

Abstract
This Letter describes a chemical lead optimization campaign directed at VU0119498, a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM) with the goal of developing a selective M1 PAM. An iterative library synthesis approach delivered a potent (M1 EC50 = 830 nM) and highly selective M1 PAM (>30 μM vs. M2-M5).
Recently, we described the identification of VU0119498, a pan Gq mAChR M1, M3, M5 positive allosteric modulator (PAM), from a functional high throughput screen (Fig. 1).1 In subsequent Letters, we described chemical lead optimization efforts based on VU0119498 (1) that delivered the first highly M5-preferring PAM (VU0238429 (2)) and a highly M5-selective PAM (VU0400265 (3)).2,3
Figure 1.

HTS lead VU0119498, a pan Gq mAChR M1, M3, M5 PAM, VU0238441, VU0238429, a highly M5-preferring PAM and VU0400265, a highly selective M5 PAM. Data represent means from at least three independent determinations with similar results using mobilization of intracellular calcium in M1-M5 CHO cells (M2 and M4 cells co-transfected with Gqi5).
Incorporation of a 5-OCF3 moiety on the isatin ring was essential for M5 PAM activity and can be viewed as a ‘molecular switch’ to modulate mAChR subtype selectivity.1-3 As we described previously, other substituents on the isatin ring led to pan mAChR PAMs with varying degree of potency and efficacy across M1-M5.2,3
Selective M1 activation is an attractive therapeutic approach for the treatment of cognitive impairment, Alzheimer's disease, schizophrenia and a number of other CNS disorders.4-14 Until recently, no highly selective M1 activators existed, and those that claimed to be highly M1 selective were either not centrally penetrant or possessed significant ancillary pharmacology which prohibited their use as probes to study M1 receptor function.15,16 We have disclosed three selective M1 activators: BQCA (4),17,18 a highly selective M1 PAM, TBPB (5) a second generation M1 allosteric agonist19-21 and VU0357017 (6), a best-in-class M1 allosteric agonist.22 While BQCA was a key compound (calcium mobilization assay M1 EC50 = 845 nM, 100% ACh Max, 100-fold left-shift of ACh CRC at 100 μM), brain penetration was acceptable, but not optimal, due presumably to the carboxylic acid moiety.17,18 Our initial report on the discovery of VU0119498 also described three other series of weak M1 PAMs, and identified that different M1 PAM chemotypes displayed different modes of activity on downstream receptor signaling.1 Thus, all allosteric M1 activation is not equivalent, and additional tool compounds representing diverse chemotypes are required to truly dissect and study M1 function in the CNS. Based on our ability to develop an M5 selective PAM from a pan Gq M1, M3, M5 PAM,2,3 we initiated an effort to optimize VU0119498 for M1 PAM activity in an attempt to add a unique chemotype to our tool kit of selective M1 activators.
Our initial optimization strategy is outlined in Figure 3, and as SAR with allosteric ligands is often shallow, we employed an iterative parallel synthesis approach. From our M5 work where we counter-screened on M1, we quickly learned that most substitutions on the isatin ring led to pan mAChR activation profiles with various degrees of potency, efficacy, and subtype-selectivity. 2,3 Thus, our first libraries employed a naked isatin core and surveyed diversity on the southern benzyl moiety.
Figure 3.

Initial optimization strategy for VU0119498, a pan Gq M1, M3, M5 PAM.
Libraries were prepared according to Scheme 1, wherein commercial indoline-2,3-dione 7 was alkylated with p-bromobenzylbromide to deliver key intermediate 8. A 10-member Suzuki library was then prepared to explore the effect of introduction of biaryl and heterobiaryl motifs into VU0119498 providing analogs 9. In parallel, 7 was alkylated with functionalized phenethyl bromides 10 to probe the effect of chain homologation in analogs 11. Compound libraries were triaged by a single point (10 μM) screen for their ability to potentiate an EC20 concentration of ACh on M1 CHO cells. SAR was extremely shallow, with only one analog 9a demonstrating robust M1 potentiation (Fig. 5). VU0365137 (9a), possessing an N-methyl pyrazole in the 4-position of the southern benzyl ring displayed an M1 EC50 of 2.3 μM, and good selectivity versus M3 and M5. Moreover, 9a afforded a ∼5-fold leftward shift of the M1 ACh CRC at 10 μM, and a larger ∼ 14-fold shift at 30 μM, with ∼30% intrinsic allosteric agonism. Intriguingly, the 5-OCF3 congener of 9a is an equipotent M5-preferring PAM,2,3 highlighting the aforementioned ‘molecular switch’ to engender M5 preference. However, it was exciting to see that we could develop an M1-preferring PAM from our initial pan Gq M1, M3, M5 PAM lead.1
Scheme 1.

