

Abstract
Eukaryotic DNA topoisomerase I (Top1) is a monomeric protein clamp that functions in DNA replication, transcription, and recombination. Opposable “lip” domains form a salt bridge to complete Top1 protein clamping of duplex DNA. Changes in DNA topology are catalyzed by the formation of a transient phosphotyrosyl linkage between the active-site Tyr-723 and a single DNA strand. Substantial protein domain movements are required for DNA binding, whereas the tight packing of DNA within the covalent Top1–DNA complex necessitates some DNA distortion to allow rotation. To investigate the effects of Top1-clamp closure on enzyme catalysis, molecular modeling was used to design a disulfide bond between residues Gly-365 and Ser-534, to crosslink protein loops more proximal to the active-site tyrosine than the protein loops held by the Lys-369–Glu-497 salt bridge. In reducing environments, Top1-Clamp was catalytically active. However, contrary to crosslinking the salt-bridge loops [Carey, J. F., Schultz, S. J., Sission, L., Fazzio, T. G. & Champoux, J. J. (2003) Proc. Natl. Acad. Sci. USA 100, 5640–5645], crosslinking the active-site proximal loops inhibited DNA rotation. Apparently, subtle alterations in Top1 clamp flexibility impact enzyme catalysis in vitro. Yet, the catalytically active Top1-Clamp was cytotoxic, even in the reducing environment of yeast cells. Remarkably, a shift in redox potential in glr1Δ cells converted the catalytically inactive Top1Y723F mutant clamp into a cellular toxin, which failed to induce an S-phase terminal phenotype. This cytotoxic mechanism is distinct from that of camptothecin chemotherapeutics, which stabilize covalent Top1–DNA complexes, and it suggests that the development of novel therapeutics that promote Top1-clamp closure is possible.
DNA topoisomerases alter DNA topology by the concerted cleavage and religation of DNA strand(s) (reviewed in refs. 1–3). The nucleophilic attack of an active-site tyrosine on a phosphodiester DNA bond generates a phosphotyrosyl linkage, providing a transient protein-linked gate through which another DNA strand or duplex may pass. A second transesterification resolves the tyrosyl–DNA linkage to restore DNA integrity. DNA topoisomerases play critical roles in most DNA transactions, including DNA replication, transcription, and recombination, as well as chromosome segregation. These enzymes also constitute the targets of clinically important anticancer and antibacterial agents (4–6). With human DNA topoisomerase I (Top1), camptothecin (CPT) analogues (topotecan and CPT-11) have shown remarkable antitumor activity against pediatric and adult malignancies (7). These agents poison Top1 by reversibly stabilizing the covalent enzyme–DNA complex, inducing S-phase-dependent cell lethality.
Type IB enzymes, including eukaryotic and vaccinia virus Top1, are distinct from other enzymes in that the active-site tyrosine becomes linked to a 3′-phosphoryl end, versus the 5′-phosphotyrosine bond formed by types IA and II enzymes (1–3). Crystallographic data indicate that the monomeric enzymes (types IA and IB) or the protein subunits that constitute the symmetrical halves of type II enzymes [DNA gyrase and DNA topoisomerase II (Top2)] form a protein clamp (1–3). Yet, type IB enzymes are again unique in that biochemical and structural data support a mechanism of DNA rotation, as opposed to the coordinated domain movements in types IA and II enzymes that are necessary for an enzyme-bridging mechanism (1, 3, 8).
The conserved central domain of eukaryotic Top1 forms the protein clamp (Fig. 1). Subdomains I and II constitute the upper cap, which is connected by a flexible hinge to the bottom part of the clamp, subdomain III. Two extended α-helices connect the clamp core with the conserved C-terminal domain, containing the active-site tyrosine. Human Top1 structures are cocrystals of a C-terminal 70-kDa fragment of the enzyme (Topo 70, a 70-kDa form of human Top1 that is missing the nonessential N terminus), or reconstituted core and active-site tyrosine domains, with duplex DNA packaged tightly into a central pore 15–20 Å in diameter (8–11). Opposable “lip” domains (one loop from subdomain I and two loops from subdomain III) interact with phosphates in the DNA and form a salt bridge to complete the circumnavigation of Top1 around the DNA. The majority of interactions involve protein side chains or primary amines with DNA phosphates, over a 10-bp stretch of DNA centered on the site of strand scission. The scarcity of base-specific contacts is consistent with the lack of Top1–DNA sequence specificity and the global activity of this enzyme in replication and transcription (1, 2). However, the tight packing of DNA within the protein clamp raises questions about the mechanisms of DNA binding and rotation that are necessary to effect changes in DNA linking number.
Fig. 1.
