton S, Malinski T. L-arginine treatment alters the kinetics of nitric oxide and superoxide release and reduces ischemia/reperfusion injury in skeletal muscle. Circulation. 1997;96:667–675. doi: 10.1161/01.cir.96.2.667. [DOI] [PubMed] [Google Scholar]
- Leccia MT, Richard MJ, Favier A, Beani JC. Zinc protects against ultraviolet A1-induced DNA damage and apoptosis in cultured human fibroblasts. Biological Trace Element Research. 1999;69:177–190. doi: 10.1007/BF02783870. [DOI] [PubMed] [Google Scholar]
- Lee SC, Lee JW, Jung JE, Lee HW, Chun SD, Kang IK, Won YH, Kim YP. Protective role of nitric oxide-mediated inflammatory response against lipid peroxidation in ultraviolet B-irradiated skin. The British Journal Of Dermatology. 2000;142:653–659. doi: 10.1046/j.1365-2133.2000.03409.x. [DOI] [PubMed] [Google Scholar]
- Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, Whitlow M, Poulos TL. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. The Journal of biological chemistry. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- Liu W, Wu S. The Differential Role of Nitric Oxide Synthases in Ultraviolet Light B-induced Apoptosis. The FASEB Journal. 2009:890–895. [Google Scholar]
- Lobner D, Canzoniero LM, Manzerra P, Gottron F, Ying H, Knudson M, Tian M, Dugan LL, Kerchner GA, Sheline CT, Korsmeyer SJ, Choi DW. Zinc-induced neuronal death in cortical neurons. Cellular And Molecular Biologyl (Noisy-le-grand) 2000;46:797–806. [PubMed] [Google Scholar]
- Lu W, Laszlo CF, Miao Z, Chen H, Wu S. The role of nitric oxide synthase in regulation of ultraviolet light-induced phosphorylation of the alpha-subunit of eukaryotic initiation factor 2. J Biol Chem. 2009 doi: 10.1074/jbc.M109.008821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan AJ, Fernandes RS, Verhaegen S, Cotter TG. Zinc inhibits UV radiation-induced apoptosis but fails to prevent subsequent cell death. International Journal Of radiation Biology. 1994;66:343–349. doi: 10.1080/09553009414551281. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parat MO, Richard MJ, Pollet S, Hadjur C, Favier A, Beani JC. Zinc and DNA fragmentation in keratinocyte apoptosis: its inhibitory effect in UVB irradiated cells. Journal of photochemistry and photobiology B, Biology. 1997;37:101–106. doi: 10.1016/s1011-1344(96)07334-4. [DOI] [PubMed] [Google Scholar]
- Parker SH, Parker TA, George KS, Wu S. The roles of translation initiation regulation in ultraviolet light-induced apoptosis. Molecular And Cellular Biochemistry. 2006;293:173–181. doi: 10.1007/s11010-006-9239-y. [DOI] [PubMed] [Google Scholar]
- Powell SR. The antioxidant properties of zinc. The Journal Of Nutrition. 2000;130:1447S–1454S. doi: 10.1093/jn/130.5.1447S. [DOI] [PubMed] [Google Scholar]
- Schindl A, Klosner G, Honigsmann H, Jori G, Calzavara-Pinton PC, Trautinger F. Flow cytometric quantification of UV-induced cell death in a human squamous cell carcinoma-derived cell line: dose and kinetic studies. Journal of photochemistry and photobiology B, Biology. 1998;44:97–106. doi: 10.1016/s1011-1344(98)00127-4. [DOI] [PubMed] [Google Scholar]
- St Croix CM, Wasserloos KJ, Dineley KE, Reynolds IJ, Levitan ES, Pitt BR. Nitric oxide-induced changes in intracellular zinc homeostasis are mediated by metallothionein/thionein. American Journal Of Physiology Lung Cellular And Molecular Physiology. 2002;282:L185–L192. doi: 10.1152/ajplung.00267.2001. [DOI] [PubMed] [Google Scholar]
- Suh SW, Hamby AM, Gum ET, Shin BS, Won SJ, Sheline CT, Chan PH, Swanson RA. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. Journal Of Cerebral Blood Flow And Metabolism. 2008;28:1697–1706. doi: 10.1038/jcbfm.2008.61. [DOI] [PubMed] [Google Scholar]
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Reviews Drug Discovery. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Taylor CG, McCutchon TL, Boermans HJ, DiSilvestro RA, Bray TM. Comparison of Zn and vitamin E for protection against hyperoxia-induced lung damage. Free Radical Biology & Medicine. 1997;22:543–550. doi: 10.1016/s0891-5849(96)00390-5. [DOI] [PubMed] [Google Scholar]
- Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiological Reviews. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal Of Immunological Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Wu S, Wang L, Jacoby AM, Kasinski K, Kubant R, Malinski T. Ultraviolet B Light-Induced Nitric Oxide/Peroxynitrite Imbalance in Keratinocytes – Implication in Apoptosis and Necrosis. Photochemistry and photobiology. 2010 doi: 10.1111/j.1751-1097.2009.00682.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka J, Kawana S, Miyachi Y. Nitric oxide inhibits ultraviolet B-induced murine keratinocyte apoptosis by regulating apoptotic signaling cascades. Free Radical Research. 2004;38:943–950. doi: 10.1080/10715760412331284807. [DOI] [PubMed] [Google Scholar]
- Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. American Journal Of Physiology Heart And Circulatory Physiology. 2009;297:H1829–H1836. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SJ, Lee DJ, Kim MO, Jung B, Kim SO, Sohn NW, Lee EH. Reduction but not cleavage of poly(ADP-ribose) polymerase during stress-mediated cell death in the rat hippocampus. Neuroreport. 2003;14:935–939. doi: 10.1097/01.wnr.0000074340.81633.f1. [DOI] [PubMed] [Google Scholar]
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. The Journal of clinical investigation. 2002;109:817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.