

Abstract
Nuclear factor-kappa B (NF-κB) is a transcription factor that plays a critical role across many cellular processes including embryonic and neuronal development, cell proliferation, apoptosis, immune responses to infection, and inflammation. Dysregulation of NF-κB signaling is associated with inflammatory diseases and certain cancers. Constitutive activation of NF-κB signaling has been found in some types of tumors including breast, colon, prostate, skin and lymphoid, hence therapeutic blockade of NF-κB signaling in cancer cells provides an attractive strategy for the development of anticancer drugs. To identify small molecule inhibitors of NF-κB signaling, we screened approximately 2,800 clinically approved drugs and bioactive compounds from the NIH Chemical Genomics Center Pharmaceutical Collection (NPC) in a NF-κB mediated β-lactamase reporter gene assay. Each compound was tested at fifteen different concentrations in a quantitative high throughput screening format. We identified nineteen drugs that inhibited NF-κB signaling, with potencies as low as 20 nM. Many of these drugs, including emetine, fluorosalan, sunitinib malate, bithionol, narasin, tribromsalan, and lestaurtinib, inhibited NF-κB signaling via inhibition of IκBα phosphorylation. Others, such as ectinascidin 743, chromomycin A3 and bortezomib utilized other mechanisms. Furthermore, many of these drugs induced caspase 3/7 activity and had an inhibitory effect on cervical cancer cell growth. Our results indicate that many currently approved pharmaceuticals have previously unappreciated effects on NF-κB signaling, which may contribute to anticancer therapeutic effects. Comprehensive profiling of approved drugs provides insight into their molecular mechanisms, thus providing a basis for drug repurposing.
Keywords: caspase 3/7, cervical cancer, IκBα phosphorylation, NCGC Pharmaceutical Collection, NF-κB signaling
1. Introduction
NF-kB is a transcription factor that stimulates the expression of its target genes in response to stimuli such as viral and bacterial antigens, UV radiation, and cytokines such as IL-2 and TNF-α [1]. The NF-κB signaling pathway has multiple points of regulation, and is implicated in numerous diseases, thus it has recently become a target for therapeutic drugs. Within the cell, the NF-κB molecule exists as a homodimer or heterodimer complexed with an inhibitory protein, IκB. Upon stimulation of the pathway, the signaling cascade results in activation of the IκB kinase (IKK), which phosphorylates IκB. IκB dissociates from the NF-κB dimer through proteasome degradation, revealing the dimer’s nuclear localization signal. NF-κB is then shuttled into the nucleus, binds the κB binding site and stimulates the production of specific proteins [2]. So far, almost 400 NF-κB targeted genes [3], such as cytokines, chemokines, cell adhesion molecules, growth factors, oncogenes, and pro-/antiapoptotic proteins have been identified [4].
Regulation of the NF-κB pathway plays a pivotal role in cellular responses to changes in the environment. The nuclear factor assists in embryonic and neuronal development, cell proliferation, apoptosis and immune responses to infection and inflammation [5]. However, dysregulation of the pathway is associated with diseases such as chronic inflammation, immunodeficiency, and cancer [6]. Under physiological conditions, the NF-κB pathway is constitutively active only in a few types of cells including neurons, B cells, and thymocytes, and is always inactive in all other cell types [7]. However, in cancer cells such as breast, colon, pancreatic, ovarian, lymphoma, and melanoma, the NF-κB pathway has been reported to be constitutively active [8]. Constitutive activity of NF-κB signaling in tumor cells is involved in cellular proliferation, blocking apoptosis, promotion of angiogenesis, and invasion and metastasis [9]. Since dysregulation of the NF-κB pathway is implicated in many types of tumors, the inhibition of this pathway by small molecule antagonists may be able to reverse or halt the growth and spread of tumors.
Several inhibitors of the NF-κB signaling may have the potential to enter clinical development, such as pristimerin [10], KINK-1 [9] and DHMEQ [11]. These compounds were reported to block tumor growth through various mechanisms such as inhibition of IKK [10] and prevention of NF-κB activation [11]. Although few of these compounds have some potential for clinical development, there is still a great opportunity for development of anticancer drug by targeting NF-κB signaling pathway.
Virtually all clinically used drugs have effects on biological systems other than those for which they were designed. This property of drugs may result in a positive outcome, as when a drug developed for one therapeutic indication finds utility in another (i.e,. “repurposing”), or a negative outcome, as when a drug has an unanticipated toxic effect. In addition, the mechanism(s) of action of many clinically used drugs is not known. Systematically profiling approved drugs may provide insights into each of these processes (Huang et al., unpublished data). To rapidly and efficiently identify currently approved drugs that can inhibit NF-κB signaling, we have screened approximately 2,800 drugs that are either marketed or have been in clinical trials from the NIH Chemical Genomics Center Pharmaceutical Collection (NPC) in a NF-κB mediated β-lactamase reporter gene assay. We utilized a quantitative high-throughput screening (qHTS) format and identified several small molecule drugs that inhibited the NF-κB signaling pathway. These compounds were further investigated in follow-up testing with regard to IκBα phosphorylation and caspase activation. These results provide mechanistic data that can inform repurposing studies, and provide a paradigm that can be applied broadly to maximize appropriate uses of currently approved drugs.
2. Materials and Methods
2.1. Cell line and culture conditions
The CellSensor™ NF-κB-bla ME180 cell line (NF-κB-bla cells) and LanthaScreen™ IκBα GripTite (clonally derived HEK-293) cell line (LanthaScreen™ IκB cells) were obtained from Invitrogen (Carlsbad, CA). ME-180 is a human cervical cancer cell line [12]; the NF-κB-bla ME 180 cell line stably expresses a β-lactamase reporter gene under the regulation of a NF-kB response element. IκB cell line contains a fusion protein consisting of the cDNA encoding for GFP and IκBα (GFP is fused to the N-terminus of full-length IκBα) under the control of a CMV promoter. Both cell lines were cultured in DMEM medium supplemented with 10% dialyzed fetal bovine serum, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1mM sodium pyruvate, 25 mM HEPES, 50 U/ml penicillin and 50 μg/ml streptomycin, and 5 μg/ml of blasticidin. NF-kB-luc2P Hek 293 cell line (Promega, Madison, WI) contains a luciferase gene (luc2P) under control of a minimal TATA promoter with multiple Nuclear Factor-kB response elements. NF-kB-luc2P Hek 293 cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (Thermo Scientific HyClone, Logan, UT), 100 U/ml penicillin, 100 μg/ml streptomycin and 50ug/ml Hygromycin B. HeLa cell line, another human cervical cancer cell line, was cultured in DMEM medium supplemented with 10% fetal bovine serum (Thermo Scientific HyClone), 50 U/ml penicillin and 50 μg/ml streptomycin. All the cell culture reagents were obtained from Invitrogen. The cultures were maintained in a 37°C incubator with 5% CO2 and under a humidified atmosphere.