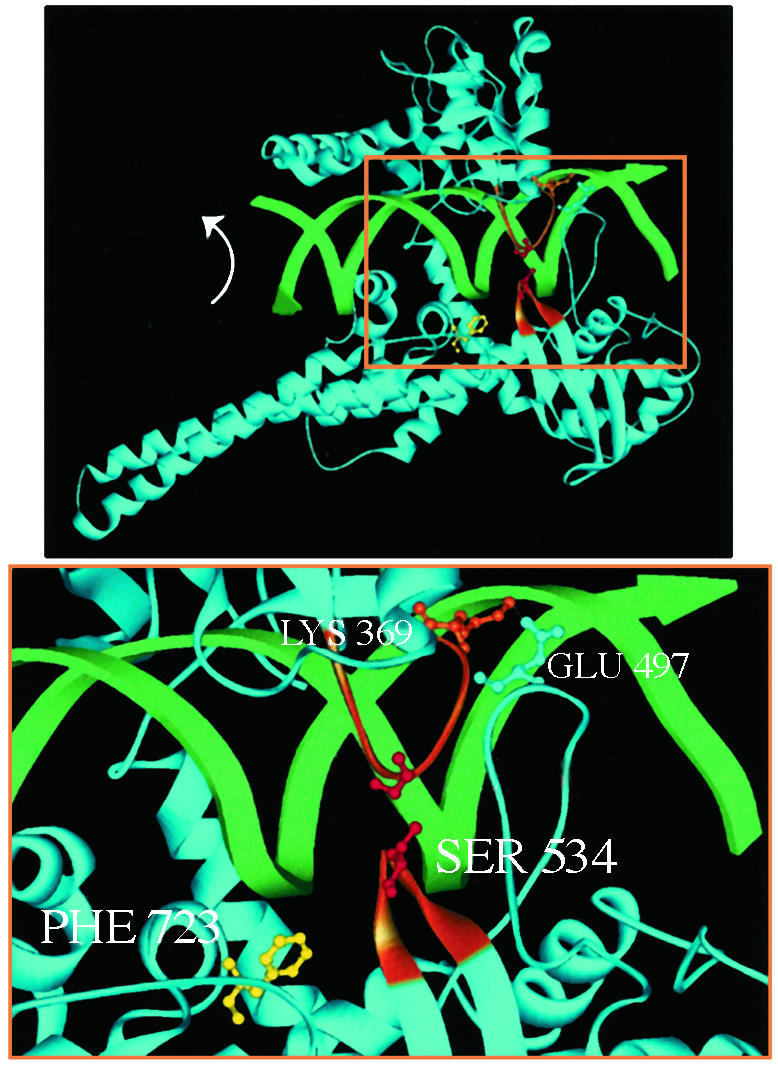
(Upper) The bilobed structure of Top1 clamped around duplex DNA. Two α-helices extend from the protein clamp to link the core with the active-site tyrosine domain. The active-site tyrosine (shown here as the Phe-723 mutant, yellow) is poised to cleave the scissile DNA strand. DNA rotation in the covalent complex is indicated (arrow). (Lower) The higher magnification of the boxed area in Upper highlights the lip domains that close the protein clamp. Two loops constitute the lower lip: one loop contains Ser-534 (orange), and the other loop contains Glu-497, which forms a salt bridge with Lys-369 in the upper lip (orange). Cysteine substitutions for Gly-365 and Ser-534 that are predicted to form a disulfide bond are shown in red.
Significant domain movements are required for the protein to bind DNA, presumably around the flexible hinge formed at the top of the helix between the cap and subdomain III (10, 12). When formed, does the clamp remain closed throughout the catalytic cycle, or is breathing of the lip domains necessary for DNA cleavage and/or rotation? Such flexibility might allow the enzyme to slide along the DNA, whereas lateral movements of the closed clamp would require rotation of Top1 around the DNA helix, much like a nut around the threads of a bolt. These issues are critical to understanding Top1 catalysis in the context of other cellular processes and are germane to more general considerations of other proteins that form rings or clamps around DNA.
Using molecular modeling, we designed a reversible disulfide bond between opposable residues (Gly-365 and Ser-534) within the lip domains to lock the Top1 clamp (Fig. 1). Contrary to a recent report on crosslinking the salt-bridge loops (13), the crosslink in our Top1-Clamp (Fig. 1) is more proximal to the active-site tyrosine and produced markedly distinct effects on enzyme catalysis. We report that DNA rotation was inhibited within locked Top1-Clamp–DNA catenanes. More surprising was the finding that Top1-Clamp expression induced yeast cell cycle arrest in the S phase and cell death. In the reducing environment of yeast, Top1-Clamp toxicity required the active-site tyrosine. However, in the more oxidizing environment of congenic glr1Δ cells, the mutant Top1Y723F-Clamp was cytotoxic in the absence of DNA cleavage and failed to induce an S-phase terminal phenotype. This mechanism of Top1 poisoning is reminiscent of ICRF-193 stabilization of the Top2 protein clamp (14), which may be exploited in the development of new therapeutics that target Top1.
Materials and Methods
Yeast Strains, Molecular Modeling, and top1 Mutants. Saccharomyces cerevisiae strains EKY3 (top1Δ) and JCW28 (top1Δ, top2ts) were described (12). MWY9 (EKY3, glr1Δ::his5+) was produced by PCR-based gene disruption.
Computer modeling with program O (version 5.10) was based on the Topo70–DNA structure (8). As described (14), cysteines were substituted for pairs of lip-domain residues, with side chains <4 Å apart. Modeling of side-chain rotamers identified two residue pairs (Gly-365/Ser-534 and His-367/Ala-499) where the distance between sulfhydryls (2–3 Å) would allow disulfide-bond formation to lock the Top1 clamp. Much larger distances between the engineered sulfhydryls and endogenous cysteines preclude intramolecular disulfide-bond formation.
