

Abstract
Background & Aims
Recurrent hepatitis C with ensuing fibrosis is the leading cause of liver allograft loss. We investigated whether histologic features in early post-transplant liver biopsies could predict the rate of fibrosis progression in this population.
Methods
From 1999 to 2007, 476 liver transplants were performed for hepatitis C at our center. We reviewed all available post transplant biopsies for these patients; patients were categorized as rapid, intermediate, or slow fibrosers based on their METAVIR fibrosis score at 24 months. Stage F0 biopsies for rapid and slow fibrosers were analyzed histologically and immunohistochemically.
Results
We identified 52 rapid fibrosers and 61 slow fibrosers in our cohort. There was a significant increase in the fibrosis progression rate in the group transplanted between 2003 to 2007 compared with 1999 to 2002. The course of fibrosis progression was determined early in the post-transplant period and the rate was constant. Rapid fibrosers had more hepatocyte apoptosis than slow fibrosers (P = 0.001) but no difference in hepatitis activity based on analysis of stage F0 biopsies. Rapid fibrosers also experienced more episodes of acute rejection following transplantation (P< 0.001). CK19 and vimentin expression on F0 stage biopsies could distinguish rapid from slow fibrosers (CK19: AUC 0.71, P = 0.0034; vimentin: P =0.0219).
Conclusions
CK19, vimentin, and hepatocellular apoptosis are promising early markers of rapid fibrosis progression in patients transplanted for hepatitis C. The rate of fibrosis progression is established early in the post-transplant period; this initial rate dictates long-term outcome.
Introduction
Hepatitis C (HCV)-related cirrhosis remains the primary indication for liver transplantation in the United States.1-3 Following transplant, the recurrence of HCV is inevitable4 and occurs within 3-4 weeks.5, 6 The natural history of HCV in immunocompromised transplant patients is different than in immunocompetent non-transplanted patients because fibrosis develops at an accelerated rate following transplantation.7-13 Progressive fibrosis is a significant cause of graft failure and post-transplantation mortality, with cirrhosis affecting between 10% and 44% of patients.3, 8, 10, 12, 14
The time course of fibrosis among HCV-infected patients is highly variable in both the non-transplant and post-transplant populations. The concept of “rapid fibrosers” emerged from the observation that a sub-population of non-transplanted HCV patients (4.6%) developed cirrhosis before age 50, progressing at a more rapid rate than the average patient.15 In the transplant setting, a subset of “rapid fibrosers” has also been recognized. Although definitions of rapid are not universally agreed upon, one study defined rapid fibrosis as a progression rate of more than 0.8 units of Ishak fibrosis stage per year.9
Alarmingly, the rate of fibrosis appears be increasing in more recently transplanted patients. Berenguer et al. showed that the median rate of fibrosis progression increased from 0.13 METAVIR fibrosis stage units/year in patients transplanted in 1988 to 0.64 stage units/year in patients transplanted in 1996.7 Although several risk factors for rapid progression have been identified, including increased donor age, certain immunosuppressive regimens, inflammation on biopsy in the first year post-transplant, elevated transaminase levels for 3 consecutive months, episodes of rejection, and histologic recurrence of HCV within 6 months, predicting a given patient's course in advance is difficult.7, 9, 12, 13, 16-24
The aim of our single-center retrospective study was to identify histologic predictors of rapid fibrosis progression. Because the ductular reaction and the expression of mesenchymal proteins by biliary epithelial cells have been associated with some forms of particularly rapid fibrosis such as biliary atresia,25 we hypothesized that increases in certain epithelial and mesenchymal markers might identify rapid progressors.
Materials and Methods
Patient Population
This study was approved by the University of Pennsylvania IRB. The cohort was composed of all HCV-infected patients who underwent liver transplant at the University of Pennsylvania between 1999-2007. During this time, 996 total liver transplants were performed, of which 476 (48%) were in patients with chronic HCV. Twenty-three patients had two transplants between 1999-2007; disease progression in each transplanted liver was examined independently. Our immunosuppression regimen was tacrolimus based, with dose reductions in the setting of renal dysfunction. Corticosteroids were used for the first 3-6 months. The use of azathioprine and mycophenolate mofetil was not routine. Protocols for antiviral therapy were standardized in 2003, after which patients did not receive antiviral therapy unless METAVIR stage F3 fibrosis was documented on biopsy, or unless they had non-genotype 1 virus or a clinical (as opposed to histologic) diagnosis of fibrosing cholestatic hepatitis (FCH). Treatment data were only available for the post-2003 cohort. Twenty-six patients in this group received treatment for FCH or non-genotype 1 virus. The conclusions from the analyses in Figs. 1, 2, and S1 were not changed by the exclusion of these cases, and they are therefore included in all analyses presented here.
Figure 1.