2.2. NCGC Pharmaceutical Collection
The NCGC Pharmaceutical Collection (NPC) was constructed in house (Huang et al., unpublished data). Briefly, the current NPC consists of 2,816 small molecule compounds, 52% of which are drugs approved for human or animal use by the United States Food and Drug Administration (FDA), 22% are drugs approved in Europe, Canada or Japan, and the remaining 25% are drugs approved in other countries or compounds that have been tested in clinical trials.
The compounds were prepared as 10mM stock solutions dissolved in dimethylsulfoxide (DMSO), except several hundred compounds prepared as 4.47 mM stock solutions, because the NPC library was assembled from several libraries, some of which contained compounds starting at 4.47 mM stock solutions. The compound were prepared first in 96 or 384 wells, and then compressed into 1,536 well plates using an Evolution P3 system (PerkinElmer, Inc., Wellesley, MA) [13]. For use in quantitative high throughput screening, each compound in the NPC compound library was prepared as a fifteen 2.23-fold dilution. After dilutions, the NPC plates were stored desiccated at room temperature for as long as 6 months when in use, or heat sealed and stored at −80°C for long-term storage [14].
2.3. Chemical compounds used in this study
MG-132 was purchased from AG Scientific Inc. (San Diego, CA). IL-1β was purchased from Invitrogen and TNF-α was from R&D Systems (Minneapolis, MN). Bithionol, cantharidin, chromomycin A3, daunorubicinum, digitoxin, emetine, narasin, ouabain, triclabendazolum, were purchased from Sigma-Aldrich (St. Louis, MO). Bortezomib, lestaurtinib, sorafenib tosylate and sunitinib malate, were purchased from LC laboratories (Boston, MA). Tribromsalan was purchased from ChemBridge Corporation (San Diego, CA). Zafirlukast was purchased from Cayman Chemical (Ann Arbor, MI). Tioconazole was purchased from Bosche Scientific (New Brunswick, NJ). Manidipine hydrochloride was purchased from Pharmeks LTD (Moscow, Russia). Ectinascidin 743 and fluorosalan were provided by the National Cancer Institute (Frederick, MD).
2.4. NF-κB β-Lactamase reporter gene assay (NF-κB bla assay) and qHTS
The NF-κB-bla cells were suspended in OPTI-MEM medium supplemented with 0.5 % dialyzed fetal bovine serum, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were dispensed at 2,000 cells/5 μl/well in 1,536-well black wall/clear bottom plates (Greiner Bio-One North America, NC) using a Flying Reagent Dispenser (FRD) (Aurora Discovery, CA) or Thermo Scientific Multidrop Combi (Thermo Fisher Scientific Inc., Waltham, MA). After the cells were incubated at 37°C overnight, 23 nl of compounds at 15 concentrations from the NPC was transferred to the assay plate by a pin tool (Kalypsys, San Diego, CA), followed by addition of either 1μl assay buffer with or without tumor necrosis factor alpha (TNF-α), a known activator [15] of the NF-κB pathway, using a FRD. One μl of 6 ng/ml TNF-α (final concentration, 1ng/ml) was used to stimulate the NF-κB pathway in order to identify the inhibitory effect of the antagonists. The final concentration of the compounds in the 6 μl assay volume ranged from 0.5 nM to 38 μM. The positive control plate format was as follows: Column 1, MG-132, a proteasome inhibitor that inhibits the NF-κB pathway [16], ranged from 1 nM to 38 μM; column 2, 20 ng/ml TNF-α (EC100); column 3, DMSO only; and columns 1, 4 to 48, 1 ng/ml TNF-α (EC80). The plates were incubated for 5 h at 37°C. One μL of LiveBLAzer™ B/G FRET substrate (Invitrogen, CA) detection mix was added and the plates incubated at room temperature for 2 h. Fluorescence intensity (405 nm excitation, 460 and 530 nm emission) was measured using an Envision plate reader (Perkin Elmer, Shelton, CT). Data was expressed as the ratio of 460nm/530nm emissions.
In the confirmation study, selected active compounds were re-tested for the inhibition of TNF-α induced NF-κB coupled β-lactamase activity in 24 point titrations with concentration ranging from 5 pM to 38 μM in the NF-κB-bla assay using the same protocol as described above except 24 point titrations were within one 1536-well plate. These compounds were also tested their inhibitory effect on NF-κB coupled β-actamase activity after the cells were stimulated by 1 ng/ml of IL-1β.
2.5. NF-κB-luciferase reporter gene assay (NF-κB luc assay)
NF-κB-luc2P HEK293 cells were dissociated with 0.05% trypsin/EDTA, resuspended in assay medium, and dispensed at 2000 cells/5 μL/well in 1536-well white wall/solid bottom plates (Greiner Bio-One North America) using a FRD. Twenty-three nL of compound was transferred to the assay plate by a pin tool (Kalypsys). One μL of medium with or without TNF-α (0.5 ng/ml) was dispensed by a FRD. After the plates were incubated 5 hrs at 37°C, 5 μL of ONE-Glo luciferase assay regent (Promega) was added. The plates incubated at room temperature for 30 min, and luminescence intensity of the plates was measured using a ViewLux plate reader (PerkinElmer).
2.6. Chemical analysis for compound purity
Analytical analysis of the ordered compounds was performed on a Waters Acquity LC/MS (Waters Corporation, Milford, MA). A 1.2 minute gradient of 2 to 100% Acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 2 minute run time at a flow rate of 0.5 mL/min. An Acquity UPLC BEH C18, 2.1× 50mm, column with a 1.7 μm particle size was used at a temperature of 45°C. Purity determination was performed using an Evaporative Light Scattering Detector and a Photo Diode Array Detector. Mass Determination was performed using a Waters Micromass ZQ mass spectrometer with electrospray. Data was analyzed using the Waters OpenLynx software. Samples which could not be identified due to poor ionization were further analyzed using an Agilent Time-Of-Flight Mass Spectrometer (TOF, Agilent Technologies, Santa Clara, CA). Mobile phase conditions were similar with the exception of 0.1% formic acid replacing trifluoroacetic acid. Confirmation of molecular formula was confirmed using electrospray ionization with the Agilent Masshunter software version B.02.