A 6×His tag and Gly-365 → Cys mutation were engineered in human TOP1 by PCR and subcloned into YEpGAL1 vectors. The Ser-534 → Cys substitution was made with the Transformer kit (Clontech). To ease discussion, the G365C, S534C double mutant is referred to as “Top1-Clamp.” ARS–CEN plasmids were generated by subcloning his-top1 sequences into YCpGAL-hTOP1. To avoid alterations in DNA binding, the His tag was replaced with the FLAG epitope in YCpGAL1-etop1 constructs by replacing a XhoI DNA fragment in YCpGAL1-ehtop1Y723F (15–18) with corresponding Top1/top1-clamp sequences.
Top1 Purification and Activity. FLAG epitope-tagged Top1 (eTop1) and eTop1-Clamp were purified from galactose-induced EKY3 cells as described (12, 16–19). Top1 protein fractions were treated with 100 mM DTT and concentrated on a phosphocellulose column. In this chromatographic step and in subsequent experiments, N2-saturated buffers prevented spontaneous oxidation of cysteine residues.
Top1 activity was assayed in plasmid DNA relaxation reactions (16–18) in the presence or absence of 100 mM DTT. To assess Top1-Clamp locking induced by N,N,N′,N′-tetramethylazodicarboxamide (diamide), Top1 proteins were incubated with 5–100 mM diamide with or without 100 mM DTT, in G-100 buffer (20 mM Tris, pH 7.5/10 mM MgCl2/0.1 mM EDTA/50 μg/ml gelatin/100 mM KCl). pHC624 plasmid DNA was then added. Aliquots were taken at 30 sec, 10 min, and 20 min and were treated with 1% SDS, and the extent of plasmid DNA relaxation was determined.
Cell Viability and Drug Sensitivity Assays. The viability of yeast cells, transformed with the indicated top1 constructs, was determined by spotting aliquots of serially diluted cultures onto selective media with or without 0.1 μg/ml CPT (18) or by assaying the number of viable cells that formed colonies on dextrose plates after 6-h induction with galactose (16, 18). Aliquots were also processed for flow cytometry (19).
Top1 and Top1-Clamp sensitivity to CPT was determined in DNA cleavage assays (18), with or without 5 mM diamide and 50 μM CPT. Cleavage products were resolved in sequencing gels and visualized with a PhosphorImager (Molecular Dynamics).
2D Gel Electrophoresis. Top1-Clamp activity in vivo was assayed in JCW28 (top1Δ, top2ts) cells as described (12, 16). Top1 preparations were incubated in vitro with relaxed pHC624 DNA in G-100 buffer and 2 μg/ml ethidium bromide (EtdBr) as indicated. After 30 min at 37°C, reactions were terminated with 1% SDS at 75°C. The phenol-extracted DNA topoisomers were resolved in 2D gels and visualized by PhosphorImager analysis of Southern blots.
Bead-Bound Top1-Clamp Activity Assays. eTop1 and eTop1-Clamp were tethered to cellulose beads by M2 antibody binding to the N-terminal FLAG tag (12). After washing with G-25 buffer (G buffer/25 mM KCl) plus 1 mM DTT, bead-bound enzymes were incubated on ice with 5 ng of relaxed plasmid DNA in G-25 buffer for 30 min. Next, 100 mM diamide was added for 2 min. Unbound DNA was eluted by centrifugation and sequential washes of low-salt buffer (G buffer/200 mM KCl/100 mM diamide) and high-salt (hs) buffer (G buffer/1 M KCl/100 mM diamide). Locked Top1-Clamp–DNA catenanes were resolved with DTT buffer (G buffer/1 M KCl/200 mM DTT). DNA molecules in the various samples were resolved in agarose gels and visualized by Southern blotting. Relative amounts of bead-bound proteins were defined by SDS/PAGE, immunoblotting with M2 antibody, and chemiluminescence.
To assay catalytic activity, locked Top1-Clamp–DNA catenanes obtained after hs washes were equilibrated with G-100 buffer plus 100 mM diamide and then treated with 2 μg/ml EtdBr plus 100 mM diamide or 200 mM DTT at 37°C for 30 min. A similarly prepared Top1 sample was incubated with 0.1 μg of relaxed DNA, 2 μg/ml EtdBr, and 200 mM DTT. Plasmid DNA was eluted with DTT buffer and treated with 1% SDS at 75°C. DNA topoisomers were resolved in 2D gels. Bead-bound proteins were resolved by SDS/PAGE.
Results
Experimental Strategy. To probe Top1 architectural features that are necessary for catalysis, we followed the lead of Roca et al. (14), who designed a reversible disulfide bond across a Top2 dimer interface. We reasoned that a similar approach could define the consequences of Top1 clamp stabilization on DNA cleavage or rotation within the covalent complex. Molecular modeling identified two pairs of residues within opposable lip domains that, when replaced by cysteine, might form a disulfide bond to lock the Top1 clamp. Several factors were considered. First, allowed rotamers of substituted cysteines would place the sulfur atoms within 2–3 Å from one another to allow disulfide-bond formation. Second, residues His-367/Ala-499, whose alteration might perturb the formation of the lip-domain salt bridge, were not considered. We focused on Gly-365 in the subdomain I loop and Ser-534 in one of the two subdomain III loops that form the lower lip (Fig. 1). The shorter 534-loop does not interact directly with the upper lip; rather, the salt bridge links the upper lip with the longer 497-loop (9). A disulfide bond between cysteines substituted for Gly-365 and Ser-534 would be in closer proximity to the active-site Tyr-723 and the noncovalently held 5′ end of the DNA than protein loops held by the salt bridge. This proximity, we posited, might be advantageous in limiting clamp flexibility and the DNA rotations needed to alter linking number. Indeed, during the completion of these studies, Carey et al. (13) reported that a disulfide bond between cysteines substituted for His-367/Ala-499 did not prevent DNA rotation. These results are considered more fully in Discussion.