a) p-bromobenzylbromide, K2CO3, KI, ACN, rt, 16 h (97%); b) RB(OH)2, Pd(Pt-Bu3)2, Cs2CO3, THF:H2O, mw, 120 °C, 20 min (15-90%); c) K2CO3, KI, ACN, rt, 16 h (50-90%).
Figure 5.
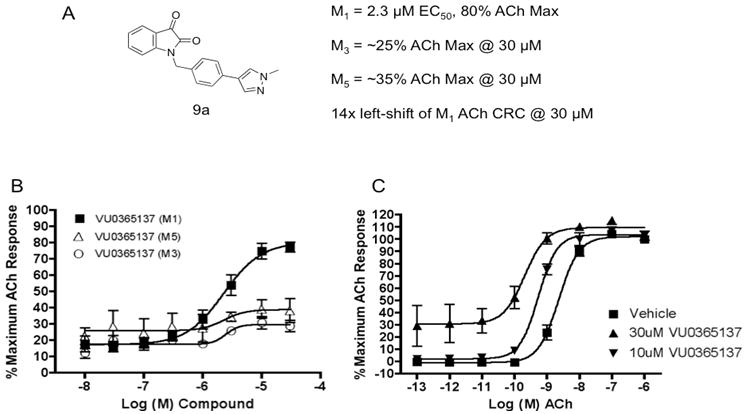
A) Structure and activity of VU0365137 (9a); B) CRCs for VU0365137 (9a) in the presence of a submaximal (∼EC20) concentration of ACh at M1, M3 and M5; C) Fold-shift experiments of the ACh CRC at M5 with both 10 μM and 30 μM concentrations of 9a, providing an approximately 5-fold and 14-fold shift, respectively. Data represent means of at least three independent determinations with similar results using mobilization of intracellular calcium in M1, M3, or M5 CHO cells.
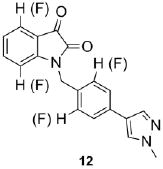
Since SAR was incredibly shallow, we next incorporated subtle changes, in the form of fluorine atoms, to the VU0365137 (9a) scaffold, as we had previously shown was productive in optimizing BQCA, 4.23 Interestingly, there was some, but highly limited SAR overlap between these two series of M1 PAMs. Following the synthetic route outlined in Scheme 1, analogs with fluorine on both the isatin scaffold and the benzyl ring were readily prepared and evaluated for their ability to potentiate an EC20 of ACh at M1. This effort was more productive (Table 1) with five of the analogs 12 displaying potentiation of M1, and two analogs provided M1 EC50s below 1 μM. Fluorine substitution was well tolerated on both the isatin core (4,7-difluoro or 7-fluoro) and on the benzyl ring (2-fluoro and 2,6-difluoro). The addition of a single fluorine atom to the 2-position of the benzyl ring delivered 12a, with an M1 EC50 of 830 nM (65% ACh Max) – comparable to BQCA (M1 EC50 = 845 nM), but without the carboxylic acid moiety. This single change afforded a three-fold increase in potency over VU0365137 (9a). A 2,6-difluorobenzyl congener 12b provides equivalent M1 potency with a slightly diminished ACh Max (60%). As fluorine content increased 12c-12e (fluoro-substitution on both the isatin core and benzyl ring) provided comparable M1 potency, but lower ACh Max (40-55%). VU0366369 (12a) was studied further (Fig. 6). Gratifyingly, 12a was found to be a highly selective M1 PAM, with minimal/no activation of M2-M5 up to 30 μM (Fig. 5A-B). However, in M1 ACh CRC fold-shift experiments, 12a as well as the di-fluoro congener 12b displayed only a subtle effect, increasing the potency of ACh by only 3× and 2× respectively, at 30 μM (Fig. 5C). The smaller fold-shift appears to correlate with the lower overall ACh Max for this series.1,15,16 Lack of correlation between PAM potency and fold-shift is commonly observed within series of mAChR allosteric modulators and underscores the importance of determining both parameters when establishing SAR.15 Nonetheless, VU366369 (12a) represents the second known chemotype to provide potent and selective M1 positive allosteric modulation.
Table 1.
Structures and activities of analogs 12.
 | ||||
|---|---|---|---|---|
| Cmpd | VU Number | Compound | M1 EC50 (μM)a | % ACh Maxa |
| 12a | 0366369 |  |
0.83 | 65 |
| 12b | 0366368 |  |
0.86 | 60 |
| 12c | 0366370 | 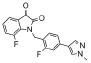 |
2.3 | 55 |
| 12d | 0366367 | 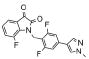 |
1.1 | 40 |
| 12e | 0366372 | 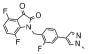 |
1.2 | 50 |
Average of at least three independent determinations. All compounds M1 EC50 >30 μM.
Figure 6.
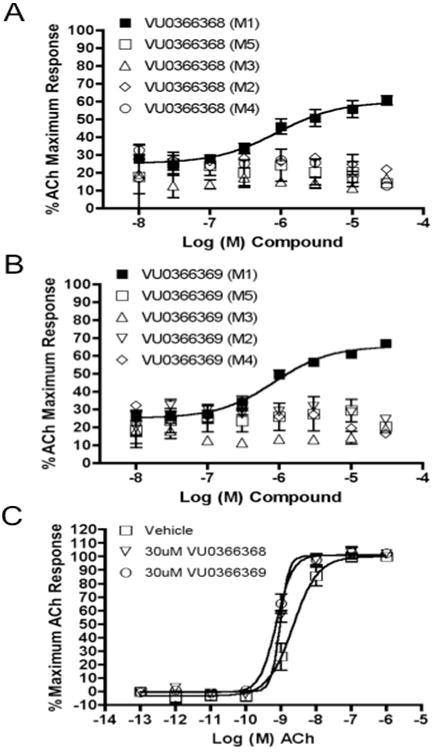
A) and B) CRCs for VU0366368 (12b) and VU0366369 (12a) in the presence of a submaximal (∼EC20) concentration of ACh at M1, M2/Gqi5, M3, M4/Gqi5 and M5; C) Fold-shift experiments of the ACh CRC at M5 with 30 μM of 12a and 12b, providing an approximately 3- and 2-fold-shift, respectively. Data represent means of at least two independent determinations with similar results using mobilization of intracellular calcium in M1, M2/Gqi5, M3, M4/Gqi5 and M5 CHO cells.
Having been able to optimize a pan Gq M1, M3, M5 PAM to deliver a potent and selective M1 PAM (VU0366369, 12a) and a potent and selective M5 PAM (VU0400265, 3),2,3 we hoped to identify ‘molecular switches’ within this chemotype that would engender M3 PAM selectivity. We began by evaluating all analogs synthesized to date, that did not potentiate an EC20 of ACh at M1 or M5, for their ability to potentiate an EC20 of ACh at M3 at a 10 μM concentration. Surprisingly, identification of an M3 PAM within this chemotype remains elusive.
Thus, optimization of a pan Gq mAChR M1, M3, M5 PAM, which previously led to the discovery of the first selective M5 PAM (VU0400265), provided VU0366369 (12a), a highly selective and potent M1 PAM. VU0366369 possesses comparable potency to BQCA and represents only the second known chemotype to provide highly selective M1 potentiation. Efforts to develop an M3 PAM from this chemotype have thus far proven unsuccessful; however, the ability to dial in or out M1 and M5 PAM activity within a single scaffold is unprecedented. Further in vitro and in vivo characterization of VU0400265 and VU0366369 is in progress with exciting results, which will be reported in due course.
Figure 2.