2.7. IκBα phosphorylation assay
The measurement of phosphorylation of GFP-IκBα in IκBα GripTite cells was described previously [17]. This assay used terbium (Tb)-labeled antibody which specifically recognizes pS32 of IκBα. When the phosphorylation of GFP-IκBα occurs, the Tb-labeled antibody binds the pS32 site of GFP fusion of IκBα, which causes the increase of a time-resolved resonance energy transfer (TR-FRET). In this assay, IκBα GripTite cells were dispensed at 3,000 cells/5 μl/well in 1536-well white/solid-bottom assay plates in OPTI-MEM medium supplemented with GlutaMAX/HEPES, 1% charcoal/ dextran stripped FBS, 0.1 NEAA, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1mM sodium pyruvate. After the cells were incubated overnight at 37°C, 23 nl of the compounds was transferred to the assay plate by a pin tool (Kalypsys, San Diego, CA), followed by addition of either 1μl assay buffer with or without 1ng/ml TNFα. The assay plates were incubated for 30 min at 37°C. Following the incubation, cells were lysed by the addition of 4 μl/well of a cell lysis buffer (Invitrogen), which contains lysis buffer (20mM Tris; 5mM EDTA; 5mM Sodium Pyrophosphate; 5mM NaF; 150mM NaCl; 2mM VO4 and 1% NP-40), 3nM Tb-labeled anti-pS32-IκBα antibody (Invitrogen) and 1% of protease and phosphatase inhibitors (Sigma). The assay plates were then incubated for 1 h at room temperature, and fluorescence intensity (340 nm excitation, 520 and 490 emission) was measured using an Envision plate reader. Data was expressed as the ratio of 520nm/490nm emissions.
For Western blot analysis, 1 × 106 IκB HEK293 cells per well were cultured in a 6-well plate. Next day the cells were treated with DMSO control, bortezomib, digitoxin, emetine, narasin or fluorosalan at 10 μM, followed by addition of TNF-α (1 ng/ml) for 1 h. Cells were lysed in a cell lysis buffer (Invitrogen) containing 1% of protease inhibitor (Sigma) for 10 min on ice. After samples were centrifuged at 14000 rpm at 4 °C, supernatants were collected and subjected to SDS-PAGE analysis on a 10% Tris-Glycine gel (Invitrogen). And then the proteins were transferred to a PVDF membrane (Invitrogen). The membrane was blotted with primary antibodies against IκBα [pS32] at 1:2000 dilution (Biosource, CA) or β-Actin at 1:5000 dilution (Sigma) as a loading control. HRP conjugated secondary antibody (1:1000 dilution; goat anti-rabbit or anti-mouse, Santa Cruz, CA) was used together with Western blot Luminol reagent kit (Santa Cruz Biotechnology, CA) to develop the membrane that was read with the ChemiDoc XRS system (Bio-Rad, Hercules, CA).
2.8. Caspase 3/7 assay
Caspase 3/7 activity was measured as previously described [18]. NF-κB-bla ME180 cells were dispensed in culture medium at 2,000 cells/5 μl/well in 1536-well white/solid-bottom assay plates. The cells were incubated a minimum of 5 h at 37°C. The compounds (23 nl/well) were added via the pin tool. The treated cells were incubated for 5 or 24 h at 37°C, followed by the addition of the Caspase-Glo 3/7 (Promega, Madision, WI) reagent at 5μl/well. After 30 min incubation at room temperature, the luminescence intensity of the assay plates was measured using a ViewLux Plate Reader.
2.9. Mitochondrial membrane potential assay
Mitochondrial membrane potential was measured by using Mitochondrial Membrane Potential Indicator (m-MPI, Codex Biosolutions, Inc. MD). Briefly the ME180 cells were dispensed at 2,000 cells/5 μl/well in 1,536-well black wall/clear bottom plates using a FRD. After the cells were treated with compounds at 37°C for 24 h, 5 μl of m-MPI reagent was added into the wells and the plates were incubated for 30min at 37°C. Fluorescence intensities (485 nm excitation/535 nm emission; 540 nm excitation/ 590 nm emission) were measured using an Envision plate reader. Data was expressed as the ratio of 535nm/590nm.
2.10. Cell viability assay
Cell viability after compound treatment was measured using a luciferase-coupled ATP quantitation assay (CellTiter-Glo viability assay, Promega) in ME180 and HeLa cells. The change of intracellular ATP content indicates the number of metabolically competent cells after compound treatment. The cells were dispensed at 1,000 cells/5 μl/well for HeLa cells and 2,000 cells/5 μl/well for ME180 cells in 1,536-well white/solid bottom assay plates using an FRD. The cells were incubated a minimum of 5 h at 37°C, followed by the addition of compounds using the pin tool. The assay plates were incubated for 5, 24, 48 or 72 h at 37°C, followed by the addition of 5μl/well of CellTiter-Glo reagent. After 30 min incubation at room temperature, the luminescence intensity of the plates was measured using a ViewLux plate reader (PerkinElmer).
2.11. LDH (lactate dehydrogenase) release assay
LDH release was measured by using Cyto-Tox-One™ Homogenous Membrane Integrity kit (Promega). The ME180 cells were dispensed at 2,000 cells/5 μl/well in 1,536-well black/clear bottom assay plates using an FRD. The cells were incubated overnight at 37°C, followed by the addition of compounds using the pin tool. The assay plates were incubated for 24 h at 37°C, followed by the addition of 5μl/well of Cyto-Tox-One™ reagent. After 10 min incubation at room temperature, the fluorescence intensity (560 nm excitation/590 emission) was measured using an Envision plate reader (PerkinElmer).
2.12. Data analysis
Data normalization and curve fitting was performed as previously described [19, 20]. Briefly, raw plate reads for each titration point were first normalized relative to TNF-α control (1 ng/ml, 100%) and DMSO only wells (basal, 0%), and then corrected by applying a pattern correction algorithm using compound-free control plates (DMSO plates). Concentration-response titration points for each compound were fitted to the Hill equation yielding concentrations of half-maximal inhibition (IC50) and maximal response (efficacy) values. Compounds from qHTS were classified into four major classes using the set of criteria listed in previous studies [19].
Compounds which showed inhibition in both the ratiometric and 460 nm readings and had curve class 1.1, 1.2, 2.1 or 2.2 with >50% efficacy in the ratiometric reading were considered as active in the NFκB bla assay. These compounds were further prioritized based on their activity in the cell viability assay after 5 h compound treatment. Compounds with class 1.1, 1.2, 2.1 or 2.2 curve with >50% efficacy in the cell viability assay were likely cytotoxic, while compounds that were class 4 were not cytotoxic, and compounds with all other curve classes were inconclusive. Compounds that were active in the NFκB bla assay and inactive in the cell viability assay were assigned the highest priority and selected for confirmation and follow up studies. A fraction of compounds that were active in the NFκB bla assay but also showed some level of cytotoxicity (active or inconclusive in the viability assay after 5 h treatment) were also included based on their relative potency in the two assays, resulting in a total of 60 compounds selected for confirmation and follow up.