The G365C and S534C mutations were engineered in epitope-tagged human Top1 in yeast vectors under the galactose-inducible GAL1 promoter. Because yeast TOP1 is nonessential, it is possible to express plasmid-borne human TOP1 in the absence of the endogenous enzyme (18, 20).
We first asked if the cysteine substitutions altered Top1-Clamp activity. To assess catalytic activity in vivo, Top1, Top1-Clamp, and Top1G365C proteins were expressed in top1Δ, top2ts yeast that constitutively expresses bacterial topA (12). In the absence of human Top1 at 35°C, the major relaxation activity is that of bacterial TopA, which relaxes only negatively supercoiled DNA. Transcription of plasmid DNA produces local domains of positive supercoils in front of transcription complexes and negative supercoils behind the complexes (21). Bacterial TopA selectively relaxes negative supercoils, so positively supercoiled plasmid DNA accumulates. This result is shown for the pGAL1 control (Fig. 2). If catalytically active Top1 is expressed also, both positive and negative supercoils are relaxed. In Top1-expressing cells (Fig. 2), enzyme activity was evidenced by a quantitative shift of positively super-coiled topoisomers to the position of negatively supercoiled DNA. Similar results were obtained with Top1-Clamp- and Top1G365C-expressing cells, indicating that these enzymes were active in vivo.
Fig. 2.

Top1-Clamp is catalytically active in vivo. Top1, Top1-Clamp, and Top1G365C activity were assayed in top1Δ, top2ts yeast expressing bacterial topA as described (12). The distribution of 2-μm DNA topoisomers was determined by Southern blotting of 2D gels. Along the topoisomer arc, positively (+) and negatively (–) supercoiled DNAs are shown. The pGAL vector was used as a negative control.
Purified Top1-Clamp activity in DNA relaxation assays was restored by DTT (Fig. 3 and data not shown). With these preparations, the specific activity of Top1-Clamp, in the presence or absence of DTT, was ≈5- to 10-fold lower than that of Top1 (data not shown). Although Top1 and Top1-Clamp catalytic activity was optimal at 100 mM KCl, a more precipitous drop in Top1-Clamp catalysis at higher salt concentrations suggested reduced DNA binding.
Fig. 3.

Top1-Clamp activity is reversibly inhibited by diamide. Equal concentrations of Top1 and Top1-Clamp were incubated in plasmid DNA relaxation assays in the presence of 5 mM diamide and 100 mM DTT as indicated. Plasmid DNA was added at time 0, and the extent of DNA relaxation was assessed after 30 sec, 10 min, and 20 min at 37°C. Lane C, untreated plasmid DNA.
Locking the Top1 Clamp. As in studies of Top2 (22), the oxidizing agent diamide was used to induce disulfide-bond formation between opposable cysteine residues. In Fig. 3, pretreatment of Top1-Clamp with diamide inhibited enzyme activity, even after prolonged periods (20 min), without affecting wild-type Top1 catalysis at concentrations of up to 100 mM. Moreover, diamide-induced inhibition of Top1-Clamp activity was reversed with DTT (Fig. 3). Diamide treatment did not induce protein–protein crosslinks because higher molecular weight Top1-Clamp species were not detected in nonreducing gels after diamide treatment (data not shown). These data are consistent with the formation of intramolecular disulfide bonds to lock the clamp closed, rather than intermolecular bonds to inhibit enzyme catalysis, and suggest that Top1 clamp closure occurs in the absence of DNA.
To address the question of DNA cleavage in the locked-clamp configuration, the CPT sensitivity of Top1-Clamp was assessed. CPT targets Top1 by reversibly stabilizing the covalent enzyme–DNA intermediate (1, 5, 6, 11). Mutation of residues, such as Gly-363, within the Top1 upper lip domain has been reported to alter enzyme sensitivity to CPT, producing a drug-resistant phenotype (12, 16, 23, 24). The structure of Top1–DNA–topotecan indicates that the 365-loop stabilizes an intercalation pocket within Top1 to allow the rotation of DNA caused by drug binding (11). These results predict that the G365C mutation would reduce enzyme sensitivity to CPT, which was borne out in DNA cleavage assays (data not shown) and in top1Δ yeast expressing Top1G365C (see Fig. 6).
Fig. 6.