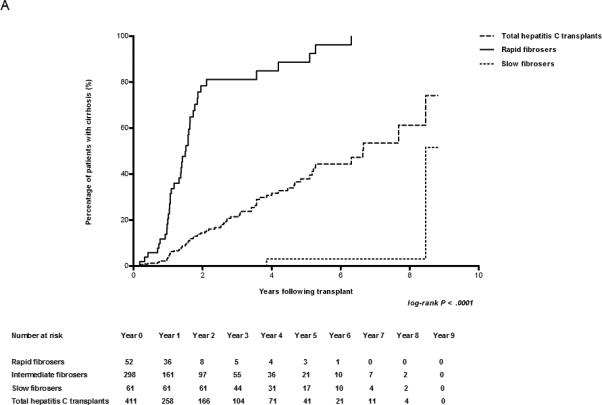
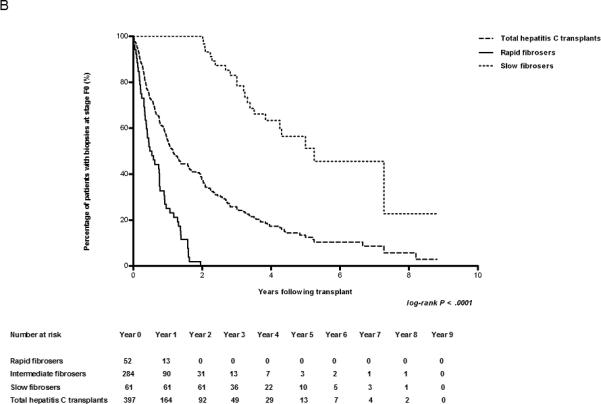
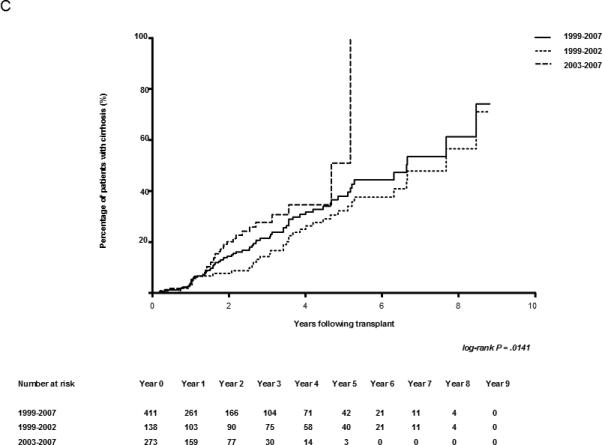
Early biopsies predict the rate of fibrosis. (A) Kaplan-Meier estimates of the development of cirrhosis (METAVIR Stage F4) in HCV-infected patients after transplant. The number of cases represented within each cohort is 440, 52, and 61 for the entire cohort, rapid fibrosers, and slow fibrosers, respectively. (B) Kaplan-Meier estimates of the development of fibrosis (METAVIR Stages F1-4) in F0 transplant livers of HCV-infected patients. Patients with baseline fibrosis on time zero biopsy were excluded. The number of cases represented within each cohort is 421, 52, and 61 for the entire cohort, rapid fibrosers, and slow fibrosers. (C) Kaplan-Meier estimates of cirrhosis progression according to transplant year in all patients undergoing liver transplantation secondary to HCV. The number of transplant cases represented is 141 for the entire cohort transplanted between 1999-2002, and 299 for the entire cohort transplanted between 2003-2007. The log-rank P value of 0.0141 reflects the comparison in cirrhosis progression among groups from 1999-2002 and 2003-2007. The total cohort includes rapid, slow, and intermediate fibrosers. The number at risk for each year is shown below the graph.
Figure 2.


ROC analysis of CK19 expression (A) and Hsp47 expression (B) in stage F0 biopsies of rapid vs. slow fibrosers.