3. Results
3.1. Screen for inhibitors of NF-κB transcriptional activity using cell-based qHTS
To identify small molecule drugs that can act as inhibitors of the NF-κB pathway, we screened the NPC using qHTS [20] in a cell-based NF-κB β-lactamase reporter gene assay (NF-κB bla assay, Fig.1). TNF-α and MG132 were used as positive controls for the screening. As shown in Fig. 2A, TNF-α induced NF-κB coupled β-lactamase activity in a concentration-dependent fashion with an EC50 of 0.31 ng/ml, while MG132 blocked TNF-α induced NF-κB coupled β-lactamase activity, with an IC50 of 0.3 μM (Fig. 2B). The inhibitory effect of the compounds was measured in the presence of 1 ng/ml (EC90 value) of TNF-α in the primary screen. The MG132 concentration response curves reproduced well in all 34 plates with an average EC50 of 0.65 ± 0.17 μM. The signal to background ratio was 6.0 ± 0.6 and Z’ factor [21] averaged 0.88 ± 0.06 for the entire screen.
Fig. 1.

Principle of the β-lactamase reporter gene assay. Binding of TNF-α to its receptor activates the IκB kinase (IKK), which phosphorylates IκB. The phosphorylated IκB dissociates from the NF-κB dimmer through proteasome degradation. NF-κB translocates across cytoplasmic/nuclear boundary into the nucleus, and then enhances the transcription of the β-lactamase reporter gene by binding to the κB binding site. β-Lactamase expression cleaves the fluorescent substrate (CCF4) containing the two fluorophores, coumarin and fluorescein. This reaction disrupts the fluorescence resonance energy transfer (FRET) from coumarin to flourescein, which produces a blue fluorescent signal at 460 nm. In the absence of β-Lactamase expression, the substrate molecule remains intact and the excitation of the coumarin enables FRET to the fluorescein moiety, producing a green signal with an emission peak at 530 nm.
Fig 2.

(A) TNFα stimulated β-lactamase activity in NF-kB bla ME180 cells; (B) MG132 concentration-dependent inhibition of β-lactamase activity in the presence of 1ng/ml TNFα in NF-kB bla ME180 cells.
Based on concentration-response curve quality in the primary screen (see Methods for details), sixty compounds were selected and re-tested in the NF-κB bla assay; activity was confirmed in 55 out of the 60 re-tested compounds, yielding a confirmation rate of 92%. Based on potency and novelty of mechanism, 20 compounds were selected for further study, and powder samples purchased from commercial vendors. The structures of all the compounds were confirmed and the purity of these compounds was 100%. Of the 20 purchased compounds, the activities of 19 compounds (Table 1) were confirmed in the TNF-α induced NF-κB bla assay. The inhibitory effect of these compounds was also measured in the presence of 1 ng/ml of IL-1β. There is a good correlation (R = 0.85) of IC50s of these compounds between the TNF-α and IL-1β treatments (Table 1). Two cardiac glycoside drugs, digitoxin and ouabain, had similar inhibitory effect on both TNF-α and IL-1β induced NF-κB bla activity (Table 1).
Table 1.
Compound potency (μM) in NFκB-bla reporter gene, NFκB-luc reporter gene, GFP-IκB phosphorylation, and caspase 3/7 assays
| Compound Name |
NFκB-bla TNF-α* IC50 |
NFκB-bla IL-1β** IC50 |
NFκB-luc TNF-α* IC50 |
GFP-IκBα phosphorylation IC50 |
Caspase 5 h EC50 |
Caspase 24 h EC50 |
|---|---|---|---|---|---|---|
| Bithionol | 11.2 | 25.5 | 47.2 | 15.8 | 12.6 | 29.9 |
| Bortezomib | 0.45 | 0.31 | 0.19 | Inactive | 0.35 | 0.05 |
| Cantharidin | 11.2 | 14.2 | 11.0 | 39.8 | 7.9 | 11.3 |
| Chromomycin A3 | 0.56 | 0.26 | 0.13 | Inactive | 0.2 | 0.08 |
| Daunorubicinum | 10.0 | 24.4 | 14.2 | Inactive | 4.0 | 1.3 |
| Digitoxin | 0.09 | 0.07 | 0.17 | Inactive | Inactive | Inactive |
| Ectinascidin 743 | 0.02 | 0.02 | 0.02 | 39.8 | 0.009 | 0.003 |
| Emetine | 2.0 | 4.2 | 0.14 | 0.31 | 1.1 | 17.0 |
| Fluorosalan | 7.9 | 24.4 | 25.3 | 2.8 | Inactive | Inactive |
| Manidipine hydrochloride | 12.6 | 16.8 | 27.3 | Inactive | Inactive | Inactive |
| Narasin | 3.6 | 4.7 | 0.3 | 3.2 | 19.9 | 44.7 |
| Lestaurtinib | 1.6 | 1.9 | 3.2 | 7.9 | 4.7 | 6.2 |
| Ouabain | 0.18 | 0.07 | 0.19 | Inactive | Inactive | Inactive |
| Sorafenib tosylate | 7.9 | 11.8 | 13.3 | 22.4 | Inactive | Inactive |
| Sunitinib malate | 35.5 | 29.5 | 21.7 | Inactive | 79.4 | 79.4 |
| Tioconazole | 39.8 | 61.2 | 54.6 | 44.7 | 79.4 | Inactive |
| Tribromsalan | 11.2 | 21.6 | Inactive | 7.9 | Inactive | Inactive |
| Triclabendazolum | 14.1 | 15.9 | 54.6 | 25.1 | 70.8 | 79.4 |
| Zafirlukast | 35.5 | 23.6 | 56.6 | 31.6 | 50.1 | 75.1 |
TNF-α was used as stimulator NFκB-bla and NFκB-luc assaysin
IL-1β was used as stimulator in NFκB-bla assay
Among the 19 confirmed compounds, ectinascidin 743 was the most potent with IC50 of 20 nM, followed by digitoxin (90 nM in the presence of TNF-α, and 70 nM in the presence of IL-1β). Several known NF-κB inhibitors were confirmed in this study, such as bortezomib, a proteasome inhibitor that blocks the NF-κB signaling pathway [22], and cantharidin, a natural product that reduces NF-κB protein levels in cells and inhibits invasion and metastasis of highly metastatic ovarian carcinoma cell line [23]. All the other compounds listed in Table 1 are potential novel inhibitors of NF-κB signaling. Next, these compounds were further evaluated in human embryonic kidney 293 cells (HEK 293) using an NF-κB luciferase reporter gene assay. Of the 19 compounds tested in the TNF-α induced NF-κB bla assay, the activities of 18 compounds were confirmed in HEK293 NF-κB luc assay with a concordance rate of 95%. The potency ranking of the compounds in these two assays was similar. Ectinascidin 743 was the most potent in the NF-κB luc and NF-κB bla assays, with IC50 values of 20 nM in both assays (Table 1).