BQCA, a highly selective M1 PAM, TBPB, a second generation M1 allosteric agonist, and VU0357017, a best-in-class M1 allosteric agonist.
Figure 4.

Representative analogs 9 comprising the first generation M1 PAM library. EC50s >10 μM.
Acknowledgments
The authors thank NIMH (1RO1 MH082867), NIH, the MLPCN (1U54 MH084659) and the Alzheimer's Association (IIRG-07-57131) for support of our Program in the development of subtype selective allosteric ligands of mAChRs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Mol Pharm. 2009;75(3):577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. doi: 10.1016/j.bmcl.2010.08.042. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Life Sci. 2001;68(2223):2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- 5.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva Nat Neurosci. 2003;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 6.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 7.Bartus RT. Exp Neurol. 2000;163:495–529. doi: 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- 8.Langmead CJ, Watson J, Reavill C. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 9.Fisher A. Neurodegener Dis. 2008;5:237–240. doi: 10.1159/000113712. [DOI] [PubMed] [Google Scholar]
- 10.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Mol Psychiatry. 2008;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 11.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 12.Scarr E, Sundrarn S, Keriakous D, Dean B. Biol Psychiatry. 2001;61:1161–1170. doi: 10.1016/j.biopsych.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich JN, Butera JA, Carrick T, Kramer A, Kowal D, Lock T, Marquis KL, Pausch MH, Popiolek M, Sun SC, Tseng E, Uveges AJ, Mayer SC. Eur J Pharmacol. 2009;605:53–56. doi: 10.1016/j.ejphar.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 14.Mirza NR, Peters D, Sparks RG. CNS Drug Rev. 2003;92:159–186. doi: 10.1111/j.1527-3458.2003.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conn PJ, Christopoulos A, Lindsley CW. Nat Rev Durg Disc. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conn PJ, Jones C, Lindsley CW. Trends in Pharm Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MW, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. J Neurosci. 2008;28:10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridges TM, Brady AE, Kennedy JP, Daniels NR, Miller NR, Kim K, Breininger ML, Gentry PR, Brogan JT, Jones JK, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:5439–5442. doi: 10.1016/j.bmcl.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller NR, Daniels NR, Bridges TM, Brady AE, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:5443–5446. doi: 10.1016/j.bmcl.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. doi: 10.1021/cn900003h. ARTICLE ASAP, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]