The catalytically inactive Top1Y723F-Clamp induces glr1Δ strain lethality. (A) Independent cultures of top1Δ or top1Δ, glr1Δ cells, transformed with the indicated YCpGAL1-top1 vector, were serially diluted 10-fold and spotted onto selective media supplemented with dextrose or galactose and 0.1μg/ml CPT as indicated. Cell viability was assessed after incubation at 30°C. (B) top1Δ or top1Δ, glr1Δ cells, transformed with the indicated YCpGAL1-top1 vector, were induced with galactose at t = 0. After 6 h, aliquots were plated on dextrose medium or processed for flow cytometry. The fold increases in the number of cells forming colonies, relative to t = 0, are an average of three independent experiments. In representative flow cytometry profiles, 1N and 2N DNA peaks are marked with single and double arrows, respectively.
In contrast, Top1-Clamp was sensitive to CPT in DNA cleavage assays (Fig. 4). CPT induced elevated levels of Top1-Clamp covalent complexes in the presence or absence of DTT. The proportionately lower levels of CPT-stabilized complexes, relative to those obtained with Top1, reflect the ≈5- to 10-fold decrease in Top1-Clamp specific activity under the reaction conditions. Although the single S534C mutation did not alter Top1 sensitivity to CPT (data not shown), substituting Cys for Ser-534 in Top1-Clamp restored the CPT sensitivity of Top1G365C. This intragenic complementation suggests that the coordinated interaction of the lip domains affects the formation of the intercalation pocket that is necessary for drug binding.
Fig. 4.

Top1-Clamp is sensitive to CPT. Top1, Top1-Clamp, and Top1Y723F were incubated in DNA-cleavage reaction mixtures with a single 3′ 32P-labeled DNA fragment (18). As indicated, reaction mixtures contained 50 μM CPT, 5 mM diamide, or 100 mM DTT. After 10 min at 37°C, covalent-complex formation was assessed in sequencing gels by PhosphorImager analysis. The asterisk indicates a high-affinity cleavage site. Arrows indicate novel cleavage sites.
In contrast to Top1, low levels of Top1-Clamp–DNA covalent complexes were observed in the absence of CPT and DTT (arrows in Fig. 4). These cleavage sites were different from those induced by CPT yet were eliminated by DTT treatment. This finding supports the notion that formation of intramolecular disulfide bonds contributes to the stability of the covalent complex. When Top1-Clamp–DNA was pretreated with diamide to induce clamp locking, before the addition of CPT, a low level of drug-stabilized complexes was still detected (Fig. 4). The inactive Top1Y723F mutant served as a negative control. Similar results were obtained in plasmid DNA nicking assays, where CPT-induced nicking of monomeric plasmid DNA was still observed with Top1-Clamp–DNA complexes that were pretreated with diamide (data not shown). Thus, CPT-induced covalent-complex formation was detectable with the locked Top1-Clamp, albeit at reduced levels.
DNA Strand Rotation Within the Locked Top1-Clamp. The previous experiments support the formation of the locked Top1 clamp in the absence of DNA and DNA cleavage within the crosslinked Top1-Clamp–DNA complex. Yet for DNA rotation to occur, the structures evident in Top1–DNA crystals dictate DNA bending to avoid substantial clashes between amino acid side chains and DNA (8–11). Given the dramatic domain movements that are necessary for Top1 binding of duplex DNA, protein flexibility might also regulate DNA rotation within the clamp core.
To address this possibility, a strategy based on previous studies of bead-bound Top1 complexes (12, 15, 16) was devised to purify catenanes between a locked Top1-Clamp and a DNA ring. However, we first established conditions to regulate Top1 catalytic activity with EtdBr. As shown in Fig. 5A, when Top1 and the unlocked Top1-Clamp were incubated with closed-circular relaxed plasmid DNA, the distribution of topoisomers in 2D gels was unaltered. EtdBr intercalation induces compensatory positive writhe within plasmid DNA, which Top1 relaxes. After terminating the reactions, the removal of EtdBr induces DNA rewinding and the accumulation of compensatory negative writhe. Top1 catalytic activity would be evidenced by a shift of relaxed DNA topoisomers to negatively supercoiled DNA (Fig. 5A Lower), which was observed with Top1 and Top1-Clamp. Thus, DNA rotation within the locked Top1-Clamp could be assessed in the presence of EtdBr after treatment with diamide.
Fig. 5.

DNA rotation is inhibited in locked Top1-Clamp–DNA complexes. (A) Top1 and Top1-Clamp were incubated with relaxed plasmid DNA in G-100 buffer. EtdBr (green) was added to induce positive supercoils. After 30 min at 37°C, reactions were terminated and plasmid DNA topoisomers were resolved in 2D gels. Relaxed (R) and negatively supercoiled (–) DNAs are indicated. (B) Top1 and Top1-Clamp, bound to cellulose beads by the M2 antibody (12), were incubated with relaxed plasmid DNA, followed by diamide. DNA in the flow-through (FT), low-salt (lo), hs, and DTT/1 M KCl (DTT) washes were resolved in an agarose gel and visualized with Southern blotting. Monomer (m), nicked (n), and dimer (d) DNAs are indicated. Anomalies in DNA migration (FT and lo) were an artifact of protein contamination. (C) Bead-bound Top1 and Top1-Clamp hs samples were equilibrated with G-100 buffer in the presence or absence of diamide. Locked Top1-Clamp (single red dot) was treated with EtdBr (green) to induce positive supercoils. The sample lacking diamide was treated with EtdBr plus DTT to unlock the clamp (double red dots). Relaxed plasmid DNA and EtdBr were added to a similar Top1 sample. After 30 min at 37°C, reactions were terminated with DTT/1 M KCl/SDS, and DNA topoisomers were resolved in 2D gels.