3.2. Effect of NF-κB inhibitors on IκBα phosphorylation
In the NF-κB signal transduction cascade, the IκB kinase (IKK) complex is activated upon stimulation by a cytokine, such as TNF-α. The activated IKK complex phosphorylates IκBα, which, in turn, dissociates from NF-κB, allowing NF-κB to translocate to the nucleus and alter gene expression. To investigate the effect of potential NF-κB inhibitors on the phosphorylation of IκBα protein, the phosphorylation assay of IκBα at proximal serine residues (serine-32) was measured in GFP-IκBα GripTite cells. As listed in Table 1, 12 out of 19 drugs inhibited IκBα phosphorylation with IC50 values ranging from 0.31 to 44.7 μM. Of these, five had IC50 values less than 10 μM, with rank order of potency of emetine (0.31 μM), fluorosalan (2.8 μM), narasin (3.2 μM), lestaurtinib (7.9 μM), and tribromsalan (7.9 μM). A further seven drugs inhibited IκB phosphorylation only at higher concentrations: bithionol (15.8 μM), sorafenib tosylate (22.4 μM), triclabendazolum (25.1 μM), zafirlukast (31.6 μM), cantharidin (39.8 μM), ectinascidin 743 (39.8 μM), and tioconazole (44.7 μM). In contrast, five compounds, some with robust activity in the NF-κB bla assay, showed no activity in IκB phosphorylation up to the highest concentration (38 μM) tested, suggesting that they inhibit the NF-κB pathway via other mechanisms. These drugs were bortezomib, chromomycin A3, daunorubicinum, digitoxin, manidipine hydrochloride, ouabain, and sunitinib malate.
To confirm their inhibitory effect on IκBα phosphorylation, bortezomib, digitoxin, emetine, narasin and fluorosalan were further examined for their effect on IκBα phosphorylation by Western blot analysis. As shown in Figure 3, emetine, narasin and fluorosalan reduced TNF-α induced IκBα phosphorylation at 10 μM, whereas bortezomib and digitoxin did not show any inhibition. The Western blot results correlated well with the data from GFP-IκBα phosphorylation TR-FRET assay.
Fig 3.
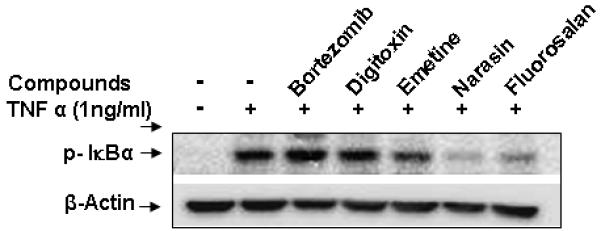
Inhibitory effect of compounds on TNFα-induced IκBα phosphorylation. Cells were treated with DMSO, bortezomib (10 μM), digitoxin (10 μM), emetine (10 μM), narasin (10 μM) and fluorosalan (10 μM) in the absence or presence of TNF-α (1ng/ml) for 1 h, followed by Western blotting analysis.
3.3. Effect of NF-κB inhibitors on caspase 3/7 activity
NF-κB is known to suppress apoptosis by inducing the expression of multiple anti-apoptotic proteins [4]. Constitutive activation of the NF-κB pathway has been reported in many types of cancer cells due to the inhibition of apoptosis by NF-κB signaling [9]. To determine the effect of these NF-κB inhibiting drugs on apoptosis, caspase 3/7 activity was measured in ME180 cells after compound treatment at 5 and 24 h. Of these 19 compounds (Table 1), ectinascidin 743 was the most potent stimulator of caspase 3/7 activity 24 h post treatment, with an EC50 value of 3 nM (Fig 4A and 5A), followed by bortezomib (50 nM, Fig 4B and 5B), chromomycin A3 (80 nM, Fig 4C and 5C), emetine (1.1 μM, Fig 4D and 5D), daunorubicinum (1.3 μM, Fig 4E and 5E), and lestaurtinib (4.7 μM, Fig 4F and 5F). Bithionol, cantharidin and narasin (Table 1) stimulated caspase 3/7 activity EC50 values of 7.9 μM, 12.6 μM and 19.9 μM, respectively, and zafirlukast (50.1 μM), triclabendazolum (70.8 μM), sunitinib malate (79.4 μM), and tioconazole (79.4 μM) showed low-potency caspase 3/7 induction, consistent with their low potency in the initial NF-κB bla assay. In contrast, digitoxin, fluorosalan, manidipine, sorafenib tosylate, ouabain, and tribromsalan had no effect on caspase 3/7 induction at 5 and 24 hours, though of note most of these drugs showed significant cell killing only at later time points (Table 2).
Fig 4.

Concentration response curves of NF-κB antagonists in NF-κB bla, IκB phosphorylation and caspase 3/7 assays. NF-κB bla activity (■) and IκB phosphorylation (▲) were measured after the cells were incubated with various concentrations of ectinascidin 743 (A), bortezomib (B), chromomycin A3 (C), emetine (D), daunorubicinum (E), and lestaurtinib (F) in the presence of 1ng/ml TNFα for 5 h (NF-κB bla assay) or 30 min (IκB phosphorylation assay). Caspase 3/7 activity (▼) was also measured in these cells after the treatment of above drugs for either 5 h or 24 h.
Fig 5.
Chemical structures of NF-kB inhibitors. (A) Ectinascidin 743; (B) Bortezomib; (C) Chromomycin A3; (D) Emetine; (E) Daunorubicinum; (F) Lestaurtinib.
Table 2.
Compound potency (μM, IC50) with regard to cytotoxicity across different treatment times in ME180 cells
| Compound Name | 5 h | 24 h | 48 h | 72 h |
|---|---|---|---|---|
| Bithionol | 70.8 | 39.8 | 26.7 | 3.4 |
| Bortezomib | Inactive | 0.45 | 0.04 | 0.04 |
| Cantharidin | Inactive | 15.0 | 11.2 | 11.2 |
| Chromomycin A3 | Inactive | 0.11 | 0.06 | 0.06 |
| Daunorubicinum | Inactive | 4.2 | 1.8 | 1.2 |
| Digitoxin | Inactive | 0.05 *** | 0.16 | 0.20 |
| Ectinascidin 743 | Inactive | 3.1 | 0.004 | 0.003 |
| Emetine | Inactive | 22.0 | 11.9 | 3.4 |
| Fluorosalan | Inactive | 9.0 | 2.7 | 1.3 |
| Manidipine hydrochloride | Inactive | Inactive | Inactive | 26.6 |
| Narasin | Inactive | 44.7 | 11.9 | 7.08 |
| Lestaurtinib | 10.0 | 6.7 | 5.6 | 4.0 |
| Ouabain | Inactive | 0.08 *** | 0.21 | 0.19 |
| Sorafenib tosylate | Inactive | Inactive | 12.6 | 7.9 |
| Sunitinib malate | Inactive | 57.7 | 75.1 | 70.8 |
| Tioconazole | 79.4 | 45.0 | 45.0 | 42.2 |
| Tribromsalan | 39.8 | 15.0 | 3.6 | 2.1 |
| Triclabendazolum | 79.4 | 70.8 | 42.2 | 28.4 |
| Zafirlukast | 44.7 | 42.2 | 37.6 | 33.5 |
Compound efficacy < 25%
Although caspase 3/7 is a major player in cellular apoptosis, recently a growing evidence indicates that the programmed cell death can be in complete absence and independent of caspase activation [24]. The disruption of electrochemical gradient across the mitochondrial membrane is one of the early events during cellular apoptosis. To investigate these drugs which lacked induction of caspase activity, the mitochondrial membrane potential was measured in the cells after 24 h treatment. Among these 6 compounds which were inactive with regard to caspase 3/7 induction, fluorosalan, tribromsalan, sorafenib tosylate, and manidipine depolarized mitochondrial membrane potential with potencies of 1.2, 1.2, 5.7, and 14.7 μM, respectively (Supplemental Table 1). However, digitoxin and ouabain had no effect on the change of mitochondrial membrane potential (Supplemental Table 1).