Protocol biopsies were instituted in 2003 and were performed once at months 4 through 6, then yearly beginning at 1 year post-transplant. All post-transplant biopsies from the entire cohort of HCV-infected patients (476 transplants; 1272 biopsies) were reviewed to categorize patients as rapid fibrosers or slow fibrosers. We defined rapid as reaching METAVIR stage F3 or F4 before or at 24 months post transplant and slow as remaining at METAVIR stage F0 or F1 at or beyond 24 months post transplant. These definitions were selected because a) biopsies are inaccurate, and both F3 and F4 represent clinically meaningful significant fibrosis; b) antiviral therapy (except as noted above) was started when patients reached F3, so therapy did not affect the categorization of a given patient; and c) the development of significant fibrosis by 24 months is a clinically meaningful indicator of rapid progression. We identified 52 cases as rapid and 61 as slow fibrosers. Twenty cases were excluded from assignment to either category because baseline fibrosis was detected in the time zero biopsy, but were still included in the study of the entire HCV transplant population. These cases were included in the analysis for Fig. 1A, but not Fig. 1B. The 274 cases (57.6%) without time zero biopsies were assumed to have received grafts without baseline fibrosis (F0). Thirty-six (7.6%) of the total population had no adequate biopsies (defined as including at least five portal tracts) and were not included in the analyses. Two hundred seventy-five cases (57.8%) did not have follow-up biopsies beyond 24 months including 37 rapid fibrosers (71%) who had stage F3 or F4 disease on the last available biopsy. There were 232 post-transplant biopsies obtained and reviewed in the rapid fibroser subgroup (20 from time zero biopsies), and 164 within the slow fibroser subgroup (14 from time zero). Analyses of the subgroup transplanted after the initiation of protocol biopsies (2003-2007) are shown in the Supplemental Data (Figs. S1, S3, S4; Tables S1, S4, S5, S7, S8) and, with the exception of vimentin immunostaining, are not significantly different from the analyses of total cases from 1999-2007. Cox regression analyses for the post-2003 subgroup could not be carried out due to insufficient numbers of patients.
Histology
All post-transplant biopsies for the 52 rapid and 61 slow fibrosers were evaluated for acute and chronic rejection (see Supplemental Methods). Thirty-two rapid and 46 slow fibrosers had METAVIR F0 or F1 fibrosis biopsies available for further study. These were evaluated histologically as described in the Supplemental Methods.
Immunohistochemistry and sirius red staining
F0 and F1 biopsies from rapid and slow fibrosers were stained with the collagen binding dye sirius red and with antibodies against Hsp47, CK19, and vimentin (see Supplemental Methods). Immunoreactivity for CK19 and Hsp47 was scored on a 0-3 scale (see Fig. 3A,B), while vimentin was scored on a binary scale (baseline =0, increased =1; see Fig. 3C,D) in a blinded fashion by the same pathologist.
Figure 3.

CK19 and vimentin immunostaining are increased in rapid compared with slow fibrosers. Representative F0 biopsies (×200) from a slow (A,C) and rapid (B,D) fibroser, immunostained for CK19 (A,B) and vimentin (C,D) expression. CK19 staining was scored as 0 (A) through 3 (B); vimentin was scored as 0 (C) or 1 (D).
Statistics
See Supplemental Methods.
Results
The characteristics of our study population are shown in Tables 1 and S1. The same transplant team remained in place during the entire study period, and post-transplant patient and graft survival were consistently above the national average (data not shown). From the 476 transplants performed secondary to HCV cirrhosis, 52 (10.92%) rapid and 61 (12.81%) slow fibrosers were identified. The mean follow-up time in years (± SD) was 2.11 (± 1.99) for the entire HCV cohort, 1.91 (± 1.39) for rapid fibrosers, and 4.20 (± 1.70) for slow fibrosers. Though the rapid fibroser population consisted of a greater percentage of women (n=14; 27%) than the slow fibroser population (n=7; 11%), this difference did not reach statistical significance (P = 0.0513). There was no statistically significant difference in known recipient race between rapid and slow fibrosers (P= 0.8894). There were statistically significant differences between the mean age at time of transplant for rapid vs. slow fibrosers (49.75 ± 8.20 for rapid vs. 52.79 ± 7.33 for slow, P=0.0399) andthe mean donor age (44.81 ± 17.96 for rapid vs. 37.81 ± 2.26 for slow, P=0.0447). There was no statistically significant difference in the percentage of patients with co-existing alcoholic liver disease or hepatitis B for rapid vs. slow fibrosers (P=0.1623 for alcoholic cirrhosis, P=0.4602 for hepatitis B).
Table 1.
Demographics of transplant recipients.
| Total Hepatitis C Transplants | Rapid Fibrosers | Slow Fibrosers | ||||
|---|---|---|---|---|---|---|
| P valuea | ||||||
| N (Liver transplants secondary to hepatitis C) | 476 | 52 | 61 | |||
| Percent of hepatitis C transplant population | 100 | 10.92 | 12.81 | |||
| Gender | ||||||
| N (%) | ||||||
| Female | 99 (20.80) | 14 (26.92) | 7 (11.48) | .0513 | ||
| Male | 377 (79.20) | 38 (73.08) | 54 (88.52) | |||
| Mean age ± SD (yr.) | 51.79 ± 7.67 | 49.75 ± 8.20 | 52.79 ± 7.33 | .0399 | ||
| Race | ||||||
| N (%) | ||||||
| Black | 80 (16.81) | 11 (21.15) | 11 (18.03) | .8894b | ||
| Hispanic | 28 (5.88) | 2 (3.85) | 2 (3.28) | |||
| White | 343 (72.06) | 37 (71.15) | 46 (75.41) | |||
| Other | 25 (5.25) | 2 (3.85) | 2 (3.28) | |||
| Co-existing alcoholic cirrhosis | 135 (28.36) | 13 (25) | 23 (37.70) | .1623 | ||
| Co-existing hepatitis B | 7 (1.47) | 1 (1.92) | 0 (0) | .4602 |
P has been calculated between rapid and slow fibrosers.