To ensure binding of full-length Top1 proteins, Top1 and Top1-Clamp were tethered to streptavidin-coated cellulose beads by a biotinylated M2 antibody that recognizes the N-terminal FLAG tag (Fig. 5B Upper). After the addition of relaxed plasmid DNA, diamide was added to lock Top1-Clamp. In Fig. 5B, successive low salt and hs washes removed the DNA from bead controls and bound Top1 effectively. In contrast, a significant amount of plasmid DNA was retained by the locked Top1-Clamp, which was recovered only by release with DTT and hs. Thus, diamide treatment locked the Top1-Clamp–DNA catenanes. Similar data were obtained with negatively supercoiled plasmid DNA, whereas immunoblot analyses indicated that equivalent amounts of Top1 and Top1-Clamp were bound to the beads (data not shown).
We then asked if DNA rotation occurs within the locked Top1-Clamp–DNA catenanes. As shown in Fig. 5C, the bead-bound samples were equilibrated with buffer plus diamide (locked clamp) or DTT (to unlock the clamp) and EtdBr was added to induce positive supercoils. No change in DNA linking number was detected in the locked Top1-Clamp. However, reversing the crosslink with DTT restored Top1-Clamp activity, producing a shift in topoisomer distribution. Relaxed plasmid DNA and EtdBr were added to bead-bound Top1 to ensure that the enzyme remained catalytically active. These results demonstrate that although the bead-bound Top1-Clamp was catalytically competent, DNA rotation was inhibited within the locked Top1-Clamp.
Top1-Clamp-Induced Cell Lethality. To assess in vivo consequences of Top1-Clamp expression, the viability and CPT sensitivity of top1Δ cells transformed with GAL1-top1 constructs were assayed in Fig. 6. top1Δ cells expressing human TOP1 exhibit a >3-log10 drop in viability in the presence of CPT, whereas cells expressing catalytically active Top1G365C were resistant to the drug. Remarkably, expression of Top1-Clamp induced cell lethality in the absence of CPT (Fig. 6A). This was consistent with low levels of DNA cleavage observed in the absence of CPT and DTT (Fig. 4).
In the reducing environment of yeast, Top1-Clamp-induced lethality was abolished by mutating the active-site Tyr-723 to Phe (Fig. 6A). Hence, DNA cleavage was a requisite step in the formation of cytotoxic lesions. To determine whether shifting the redox potential by increasing oxidized glutathione would enhance Top1Y723F-Clamp toxicity, the Top1 constructs were expressed in a congenic strain deleted for glutathione reductase (Glr1). The ratio of reduced to oxidized glutathione drops ≈20-fold in glr1Δ cells (25). In Fig. 6A, expression of the catalytically inactive Top1Y723F-Clamp induced a >3-log10 drop in top1Δ, glr1Δ viability, whereas the viability and CPT sensitivity of cells expressing other Top1 constructs were unaffected. However, this analysis is complicated by the slow-growth phenotype induced by Top1Y723F. Hence, the effects of Tyr-723 on cell viability were assessed further in a colony-forming assay to obviate concerns about colony size.
As shown in Fig. 6B, galactose-induced expression of Top1Y723F had no effect on cell viability relative to Top1. In contrast, Top1-Clamp expression induced a significant reduction in the number of viable cells and an accumulation in the late S and G2 phases of the cell cycle. Microscopic examination of 4′,6-diamidino-2-phenylindole-stained cells determined that 66% of the cells accumulated with a large budded phenotype and a single DNA mass. This terminal phenotype has been reported for CPT-treated cells expressing TOP1 (26). Top1-Clamp cytotoxicity was exacerbated in top1Δ, glr1Δ cells, shifting the cell cycle distribution to the early S phase (Fig. 6B), with >71% large budded cells with a single DNA mass (data not shown). Compared with wild-type cells expressing Top1-Clamp, Top1Y723F-Clamp in top1Δ, glr1Δ cells induced a similar decrease in cell viability (0.4 versus 0.5 colony-forming units, respectively; Fig. 6B). However, Top1Y723F-Clamp-induced lethality did not coincide with a significant alteration in cell cycle, and only 30% of the cells exhibited a large budded phenotype. Values of ≈20% were obtained with Top1-expressing cells. These data indicate that enhanced Top clamp locking, induced by oxidized glutathione, produces cytotoxic lesions in the absence of DNA cleavage. This mechanism of Top1 poisoning is distinct from that of known chemotherapeutics, which target Top1 by stabilizing the covalent complex. Further, these results indicate that the formation of the phosphotyrosyl linkage potentiates lethality during the S phase, possibly by enhancing the formation of replication fork-associated lesions.