3.4. Effect of NF-κB inhibitors on cancer cell viability
To investigate the effect of these NF-κB inhibitors on cancer cell viability, the intracellular ATP content of two human cervical cancer cell lines, ME180 and HeLa, was measured after the treatment of the compounds for 5 to 72 h. After 5 h treatment, most compounds showed minimal or no inhibitory effect on cell growth (Tables 2 and 3), except digitoxin and ouabain shown significant inhibition in HeLa cells (Table 3). After 24 – 72 h treatment, 14 (IC50 ≤ 11.2 μM) of the 19 compounds significantly inhibited the growth of ME 180 cells (Table 2). Among these 14 compounds, ectinascidin 743 was the most potent with an IC50 value of 3 nM, followed by bortezomib (40 nM) and chromomycin A3 (60 nM) after 48 to 72 h treatment. However, manidipine, sunitinib malate, tioconazole, triclabendazolum and zafirlukast showed relatively low potency with respect to ME180 cell growth (Table 2). Similar pattern of compound inhibition also observed in HeLa cells (Table 3). A comparison of the activities of these compounds revealed similarities between compound potencies in both ME180 and HeLa cell viability assays after 24 – 72 hour treatment and the NF-κB bla assay. As shown in Fig. 6, the correlation coefficient (R) of the 19 compound potencies in NF-κB bla assay and the cell viability of ME180 cells or HeLa cells after 72 hour treatment are 0.9 (Fig 6A) and 0.91 (Fig 6B), respectively. These data suggest that there is a good correlation between cancer cell death caused by these compounds and NF-κB inhibition.
Table 3.
Compound potency (μM, IC50) with regard to cytotoxicity across different treatment times in HeLa cells
| Compound Name | 5 h | 24 h | 48 h | 72 h |
|---|---|---|---|---|
| Bithionol | 79.3 | 14.6 | 4.3 | 4.6 |
| Bortezomib | Inactive | 13.6 | 0.02 | 0.02 |
| Cantharidin | Inactive | 7.6 | 6.6 | 6.6 |
| Chromomycin A3 | Inactive | 0.13 | 0.10 | 0.04 |
| Daunorubicinum | Inactive | 15.8 | 5.8 | 2.4 |
| Digitoxin | 0.05 | 0.05 | 0.04 | 0.05 |
| Ectinascidin 743 | Inactive | 0.01 | 0.007 | 0.005 |
| Emetine | Inactive | 1.8 | 0.22 | 0.16 |
| Fluorosalan | Inactive | 3.1 | 2.1 | 3.3 |
| Manidipine hydrochloride | Inactive | 12.2 | 14.7 | 13.8 |
| Narasin | 20.2 | 12.2 | 4.6 | 0.1 |
| Lestaurtinib | Inactive | 5.2 | 0.86 | 0.83 |
| Ouabain | 0.09 | 0.06 | 0.05 | 0.05 |
| Sorafenib tosylate | Inactive | 17.7 | 11.7 | 8.3 |
| Sunitinib malate | Inactive | 68.1 | 32.8 | 31.6 |
| Tioconazole | 76.7 | 71.0 | 49.2 | 30.7 |
| Tribromsalan | Inactive | 3.4 | 2.8 | 4.2 |
| Triclabendazolum | 50.2 | 40.3 | 31.6 | 29.4 |
| Zafirlukast | Inactive | 68.1 | 29.3 | 29.3 |
Fig 6.

Potency correlation of 19 NF-kB inhibitors between NF-kB bla assay after 5 h treatment and cell viability assays of ME180 cells (A) and HeLa cells (B) after 72 h treatment.
To further study the effect of compounds on cell death, LDH release which is an indicator of cell necrosis, was measured after 24 h compound treatment in ME180 cells. As shown in Supplemental Table 1, most drugs significantly induced LDH release. Ectinascidin 743 was the most potent with an IC50 value of 20 nM, followed by chromomycin A3 (90 nM) after 24 h treatment. These results suggested that the drugs induce cell death via triggering cell necrosis.
4. Discussion
There are two major NF-κB signaling pathways, canonical pathway (or classical) and the non-canonical pathway (or alternative pathway) [25]. The canonical pathway is mainly activated by TNF-α, IL-1 and LPS. Non-canonical pathway is activated by LTα/β, CD40 ligand and Blys/BAFF, but not by TNF-α, IL-1 and LPS [25]. In the present study, we have identified many drugs in human clinical use that inhibit the canonical NF-κB pathway, by screening a comprehensive collection of approximately 2,800 small molecule drugs that have been approved by regulatory agencies for human use or clinical trials. Some of these drugs are known or suspected NF-κB inhibitors used in the treatment of cancer, but others are used for other indications where NF-κB activity would be neither expected nor necessarily desired. Virtually all of the drugs identified as NF-κB inhibitors produced cytotoxicity on cervical cancer cells, in similar rank order of potency to their NF-κB inhibition. These drugs also induced LDH release from cervical cancer cells indicating the cell death mostly via cell necrosis. Approximately over half of the drugs activated caspase 3/7 and disrupted mitochondrial membrane potential, and only a subset inhibited IκBα phosphorylation, suggesting additional mechanisms for NF-κB inhibition. Among the 19 NF-κB inhibiting drugs identified, ectinascidin 743, digitoxin, ouabain, bortezomib and chromomycin A3 were the most potent NF-κB inhibitors, and ectinascidin 743, bortezomib, chromomycin A3, emetine, daunorubicinum, and lestaurtinib were most potent in the caspase 3/7 and cytotoxicity assays. Emetine, fluorosalan, narasin, lestaurtinib, tribromsalan and bithionol were shown to inhibit IκBα phosphorylation. These results provide new information on activities and mechanisms of action of approved drugs, which may suggest mechanisms of potential novel applications in cancer treatment.