P has been calculated between rapid and slow fibrosers only for known recipient race.
Mean age was compared using a student t-test; the remaining data was compared using Chi Square or Fisher's exact test.
The rate of progression to cirrhosis (METAVIR F4) for rapid versus slow fibrosers is illustrated in Figs. 1A and S1A, with the rate of progression for the total cohort shown for comparison. By year 8 following transplantation, 61% of the total population (rapid, slow, and intermediate groups) with biopsies available to that point had cirrhosis. By year 2, 78% of rapid fibrosers had cirrhosis; in contrast, only 4% of slow fibrosers were cirrhotic as late as year 8. By the end of the first year post-transplant, the rate of cirrhosis approximated 20, 3.6, and 0 (percent with cirrhosis/time in years) for rapid fibrosers, the entire cohort, and slow fibrosers, respectively. Based on these Kaplan-Meier data, the median time to cirrhosis (F4) was 1.5 years for rapid fibrosers, 6.6 years for the total cohort, and could not be calculated for slow fibrosers due to an insufficient number of events. At year 8.5, we noted a sharp rise in the percentage of slow fibrosers with cirrhosis due to a single atypical patient who developed mild portal fibrosis at year 7 and cirrhosis at year 8.5 in the absence of any significant co-morbidities. Exclusion of this patient from the analysis did not significantly affect our results.
Analyzing the rate of progression from F0 to any stage of fibrosis (F1-F4) also demonstrated that the degree of fibrosis on biopsies within the first year is predictive of the rate of subsequent fibrosis progression. We used the Kaplan-Meier method to calculate the probability of remaining at F0 at years 2, 4, 6, and 8 if the year 1 post-transplant biopsy was at stage F0. For the total HCV cohort biopsied the probabilities were 0.69, 0.32, 0.19, and 0.05, respectively (Figs. 1B; see also S1B). By definition, the probability of a rapid fibroser having no fibrosis beyond year 2 is 0. Slow fibrosers have probabilities of being at stage F0 at years 2, 4, 6, and 8 of 1, 0.63, 0.45, and 0.23, respectively. If the single atypical slow fibroser is excluded from this analysis, the probability of a slow fibroser remaining F0 at year 8 is 0.43. These findings suggest that rapid or slow fibrosis progression is determined early, and that these classifications are maintained in the years following transplantation.
Stratification by transplant era (1999-2002 vs. 2003-2007) demonstrated a significant increase in the rate of fibrosis progression in more recent years (Fig. 1C; log-rank P = 0.0160). The median time to cirrhosis for HCV patients was 7.68 years for patients transplanted between 1999-2002 vs. 4.67 years for patients transplanted between 2003-2007. In the unadjusted Cox analysis of the entire cohort, the risk of developing cirrhosis was 94% higher in the group transplanted from 2003-2007 (HR 1.94, 95% CI 1.14 – 3.32).
Rapid fibrosers had more episodes of acute and chronic rejection than slow fibrosers (Table 2). An adjusted Cox regression analysis was performed to determine whether donor age, transplant era, and rejection type and severity could explain the differences in progression between rapid and slow fibrosers (Table 2). Before adjusting for these factors, rapid fibrosers, not unexpectedly, were found to be more likely to progress to cirrhosis than slow fibrosers (HR 132.30, 95% CI 17.64 – 950.73). Adjustment for donor age, recipient age, and transplant era did not significantly change the differences noted between rapid and slow fibrosers. An interaction between these factors was not found. The findings were similar when intermediate fibrosers were included in the analysis (Table S2). When severity and frequency of acute rejection episodes and an assessment of chronic rejection were added to the model, there was only a modest change in the hazard ratio (HR 115.07, 95% CI 15.19 – 871.51). While the variability of these estimates was large given the small sample sizes of the rapid and slow fibroser groups, it does suggest that factors such as donor age, recipient age, transplant era, and rejection episodes influence progression to cirrhosis but do not completely explain the differences in rates of progression.
Table 2.
