

Abstract
Newcastle disease virus (NDV) has been previously shown to possess oncolytic activity, causing specific lysis of cancerous but not normal cells. Here we show that despite these findings, the oncolytic efficiency of naturally occurring NDV strains can still be relatively low, as many tumors exhibit strong innate immune responses that suppress viral replication and spread. To overcome this problem, we generated a recombinant fusogenic NDV expressing influenza NS1 protein, a protein exhibiting interferon (IFN)-antagonist and antiapoptotic functions in human and mouse cells. Interestingly, the resultant virus was dramatically enhanced in its ability to form syncytia, lyse a variety of human and mouse tumor cell lines, and suppressed the induction of the cellular IFN responses. Using the aggressive syngeneic murine melanoma model, we show that the NDV-NS1 virus is more effective than virus not expressing NS1 in clearing the established footpad tumors and results in higher overall long-term animal survival. In addition, mice treated with NDV-NS1 exhibited no signs of toxicity to the virus and developed tumor-specific cytotoxic T lymphocyte (CTL) responses. These findings demonstrate that modulation of innate immune responses by NDV results in enhancement of its oncolytic properties and warrant further investigation of this strategy in design of oncolytic NDV vectors against human tumors.
Introduction
Newcastle disease virus (NDV) is a member of the Avulavirus genus in the Paramyxoviridae family, which has been shown to infect a number of avian species.1 NDV possesses a single-stranded RNA genome in a negative sense and does not undergo recombination with the host genome or with other viruses.1 Naturally occurring NDV has been reported to be an effective oncolytic agent in a variety of animal tumor models.2 It has been used in vaccination with tumor cell oncolysates in people with head and neck squamous cell carcinomas,3 tumors of digestive tract,4 glioblastoma multiforme,5,6 malignant melanoma,7,8,9 colorectal carcinoma,10,11 and other advanced cancers. Finally, several of the naturally occurring strains of NDV have been used in multiple clinical trials against advanced human cancers.2,12,13,14,15,16
The success of naturally occurring strains of NDV in these clinical trials for advanced human cancers was only marginal.13,14,15 The results of these studies prompt investigation of potential mechanisms of tumor resistance to oncolytic NDV therapy and warrant the development of more effective oncolytic NDV strains.
Several groups, including ours, have succeeded in establishing a reverse genetics system for this virus,17,18,19 which allowed for genomic manipulation and construction of vaccines and oncolytic virus vectors. The two major goals of this study were to evaluate the NDV for oncolytic efficacy in various human tumor cells and to introduce modifications into the NDV genome that would allow it to overcome tumor resistance to oncolytic therapy.
It has been proposed that the tumor-selective lytic properties of the NDV are dependent on the defective activation of the type I interferon (IFN) response in tumor cells. Recent studies have also shown that tumor cells respond differently to IFN-β treatment than primary cells. Pretreatment of HT-1080 human fibrosarcoma cells with IFN-β did not affect killing of these tumor cells by NDV, while pretreatment of human skin fibroblasts suppressed replication of the virus in these primary cells, due to defects in the tumor cell IFN signal transduction pathway.20,21
In accord with these findings, our previous studies indicated that the restriction of the NDV to the avian host is mediated in part by the NDV V protein.22 NDV V protein in avian cells functions as a type I IFN antagonist by degrading the STAT1 protein.22,23 However, this effect was absent in the mammalian tumor cells, which resulted in decreased NDV replication.22,24 The results of these findings suggested that at least some tumors are still capable of mounting effective antiviral responses, which may partially explain the observed resistance of tumors to the oncolytic NDV therapy.
Based on these findings, we hypothesized that the oncolytic activity of NDV can be enhanced by improving the suppression of the innate immune response by the virus. We achieved this effect by introducing into the NDV genome a sequence encoding the influenza virus NS1 protein, a 27 kDa nonstructural protein which was extensively characterized for its ability to suppress the induction of type I IFN response and apoptosis in the virus-infected mammalian cells.25,26,27,28,29 In agreement with our hypothesis, NDV expressing NS1 protein was an effective oncolytic agent and replicated efficiently in a variety of human tumor cell lines. Moreover, the generated virus was effective in treatment of a mouse model of aggressive malignant melanoma, while maintaining the safety profile of the naturally occurring NDV strains. The results of these studies warrant the investigation of this new recombinant vector in therapy of human malignant tumors.