Ectinascidin 743, a marine-derived alkaloid, is approved by the European Commission and US FDA for the treatment of ovarian cancer [26]. It is also in phase II clinical trials for the treatment of prostate and breast cancers, and pediatric sarcomas [26]. There are three tetrahydroisoquinoline moieties in ectinascidin 743 structure. The central carbinolamine of three moieties of ectinascidin 743 covalently binds to DNA by alkylating the N2 aminogroup of quanines at specific sequences, which induces the cancer cells to die [26]. Our data suggest that inhibition of NF-κB signaling, activation of caspase 3/7 and induction of LDH release are mechanisms by which ectinascidin 743 acts.
Chromomycin A3, a naturally occurring antibiotic isolated from S. griseus [27], was also identified as an NF-κB inhibitor from this study. The anticancer property of chromomycin A3 includes inhibiting DNA-dependent RNA synthesis and is clinically used for the treatment of various diseases including chronic myelogenous leukemia, testicular carcinoma and Paget’s disease [27]. We found that chromomycin A3 is similar to ectinascidin 743 in its activation of caspase 3/7, induction of LDH release and growth inhibition of cervical cancer cells. These compounds inhibited the NF-κB signaling pathway in a concentration-dependent manner, but had weak or no effect on IκBα phosphorylation, which suggests that these drug may affect other targets such as degradation of IκBα.
Bortezomib, a proteasome inhibitor, has been approved for the treatment of relapse/refractory multiple myeloma and mantle-cell lymphoma [28]. Bortezomib’s inhibitory effect on myeloma cell growth and survival is mediated through NF-κB signaling because it inhibits the 26S subunit of the proteasome which leads to the inability of IκB degradation and NF-κB activation [28]. This mechanism of action of bortezomib is confirmed in our study where bortezomib inhibited the NF-κB signaling pathway, but had no effect on IκBα phosphorylation.
Emetine, a crystalline alkaloid derived from ipecac root, is known as a protein synthesis inhibitor and DNA interacting agent [29] and is clinically used in the treatment of protozoan infection. Recently the anti-cancer properties of emetine have been shown. Boon-Unge et al [30] reported that the specific mechanism of emetine is to target malignant cells via up regulation of the Bcl-xS splicing variant which promotes apoptosis. We also found that emetine inhibited both NF-κB signaling and IκBα phosphorylation, as well as induced caspase 3/7 activity and had cytotoxicity on in cervical cancer cells.
Several tyrosine kinase inhibitors, such as lestaurtinib, sorafenib tosylate and sunitinib malate, were identified as NF-κB antagonists in this study. These compounds have a broad spectrum of kinase inhibition and are shown to inhibit vascular endothelial growth factor recept and FMS-like tyrosine kinase 3 (FLT3) [31, 32]. Lestaurtinib (CEP-701), sorafenib (BAY-43-9006) and sunitinib (SU-11248) have been in phase I-II clinical trials for treatment of acute myeloid leukemia by targeting VEFGR and FLT3 [32]. In this study we found that lestaurtinib is the most potent NF-κB blocker among these three tyrosine kinase inhibitors. The inhibitory effect of lestaurtinib on NF-κB signaling is via the inhibition of IκBα phosphorylation. In human cervical cancer cells, lestaurtinib induced apoptosis by activating caspase 3/7 and suppressed the growth of these cancer cells. By contrast, sorafenib tosylate had only a moderate inhibitory effect on NF-κB signaling and IκBα phosphorylation. Sorafenib tosylate disrupted mitochondrial membrane potential, but did not activate caspase 3/7, suggesting that the apoptosis induced by this drug may be independent of caspase activation.
Two cardiac glycoside drugs, digitoxin and ouabain, were found to inhibit NF-κB signaling in the NF-κB bla assay and were potent inhibitors of cancer cell growth, but had no effect on IκBα phosphorylation and did not induce caspase 3/7(Table 1). These drugs also had no effect on mitochondrial membrane potential (Supplemental Table 1), suggesting that these drugs may not induce cellular apoptosis. We also found that digoxin, another cardiac glycoside drug in the NPC, inhibited NF-κB signaling in the NF-κB bla assay (data not shown). These results suggest that these drugs may affect other targets in the NF-κB signaling pathway. It has been recently demonstrated that digitoxin and related cardiac glycoside drugs commonly act through blocking TNF-α-dependent binding of the TNF receptor to the TNF receptor-associated death domain in the TNF-α/ NF-κB signaling pathway [33]. Clinically, cardiac glycosides have been used to treat congestive heat failure and arrhythmias via the mechanism of inhibiting Na+/K+ ATPase, which results in increased intracellular calcium concentration [34]. Additionally, these cardiac glycoside drugs had an inhibitory effect on tumor cell proliferation in many types of cancers including lung, breast, colon, prostate and liver [34]. The potential anti-cancer mechanisms of these drugs have been linked to the inhibition of HIF-1α (hypoxia inducible factor 1α) synthesis in Hep3B cells, human liver carcinoma cells [35], the reduction of the p53 levels via the inhibition of p53 protein synthesis in lung cancer cells [36], the inhibition of activation of the TNF-α/ NF-κB signaling pathway [33], and the inhibition of DNA topoisomerases I and II in breast cancer MCF-7 cells [37],
There is considerable ongoing interest in the development of NF-κB inhibitors for cancer treatment, and many new chemical entities that work on various nodes in the NF-κB pathway have been or are being developed. However, the long, expensive, and unpredictable process of new drug development has led to increasing interest in finding new clinical applications for drugs already approved for another indication. In the present study, we have identified several NF-κB inhibitors previously approved for other clinical uses, and several anti-cancer agents not previously appreciated to inhibit the NF-κB pathway. Our identification of novel NF-κB inhibitors among the existing pharmacopeia provides insight into the mechanisms of therapeutic action of existing drugs, as well as suggesting possible new uses for these drugs in diseases characterized by NF-κB pathway overactivity.