Hazard ratios for time to cirrhosis in rapid and slow fibrosers.
| Variable | Unadjusted Hazard Ratio (95% CI) | P Value | Adjusted Hazard Ratio (95% CI)† | P Value |
|---|---|---|---|---|
| Fibrosis Progression | ||||
| Slow | Reference | Reference | ||
| Rapid | 132.30 (17.64-950.73) | < 0.001 | 111.19 (14.64-844.47) | <0.001 |
| Gender | ||||
| Male | Reference | Reference | ||
| Female | 2.40 (1.23-4.66) | 0.010 | 1.15 (0.50-2.65) | 0.736 |
| Recipient Agea | 0.97 (0.94-1.01) | 0.123 | 1.00 (0.95-1.04) | 0.846 |
| Recipient Race | ||||
| White | Reference | |||
| Black | 1.11 (0.52-2.36) | 0.783 | ||
| Other | 1.37 (0.48-3.91) | 0.559 | ||
| Transplant Era | ||||
| 1992-2002 | Reference | Reference | ||
| 2003-2007 | 2.18 (1.09-4.37) | 0.027 | 1.20 (0.50-2.91) | 0.684 |
| Donor Agea | 1.02 (1.00-1.04) | 0.030 | 1.02 (0.99-1.04) | 0.127 |
| Acute Rejection Scoreb | 2.55 (1.38-4.71) | 0.003 | 1.17 (0.56-2.44) | 0.669 |
| Acute Rejection Episodesc | 1.55 (1.15-2.09) | 0.004 | ||
| Chronic Rejectiond | 3.55 (1.87-6.73) | <0.001 | 1.43 (0.65-3.14) | 0.378 |
| Alcohol | 0.74 (0.37-1.48) | 0.399 |
Adjusted model includes fibrosis progression, gender, recipient age, transplant era, donor age, rejection score modeled as a time varying covariate and chronic rejection modeled as a dichotomous variable.
Hazard ratio corresponds to a one year increase in age.
Acute rejection score is modeled as a time varying covariate with the severity of the episode noted at each biopsy being reflected at that time point.
Hazard ratio corresponds to a one unit increase in the number of acute rejection episodes.
Hazard ratio represents the risk of progression to cirrhosis if a subject ever had chronic rejection noted on a liver biopsy throughout the study period.
We carried out a detailed histologic analysis to identify features on F0 and F1 stage biopsies predictive of rapid fibrosis progression. Using the last available stage F0 biopsies and F1 biopsies for each case, we analyzed multiple histologic parameters, including inflammatory activity and apoptosis (Tables 3A, S4). F0 biopsies of rapid fibrosers demonstrated more hepatocellular apoptosis (mean apoptotic cells/hpf: 0.30 ± 0.38 rapid fibrosers vs. 0.08 ± 0.16 slow fibrosers, P = 0.0012) than those of slow fibrosers, consistent with research showing that apoptosis induces activation of fibrogenic cells.26 There was no difference between rapid and slow fibrosers in any of the other histological parameters. There were no significant differences in F1 biopsies between the two groups (Tables S3, S5).
Table 3A.
Histologic variables in stage F0 biopsies of rapid and slow fibrosers.
| Rapid Fibrosers | Slow Fibrosers | P value | ||
|---|---|---|---|---|
| N with available stage 0 biopsy for review | 26 | 44 | ||
| Portal tract density | Median (25-75 percentiles) | 1 (0.75-2.0) | 1 (1.0-2.0) | .7433 |
| Portal tract activity | Median (25-75 percentiles) | 1 (0-2.0) | 0.5 (0-1.0) | .3442 |
| Lobular activity | Median (25-75 percentiles) | 1 (0-2.0) | 1 (0.25-2.0) | .9125 |
| N with steatosis | ||||
| Present (%) | 4 (15.38) | 16 (36.36) | .0992 | |
| Absent (%) | 22 (84.62) | 28 (63.64) | ||
| N with plasma cells | ||||
| Present (%) | 2 (7.69) | 3 (6.82) | 1 | |
| Absent (%) | 24 (92.31) | 41 (93.18) | ||
| Apoptotic cells/high power field | Mean ± SD | 0.30 ± 0.38 | 0.08 ± 0.16 | .0012 |
The first 3 parameters were scored from 0-3, with 3 being the highest score.
To identify other histologic predictors of rapid vs. slow fibrosis progression, we stained the stage F0 and F1 biopsies of rapid and slow fibrosers for expression of the collagen chaperone Hsp47,27 the epithelial cell marker CK19, and the mesenchymal marker vimentin (Figs. 2, 3, S2, S3, and S4). CK19 expression was significantly greater in the F0 (but not F1) biopsies of rapid versus slow fibrosers (Stage 0: Fig. 2A, P = 0.0034, Stage F1: Fig. S2A; see also Figs. 3, S3A, S4A). Note that there was no acute inflammation, cholestasis, or other features of biliary injury. In contrast, immunostaining with Hsp47 revealed no difference in expression between rapid and slow fibrosers (Stage 0: Fig. 2B, P = 0.1278; Stage F1: Fig. S2B; see also Figs. S3B, S4B). Similarly, sirius red staining for collagen did not distinguish between rapid and slow fibrosers (Stage 0: Table 3B, P=0.1358; Stage F1: Table S6; see also Tables S7, S8). Increased vimentin staining was predictive of rapid fibrosis on F0 but not F1 biopsies from the rapid and slow fibroser groups from 1999-2007 (Stage 0: Table 3B, 81% of rapid fibrosers vs. 52% of slow fibrosers, P= 0.022, Stage 1: Table S6; see also Fig. 3 and Tables S7, S8). Vimentin staining was not significantly different between rapid and slow fibrosers transplanted from 2003 onward, likely due to the small sample size (Table S7, P = 0.2691).