Results
Recombinant NDV is an effective oncolytic agent against a panel of tumor cell lines
For the present studies, we used the attenuated NDV of Hitchner B1 strain (NDV(B1)). We previously showed that modification of the NDV F protein to a fusogenic type by introduction of a polybasic protease cleavage site allowed for efficient formation of syncytia in the infected cells, which enhanced the viral oncolytic activity both in vitro and in vivo.24 To further explore the oncolytic potential of the fusogenic NDV, we selected a number of tumor cell lines from a variety of cancer types and infected them with NDV(B1) and NDV(F3aa) viruses at multiplicity of infection (MOI) 0.1. These included human pancreatic, breast, thyroid, head and neck, and gastric cancers, as well as human and murine malignant melanoma cell lines. The results of this study are summarized in Figure 1a, with representative cancer types shown. NDV was effective against the majority of tumor cell types, with NDV(F3aa) being significantly more cytolytic than the parental nonfusogenic NDV(B1) virus for the majority of cell lines. Infection of the same cell lines with NDV(F3aa) virus expressing GFP (NDV(F3aa)-GFP) revealed that the virus effectively formed large syncytia, which was likely responsible for its enhanced cytolytic activity (Figure 1b).
Figure 1.

Recombinant Newcastle disease virus (NDV) with modified fusion protein induces enhanced cytolysis in multiple human cancer cell lines. (a) Cell lines (5 × 105 cells) were infected at multiplicity of infection (MOI) 0.1 in triplicate and lactate dehydrogenase release assays were performed at 24, 48, and 72 hours. Percentage of cells surviving at 24, 48, and 72 hours is shown. (b) Syncytia formation by the NDV(F3aa) virus. Cell lines tested in a were infected with NDV(B1)-GFP and NDV(F3aa)-GFP at MOI 0.01, stained with Hoechst after 24 hours, and images were taken under fluorescent microscope. Representative images from Panc-1 and Scc-25 cells are shown (green: GFP; blue: Hoechst).
NDV induces antiviral signaling in cancer cells
The results of these studies suggested that the NDV(F3aa) virus may be an attractive candidate for oncolytic virotherapy of a variety of human malignancies. However, previous studies showed that despite its effective oncolytic activity in CT26 cells both in vitro and in vivo, the NDV(F3aa) virus still failed to cause complete tumor regressions in the CT26 murine syngeneic flank tumor model.24,30 We speculate that effective viral replication in tumors is limited by the host and tumor factors, such as the induction of type I IFN. Indeed, it was previously shown that NDV restriction to the avian host is mediated in part by inability of the virus to overcome the antiviral response in human cells, but not in avian cells.22 To test whether the NDV(B1) and NDV(F3aa) induce type I IFN in NDV-susceptible human cells, we performed an IFN bioassay, similar to the methods described previously (see Materials and Methods section and Figure 2a). As can be seen from Figure 2b, infection of Panc-1 cells with NDV(B1) and NDV(F3aa) viruses led to the induction of antiviral cytokines at the levels comparable to 1,000 U/ml of IFN-β, which was sufficient to suppress NDV-GFP replication. These results indicate that even in an apparently NDV-susceptible cancer cell line, the induction of type I IFN may suppress viral replication, limiting the viral oncolytic efficacy.
Figure 2.