Supplementary Material
Acknowledgments
We thank Darryl Leja for illustrations. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chen F, Bower J, Demers JM, Shi X. Upstream Signal Transduction of NF-kB Activation. Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2002;6:345–69. [Google Scholar]
- [2].Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–4. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- [3].Ahn KS, Aggarwal BB. Transcription factor NF-kappaB: a sensor for smoke and stress signals. Ann N Y Acad Sci. 2005;1056:218–33. doi: 10.1196/annals.1352.026. [DOI] [PubMed] [Google Scholar]
- [4].Lee CH, Jeon YT, Kim SH, Song YS. NF-kappaB as a potential molecular target for cancer therapy. Biofactors. 2007;29:19–35. doi: 10.1002/biof.5520290103. [DOI] [PubMed] [Google Scholar]
- [5].Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- [6].Kumar A, Takada Y, Boriek AM, Aggarwal BB. Nuclear factor-kappaB: its role in health and disease. J Mol Med. 2004;82:434–48. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- [7].Baldwin AS., Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- [8].Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005;24:301–13. doi: 10.1007/s10555-005-1579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Schon M, Wienrich BG, Kneitz S, Sennefelder H, Amschler K, Vohringer V, et al. KINK-1, a novel small-molecule inhibitor of IKKbeta, and the susceptibility of melanoma cells to antitumoral treatment. J Natl Cancer Inst. 2008;100:862–75. doi: 10.1093/jnci/djn174. [DOI] [PubMed] [Google Scholar]
- [10].Tiedemann RE, Schmidt J, Keats JJ, Shi CX, Zhu YX, Palmer SE, et al. Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-kappaB with antimyeloma activity in vitro and in vivo. Blood. 2009;113:4027–37. doi: 10.1182/blood-2008-09-179796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yamamoto M, Horie R, Takeiri M, Kozawa I, Umezawa K. Inactivation of NF-kappaB components by covalent binding of (-)-dehydroxymethylepoxyquinomicin to specific cysteine residues. J Med Chem. 2008;51:5780–8. doi: 10.1021/jm8006245. [DOI] [PubMed] [Google Scholar]
- [12].Sykes JA, Whitescarver J, Jernstrom P, Nolan JF, Byatt P. Some properties of a new epithelial cell line of human origin. J Natl Cancer Inst. 1970;45:107–22. [PubMed] [Google Scholar]
- [13].Yasgar A, Shinn P, Jadhav A, Auld D, Michael S, Zheng W, et al. Compound Management for Quantitative High-Throughput Screening. JALA Charlottesv Va. 2008;13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xia M, Huang R, Witt KL, Southall N, Fostel J, Cho MH, et al. Compound cytotoxicity profiling using quantitative high-throughput screening. Environ Health Perspect. 2008;116:284–91. doi: 10.1289/ehp.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–60. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- [16].Fiedler MA, Wernke-Dollries K, Stark JM. Inhibition of TNF-alpha-induced NF-kappaB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol. 1998;19:259–68. doi: 10.1165/ajrcmb.19.2.3149. [DOI] [PubMed] [Google Scholar]
- [17].Robers MB, Horton RA, Bercher MR, Vogel KW, Machleidt T. High-throughput cellular assays for regulated posttranslational modifications. Anal Biochem. 2008;372:189–97. doi: 10.1016/j.ab.2007.09.012. [DOI] [PubMed] [Google Scholar]
- [18].Huang R, Southall N, Cho MH, Xia M, Inglese J, Austin CP. Characterization of diversity in toxicity mechanism using in vitro cytotoxicity assays in quantitative high throughput screening. Chem Res Toxicol. 2008;21:659–67. doi: 10.1021/tx700365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–8. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xia M, Huang R, Guo V, Southall N, Cho MH, Inglese J, et al. Identification of compounds that potentiate CREB signaling as possible enhancers of long-term memory. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0813020106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- [22].Sung MH, Bagain L, Chen Z, Karpova T, Yang X, Silvin C, et al. Dynamic effect of bortezomib on nuclear factor-kappaB activity and gene expression in tumor cells. Mol Pharmacol. 2008;74:1215–22. doi: 10.1124/mol.108.049114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].He TP, Mo LE, Liang NC. [Inhibitory effect of cantharidin on invasion and metastasis of highly metastatic ovarian carcinoma cell line HO-8910PM] Ai Zheng. 2005;24:443–7. [PubMed] [Google Scholar]
- [24].Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- [25].Karin M, Yamamoto Y, Wang QM. The IKK NF-kappa B system: a treasure trove for drug development. Nat Rev Drug Discov. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- [26].Schoffski P, Dumez H, Wolter P, Stefan C, Wozniak A, Jimeno J, et al. Clinical impact of trabectedin (ecteinascidin-743) in advanced/metastatic soft tissue sarcoma. Expert Opin Pharmacother. 2008;9:1609–18. doi: 10.1517/14656566.9.9.1609. [DOI] [PubMed] [Google Scholar]
- [27].Calabresi PCB. Chemotherapy of neoplastic diseases. McGraw-Hill; New York: 1996. [Google Scholar]
- [28].Pascal L, Gay J, Willekens C, Wemeau M, Balkaran S, Robu D, et al. Bortezomib and Waldenstrom’s macroglobulinemia. Expert Opin Pharmacother. 2009;10:909–16. doi: 10.1517/14656560902800160. [DOI] [PubMed] [Google Scholar]
- [29].Wink MST, Latz-Bruning B. Modes of action of allelochemical alkaloids: interaction with neuroreceptors, DNA, and other molecular targets. J Chem Ecology. 1998;24:1881–937. [Google Scholar]
- [30].Boon-Unge K, Yu Q, Zou T, Zhou A, Govitrapong P, Zhou J. Emetine regulates the alternative splicing of Bcl-x through a protein phosphatase 1-dependent mechanism. Chem Biol. 2007;14:1386–92. doi: 10.1016/j.chembiol.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mori S, Cortes J, Kantarjian H, Zhang W, Andreef M, Ravandi F. Potential role of sorafenib in the treatment of acute myeloid leukemia. Leuk Lymphoma. 2008;49:2246–55. doi: 10.1080/10428190802510349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tickenbrock L, Muller-Tidow C, Berdel WE, Serve H. Emerging Flt3 kinase inhibitors in the treatment of leukaemia. Expert Opin Emerg Drugs. 2006;11:153–65. doi: 10.1517/14728214.11.1.153. [DOI] [PubMed] [Google Scholar]
- [33].Yang Q, Huang W, Jozwik C, Lin Y, Glasman M, Caohuy H, et al. Cardiac glycosides inhibit TNF-alpha/NF-kappaB signaling by blocking recruitment of TNF receptor-associated death domain to the TNF receptor. Proc Natl Acad Sci U S A. 2005;102:9631–6. doi: 10.1073/pnas.0504097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- [35].Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–86. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang ZZM, Li Z, Li R, Jia L, Xiong X, Southall N, Wang S, Xia M, Austin CP, Zheng W, Xie Z, Sun Y. Cardiac glycosides reduce the p53 levels via inhibiting p53 protein synthesis: rescued by Src and MAPK inhibitors. Cancer Res. 2009;69:6556–64. doi: 10.1158/0008-5472.CAN-09-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bielawski K, Winnicka K, Bielawska A. Inhibition of DNA topoisomerases I and II, and growth inhibition of breast cancer MCF-7 cells by ouabain, digoxin and proscillaridin A. Biol Pharm Bull. 2006;29:1493–7. doi: 10.1248/bpb.29.1493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.