Table 3B.
Comparison of vimentin and Sirius red staining in F0 stage biopsies of rapid versus slow fibrosers.
| Stain | Rapid Fibrosers | Slow Fibrosers | P value | |
|---|---|---|---|---|
| Sirius red | Mean ± SD | 5501 ± 4608 | 4242 ± 2242 | .1358 |
| Vimentin | N with score = 0 | 5 | 21 | .0219 |
| N with score = 1 | 21 | 23 |
Discussion
In summary, we have carried out the largest single-institution retrospective study of HCV patients post-transplant to date. We show that the rate of fibrosis on a population basis is constant throughout the post-transplant period and that the course of fibrosis is determined early post-transplant. Although several studies (incorporating patients transplanted from 1988-2002) have demonstrated that the presence or absence of significant fibrosis on a biopsy at one year post-transplant was predictive of graft survival and the development of cirrhosis, the predictive value was less dramatic than in our population.9, 12, 13 This may be due to an increasing rate of fibrosis in patients transplanted more recently, as in our study. We found that as early as six months post transplant, histologic parameters alone can identify which patients are fated for rapid fibrosis progression. The presence of fibrosis on a biopsy in the first year predicted continued rapid progression, while the absence of fibrosis in the first year predicted that a patient would have little or no fibrosis at year 8, findings that have important implications for counseling patients as well as starting antiviral and, potentially, antifibrotic therapy. This work overall demonstrates that fibrosis in early post-transplant biopsies is highly predictive of long-term outcome and has led us to hypothesize that there are critical molecular and cellular events with long-lasting effects put into play early. The data support the use of protocol biopsies at one year and potentially as early as six months after transplant in HCV patients, although prospective trials that include clinical decompensation as an outcome are required before making a definitive recommendation.
Consistent with other studies,3, 7, 8, 12, 14, 18, 22 we found that the rate of fibrosis progression in HCV patients post transplant has increased significantly over the last decade, with the risk of developing cirrhosis being 94% higher in the group transplanted between 2003-2007 than in the group transplanted between 1999-2002. Although our study was not designed to identify clinical causes of increased progression, the literature suggests that a combination of declining organ quality, altered viral subspecies, and changes in immunosuppressive regimens may be responsible. Donor age is clearly associated with increased fibrosis progression,7-9, 12, 14, 18, 22-24 and the increased use of older donor allografts to expand the organ pool has likely contributed to the increased rate of fibrosis we observed. Cox regression analysis suggests, however, that donor age contributes to but does not entirely explain the recent increase in fibrosis progression in our population.
Post-transplant immunosuppressive regimens have been implicated in fibrosis progression, with associations between fibrosis and tacrolimus, mycophenolate mofetil, and the number of steroid boluses.7, 12, 14, 23 We found that the number of acute rejection episodes was associated with fibrosis progression, as has been previously reported.13, 24 Surprisingly, we did not find that rapid fibrosis was associated with inflammation, in contrast to the findings of other studies.9, 13, 16, 18 The reason for this inconsistency is not clear, but might be attributable to differences in immunosuppressive practices or anti-viral management at our center compared with others as well as differences in study design including the examination of earlier stage (F0) biopsies in our study.
One of our most intriguing findings is an increase in expression of the biliary epithelial cell marker CK19 in rapid compared with slow fibrosers. This suggests a possible role for bile ductular proliferation in fibrosis progression post-transplant. Clouston and colleagues have previously shown a correlation between the extent of the ductular reaction and the stage of fibrosis in untransplanted HCV patients.28 Similarly, a vigorous ductal reaction has been associated with features of the epithelial to mesenchymal transition (EMT) in a variety of diseases with typically rapid rates of fibrosis progression.25 Interestingly, we found that expression of the mesenchymal marker vimentin was greater in the stage F0 biopsies of rapid fibrosers than in slow fibrosers. Vimentin appears to stain a combination of parenchymal cells (likely hepatic stellate cells), portal mesenchymal cells, and some (although not all) biliary epithelial cells (Fig. 3). Neither sirius red staining nor Hsp47 expression correlated with fibrosis progression in our study, suggesting that the F0 biopsies of rapid fibrosers represent a stage before the onset of early collagen synthesis. The increased vimentin staining may therefore reflect both early biliary injury (the ductular reaction) as well as increased hepatic stellate cell activation preceding matrix deposition.