Discussion
Biochemical and structural data demonstrate that Top1 binds duplex DNA as a protein clamp and catalyzes the relaxation of supercoiled DNA by forming a covalent phosphotyrosyl intermediate (1, 2). In cocrystals of Top1 in covalent and noncovalent complexes with DNA, 10 bp of DNA centered on the site of single-strand scission are packed tightly in the Top1 protein ring (8–11). Although these structures support a mechanism of DNA rotation to effect changes in DNA linking number, they fail to clarify how this rotation is achieved within the context of the covalent complex. The crystallographic data dictate substantial distortion of helical DNA to avoid clashes with amino acid side chains (8–11). Alternatively, the dramatic domain movements that are necessary for clamp binding of duplex DNA suggest that similar protein flexibility may facilitate DNA rotation within the clamp core. Given the inhibition of DNA rotation observed with locked Top1-Clamp–DNA catenanes (Fig. 5), our results support a model of limited protein flexibility rather than DNA bending to effect changes in DNA linking number. Contrary results were recently reported (13), in which a disulfide bond introduced at residues His-367/Ala-499 failed to restrict DNA rotation. Several aspects of these studies are worth consideration.
First, a disulfide linkage between cysteines at residues Gly-365 and Ser-534 (534-loop) is in closer proximity to the active-site tyrosine than a crosslink between residues His-367 and Ala-499 in the more distal 497-loop (Fig. 1). This active-site proximal crosslink could induce more severe limitations on the flexibility of the loop structures needed for DNA rotation. Second, tethering the 497-loop would not necessarily preclude 534-loop movement. Whether the N-terminal residues missing in Topo70 constructs (13) also contribute to DNA binding and rotation within the covalent complex has yet to be determined. Nevertheless, it is clear from these studies that relatively subtle alterations in Top1 clamp structure produce distinct effects on enzyme catalysis.
Another interesting aspect of this study was the CPT sensitivity of the Top1-Clamp enzyme. The CPT resistance of Top1G365C was predictable in vivo and in vitro (Fig. 6 and data not shown). The ternary Topo70–DNA–topotecan structure illustrates the contribution of the Arg-362, Gly-363, and Arg-364 residues to the formation of an intercalation pocket in Top1 to accommodate the rotation of the nonscissile DNA strand that is induced by drug binding (11). Although Gly-365 mutations have not been reported, the CPT resistance of Top1G365C was consistent with other substitutions within this protein loop (12, 16, 23, 24). The S534C mutation complemented Top1G365C to restore the CPT sensitivity of Top1-Clamp. Based on the structure of vaccinia virus Top1 (27), Yang and Champoux (28) suggested that the interaction of opposable lip domains induces the appropriate orientation of Tyr-723 within the active site to effect DNA cleavage. Our results extend these considerations and argue that compensatory changes in the 534-loop restore the formation of the intercalation pocket, which was abolished by the G365C mutation.
Interactions between intercalation pocket residues (Arg-362, Gly-363, and Arg-364) and the nonscissile DNA strand also provide a structural basis for CPT-induced inhibition of DNA rotation (11). However, the same mechanism may apply to the lack of detectable strand rotation within the locked Top1-Clamp. This model would be consistent with the CPT sensitivity of the Top1-Clamp–DNA complex (Fig. 4) as well as with the view that the locked Top1 clamp represents a normally transient intermediate in the Top1 catalytic cycle, much like CPT-stabilized covalent complexes.
Perhaps most remarkable were the distinct terminal phenotypes induced by Top1-Clamp and the catalytically inactive Top1Y723F-Clamp. In the reducing environment of wild-type yeast, Top1-Clamp expression produced an S-phase terminal phenotype. The viability of cells expressing either the single G365C or S534C mutants or Top1Y723F-Clamp argues against lethality because of promiscuous disulfide-bond formation with other proteins. Indeed, other cysteines within Top1 are buried within the protein structure or lie sufficiently far away from the lip domains that only intermolecular linkages are possible. Because protein complexes were not detected in nonreducing gels, the formation of intramolecular disulfide bonds between the engineered cysteine substitutions appears most likely. These data also argue that covalent complex formation contributes to the cytotoxic mechanism of Top1-Clamp.
In contrast, shifting in vivo conditions to a more oxidizing environment in glr1Δ cells converted the catalytically inactive Top1Y723F-Clamp into a cellular poison (Fig. 6). Hence, stabilizing the Top1 clamp in the absence of DNA cleavage was sufficient to induce cell death. Although the lack of an S-phase terminal phenotype suggests the formation of distinct DNA lesions, the induction of different repair and checkpoint pathways by covalent versus noncovalent Top1 clamps has yet to be investigated. The non-S-phase lethality induced by Top1Y723F-Clamp may relate to the replication-independent cytotoxicity of CPT at high drug concentrations and transcription-mediated DNA damage (29). This effect is reminiscent of the cytotoxic action of ICRF-193 in stabilizing the Top2 protein clamp on DNA (14) and represents a unique mechanism of poisoning Top1, which may be exploited in the development of novel chemotherapeutics.
Acknowledgments
We thank J. Berger and J. C. Wang for insightful comments and G. Ughetto for help with the modeling. This work was supported by National Institutes of Health Grants CA58755 (to M.-A.B.) and CA21765; Ministero dell'Instruzione, dell'Università e della Ricerca Cofinanziamento 2001, Ministero della Salute, Genomica Funzionale Consiglio Nazionale delle Ricerche, and Associazione Italiana per la Ricerca Sul Cancro (to P.B.); and American Lebanese Syrian Associated Charities.
Abbreviations: EtdBr, ethidium bromide; CPT, camptothecin; hs, high salt; Top1/2, DNA topoisomerase I/II.