One limitation of our study was the use of biopsies to stage fibrosis. Although they are the current standard, biopsies are notably inaccurate. Additionally, the greater number of biopsies performed in rapid fibrosers compared with slow fibrosers, particularly before the initiation of protocol biopsies, is a potential indicator of bias, as it suggests that the rapid fibroser group was monitored more closely. Our study was retrospective and our findings, particularly that CK19 and vimentin may be clinically relevant predictors of rapid fibrosis progression, need to be confirmed in a prospective trial. Although this study was designed to identify histological and not clinical predictors of rapid progression, a prospective trial with collection of detailed clinical data would provide additional insight into fibrosis in this population and would demonstrate whether the findings from our single center are generalizable to other centers.
In conclusion, our study of fibrosis in the adult population after liver transplantation for HCV has important biological and clinical implications, including that post-transplant fibrosis results from progressive cellular processes set into motion early. Biopsies of patients within the first year warrant particular attention, as the presence or absence of fibrosis and hepatocellular apoptosis are predictive of fibrosis progression in subsequent years. As compared with patients who have fibrosis on a biopsy in the first year, patients without fibrosis after one year are less likely to develop subsequent fibrosis. Patients with immunoreactivity for CK19 and vimentin on stage 0 biopsies, episodes of rejection, and higher donor age are particularly worrisome and therefore may warrant more intense management.
Supplementary Material
Acknowledgements
We gratefully acknowledge the assistance and contributions of Drs. George Makar, Colleen Brensinger, Abraham Shaked, Kim Olthoff, Gerald Wertheim, Sriram Venneti, Rosalyn Diaz, and Helen Zhu.
Grant support: NIH grant DK-58123 to R.G.W. Pathology and Laboratory Medicine intradepartmental grant support to E.E.F.
Abbreviations
- CK19
cytokeratin 19
- HCV
hepatitis C virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No conflicts of interest exist.
References
- 1.United Network for Organ Sharing Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients. 2007.
- 2.Charlton M, Seaberg E, Wiesner R, et al. Predictors of patient and graft survival following liver transplantation for hepatitis C. Hepatology. 1998;28:823–30. doi: 10.1002/hep.510280333. [DOI] [PubMed] [Google Scholar]
- 3.Feray C, Caccamo L, Alexander GJ, et al. European collaborative study on factors influencing outcome after liver transplantation for hepatitis C. European Concerted Action on Viral Hepatitis (EUROHEP) Group. Gastroenterology. 1999;117:619–25. doi: 10.1016/s0016-5085(99)70454-3. [DOI] [PubMed] [Google Scholar]
- 4.Wright TL, Donegan E, Hsu HH, et al. Recurrent and acquired hepatitis C viral infection in liver transplant recipients. Gastroenterology. 1992;103:317–22. doi: 10.1016/0016-5085(92)91129-r. [DOI] [PubMed] [Google Scholar]
- 5.Ballardini G, De Raffele E, Groff P, et al. Timing of reinfection and mechanisms of hepatocellular damage in transplanted hepatitis C virus-reinfected liver. Liver Transpl. 2002;8:10–20. doi: 10.1053/jlts.2002.30141. [DOI] [PubMed] [Google Scholar]
- 6.Konig V, Bauditz J, Lobeck H, et al. Hepatitis C virus reinfection in allografts after orthotopic liver transplantation. Hepatology. 1992;16:1137–43. doi: 10.1002/hep.1840160506. [DOI] [PubMed] [Google Scholar]
- 7.Berenguer M, Ferrell L, Watson J, et al. HCV-related fibrosis progression following liver transplantation: increase in recent years. J Hepatol. 2000;32:673–84. doi: 10.1016/s0168-8278(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 8.Berenguer M, Prieto M, Rayon JM, et al. Natural history of clinically compensated hepatitis C virus-related graft cirrhosis after liver transplantation. Hepatology. 2000;32:852–8. doi: 10.1053/jhep.2000.17924. [DOI] [PubMed] [Google Scholar]
- 9.Firpi RJ, Abdelmalek MF, Soldevila-Pico C, et al. One-year protocol liver biopsy can stratify fibrosis progression in liver transplant recipients with recurrent hepatitis C infection. Liver Transpl. 2004;10:1240–7. doi: 10.1002/lt.20238. [DOI] [PubMed] [Google Scholar]
- 10.Gane EJ, Portmann BC, Naoumov NV, et al. Long-term outcome of hepatitis C infection after liver transplantation. N Engl J Med. 1996;334:815–20. doi: 10.1056/NEJM199603283341302. [DOI] [PubMed] [Google Scholar]
- 11.Myers RP, Patel K, Pianko S, et al. The rate of fibrosis progression is an independent predictor of the response to antiviral therapy in chronic hepatitis C. J Viral Hepat. 2003;10:16–22. doi: 10.1046/j.1365-2893.2003.00387.x. [DOI] [PubMed] [Google Scholar]
- 12.Neumann UP, Berg T, Bahra M, et al. Fibrosis progression after liver transplantation in patients with recurrent hepatitis C. J Hepatol. 2004;41:830–6. doi: 10.1016/j.jhep.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 13.Prieto M, Berenguer M, Rayon JM, et al. High incidence of allograft cirrhosis in hepatitis C virus genotype 1b infection following transplantation: relationship with rejection episodes. Hepatology. 1999;29:250–6. doi: 10.1002/hep.510290122. [DOI] [PubMed] [Google Scholar]
- 14.Berenguer M, Prieto M, San Juan F, et al. Contribution of donor age to the recent decrease in patient survival among HCV-infected liver transplant recipients. Hepatology. 2002;36:202–10. doi: 10.1053/jhep.2002.33993. [DOI] [PubMed] [Google Scholar]
- 15.Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 1997;349:825–32. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 16.Baiocchi L, Angelico M, Petrolati A, et al. Correlation between liver fibrosis and inflammation in patients transplanted for HCV liver disease. Am J Transplant. 2008;8:673–8. doi: 10.1111/j.1600-6143.2007.02107.x. [DOI] [PubMed] [Google Scholar]
- 17.Guido M, Fagiuoli S, Tessari G, et al. Histology predicts cirrhotic evolution of post transplant hepatitis C. Gut. 2002;50:697–700. doi: 10.1136/gut.50.5.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Machicao VI, Bonatti H, Krishna M, Aqel BA, Lukens FJ, Nguyen JH, Rosser BG, Satyanarayana R, Grewal HP, Hewitt WR, Harnois DM, Crook JE, Steers JL, Dickson RC. Donor age affects fibrosis progression and graft survival after liver transplantation for hepatitis C. Transplantation. 2004;77:84–92. doi: 10.1097/01.TP.0000095896.07048.BB. [DOI] [PubMed] [Google Scholar]
- 19.Pelletier SJ, Iezzoni JC, Crabtree TD, et al. Prediction of liver allograft fibrosis after transplantation for hepatitis C virus: persistent elevation of serum transaminase levels versus necroinflammatory activity. Liver Transpl. 2000;6:44–53. doi: 10.1002/lt.500060111. [DOI] [PubMed] [Google Scholar]
- 20.Sreekumar R, Gonzalez-Koch A, Maor-Kendler Y, et al. Early identification of recipients with progressive histologic recurrence of hepatitis C after liver transplantation. Hepatology. 2000;32:1125–30. doi: 10.1053/jhep.2000.19340. [DOI] [PubMed] [Google Scholar]
- 21.Testa G, Crippin JS, Netto GJ, et al. Liver transplantation for hepatitis C: recurrence and disease progression in 300 patients. Liver Transpl. 2000;6:553–61. doi: 10.1053/jlts.2000.9741. [DOI] [PubMed] [Google Scholar]
- 22.Wali M, Harrison RF, Gow PJ, et al. Advancing donor liver age and rapid fibrosis progression following transplantation for hepatitis C. Gut. 2002;51:248–52. doi: 10.1136/gut.51.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter T, Dumortier J, Guillaud O, et al. Factors influencing the progression of fibrosis in patients with recurrent hepatitis C after liver transplantation under antiviral therapy: a retrospective analysis of 939 liver biopsies in a single center. Liver Transpl. 2007;13:294–301. doi: 10.1002/lt.21000. [DOI] [PubMed] [Google Scholar]
- 24.Yilmaz N, Shiffman ML, Stravitz RT, Sterling RK, Luketic VA, Sanyal AJ, Mills AS, Contos MJ, Coterell A, Maluf D, Posner MP, Fisher RA. A prospective evaluation of fibrosis progression in patients with recurrent hepatitis C virus following liver transplantation. Liver Transpl. 2007;13:975–83. doi: 10.1002/lt.21117. [DOI] [PubMed] [Google Scholar]
- 25.Diaz R, Kim JW, Hui JJ, et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum Pathol. 2008;39:102–15. doi: 10.1016/j.humpath.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 26.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–54. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagata K, Saga S, Yamada KM. Characterization of a novel transformation-sensitive heat-shock protein (HSP47) that binds to collagen. Biochem Biophys Res Commun. 1988;153:428–34. doi: 10.1016/s0006-291x(88)81242-7. [DOI] [PubMed] [Google Scholar]
- 28.Clouston AD, Powell EE, Walsh MJ, et al. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology. 2005;41:809–18. doi: 10.1002/hep.20650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.