References
- 1.Champoux, J. J. (2001) Annu. Rev. Biochem. 70, 369–413. [DOI] [PubMed] [Google Scholar]
- 2.Wang, J. C. (2002) Nat. Rev. Mol. Cell Biol. 3, 430–440. [DOI] [PubMed] [Google Scholar]
- 3.Keck, J. L. & Berger, J. M. (1999) Nat. Struct. Biol. 6, 900–902. [DOI] [PubMed] [Google Scholar]
- 4.Burden, D. A. & Osheroff, N. (1998) Biochim. Biophys. Acta 1400, 139–154. [DOI] [PubMed] [Google Scholar]
- 5.Li, T. K. & Liu, L. F. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 53–77. [DOI] [PubMed] [Google Scholar]
- 6.Kohn, K. W. & Pommier, Y. (2000) Ann. N.Y. Acad. Sci. 922, 11–26. [DOI] [PubMed] [Google Scholar]
- 7.Rodriguez-Galindo, C., Radomski, K., Stewart, C. F., Furman, W., Santana, V. M. & Houghton, P. J. (2000) Med. Pediatr. Oncol. 35, 385–402. [DOI] [PubMed] [Google Scholar]
- 8.Stewart, L., Redinbo, M. R., Qiu, X., Hol, W. G. & Champoux, J. J. (1998) Science 279, 1534–1541. [DOI] [PubMed] [Google Scholar]
- 9.Redinbo, M. R., Stewart, L., Kuhn, P., Champoux, J. J. & Hol, W. G. (1998) Science 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 10.Redinbo, M. R., Stewart, L., Champoux, J. J. & Hol, W. G. (1999) J. Mol. Biol. 292, 685–696. [DOI] [PubMed] [Google Scholar]
- 11.Staker, B. L., Hjerrild, K., Feese, M. D., Behnke, C. A., Burgin, A. B., Jr., & Stewart, L. (2002) Proc. Natl. Acad. Sci. USA 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hann, C. L., Carlberg, A. L. & Bjornsti, M.-A. (1998) J. Biol. Chem. 273, 31519–31527. [DOI] [PubMed] [Google Scholar]
- 13.Carey, J. F., Schultz, S. J., Sission, L., Fazzio, T. G. & Champoux, J. J. (2003) Proc. Natl. Acad. Sci. USA 100, 5640–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roca, J., Berger, J. M., Harrison, S. C. & Wang J. C. (1996) Proc. Natl. Acad. Sci. USA 93, 4057–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hann, C., Evans, D. L., Fertala, J., Benedetti, P., Bjornsti, M.-A. & Hall, D. J. (1998) J. Biol. Chem. 273, 8425–8433. [DOI] [PubMed] [Google Scholar]
- 16.Fiorani, P., Amatruda, J. F., Silvestri, A., Butler, R. H., Bjornsti, M.-A. & Benedetti, P. (1999) Mol. Pharmacol. 56, 1105–1115. [DOI] [PubMed] [Google Scholar]
- 17.Fertala, J., Vance, J. R., Pourquier, P., Pommier, Y. & Bjornsti, M.-A. (2000) J. Biol. Chem. 275, 15246–15253. [DOI] [PubMed] [Google Scholar]
- 18.Woo, M. H., Vance, J. R., Marcos, A. R., Bailly, C. & Bjornsti, M.-A. (2002) J. Biol. Chem. 277, 3813–3822. [DOI] [PubMed] [Google Scholar]
- 19.Megonigal, M. D., Fertala, J. & Bjornsti, M.-A. (1997) J. Biol. Chem. 272, 12801–12808. [DOI] [PubMed] [Google Scholar]
- 20.Bjornsti, M.-A. (2002) Cancer Cells 2, 267–273. [DOI] [PubMed] [Google Scholar]
- 21.Giaever, G. N. & Wang, J. C. (1988) Cell 55, 849–856. [DOI] [PubMed] [Google Scholar]
- 22.Lindsley, J. E. (1996) Proc. Natl. Acad. Sci. USA 93, 2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benedetti, P., Fiorani, P., Capuani, L. & Wang, J. C. (1993) Cancer Res. 53, 4343–4348. [PubMed] [Google Scholar]
- 24.Rubin, E., Pantazis, P., Bharti, A., Toppmeyer, D., Giovanella, B. & Kufe, D. (1994) J. Biol. Chem. 269, 2433–2439. [PubMed] [Google Scholar]
- 25.Trotter, E. W. & Grant, C. M. (2003) EMBO Rep. 4, 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kauh, E. A. & Bjornsti, M.-A. (1995) Proc. Natl. Acad. Sci. USA 92, 6299–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng, C., Kussie, P., Pavletich, N. & Shuman, S. (1998) Cell 92, 841–850. [DOI] [PubMed] [Google Scholar]
- 28.Yang, Z. & Champoux, J. J. (2002) J. Biol. Chem. 277, 30815–30823. [DOI] [PubMed] [Google Scholar]
- 29.Liu, L. F., Desai, S. D., Li, T. K., Mao, Y., Sun, M. & Sim, S. P. (2000) Ann. N.Y. Acad. Sci. 922, 1–10. [DOI] [PubMed] [Google Scholar]