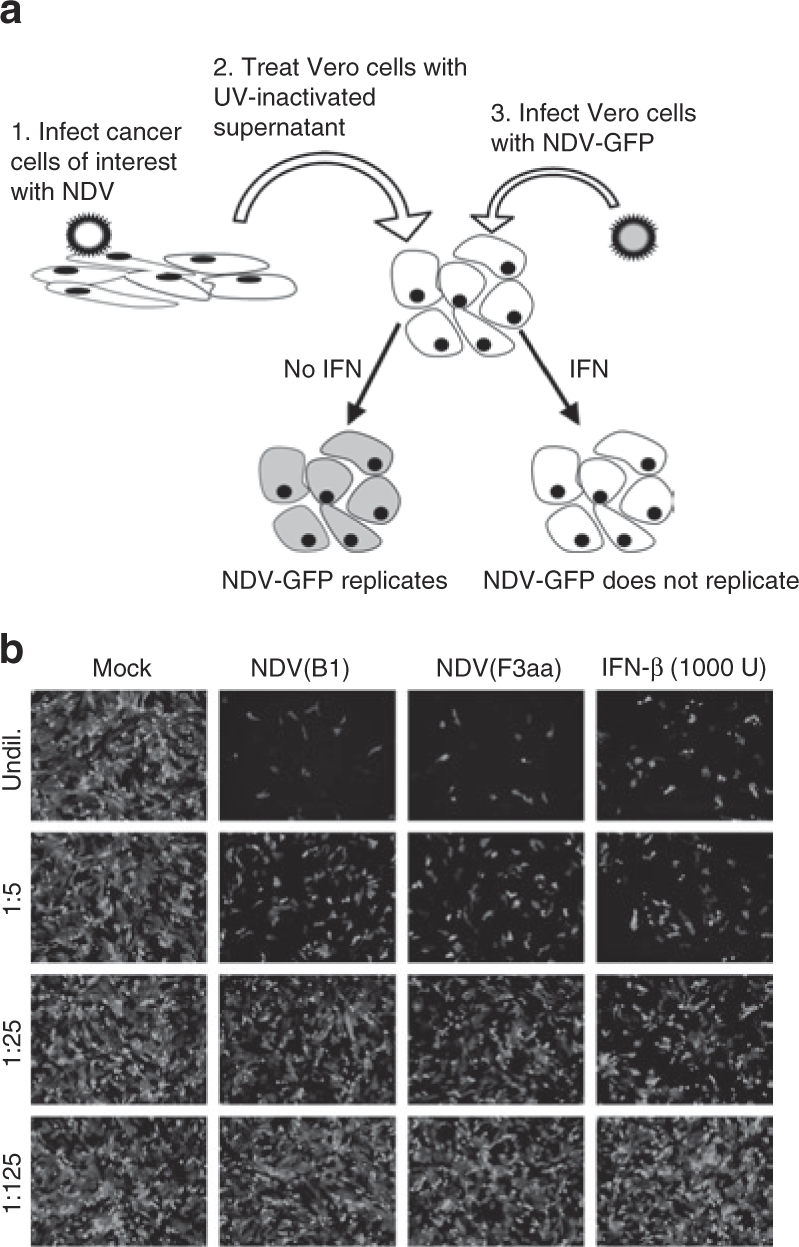
Newcastle disease virus (NDV) induces antiviral response in the infected cancer cells. (a) Schematic diagram of the bioassay for interferon (IFN) production. Panc-1 cells were infected with NDVs at multiplicity of infection (MOI) 0.1. Supernatants were collected at 24 hours, and any virus present was UV inactivated. Fresh Vero cells were treated with the inactivated supernatants and then infected with NDV-GFP at MOI of 0.1. (b) Antiviral activity in the infected Panc-1 supernatants. Supernatants were diluted in fivefold series. Supernatants from noninfected cells and recombinant human IFN-β were used as negative and positive controls respectively. NDV-GFP-infected Vero cells were examined by fluorescence microscopy.
We hypothesized that repression of IFN induction during NDV infection would permit for better viral replication in tumors, while allowing to maintain the therapeutic safety margin due to other factors playing a role in the NDV attenuation and host restriction. We proceeded to construct an oncolytic NDV expressing influenza virus NS1 protein, which was previously shown to be effective in down-modulating the cellular innate immune response in human cells.
Generation of NDV expressing influenza NS1 protein
Using the NDV(F3aa) virus as a backbone, we generated a virus expressing the NS1 protein of the influenza virus PR8 strain according to the methods described previously18 (Figure 3a). Immunofluorescent labeling of the infected Vero cells confirmed that the NS1 protein was expressed by the NDV(F3aa)-NS1 virus and not by the parental NDV(F3aa) or NDV(B1) strains (Figure 3b). Of note, all of the cells infected with NDV(F3aa)-NS1 showed expression of the protein. The expression was maintained after 10 viral passages in embryonated chicken eggs, confirming the stability of the recombinant virus (data not shown). To confirm that the NS1 protein was expressed to high levels, we performed a time course of NDV infection in Vero cells and analyzed the induction of NS1 expression within the period of one viral cycle. As can be seen from Figure 3c, at MOI 0.1 NS1 could be detected as early as 9 hours postinfection.
Figure 3.

Generation of NDV(F3aa)-NS1 virus. (a) Schematic diagram of the generated NDV(F3aa)-NS1 virus genome. (b) Expression of the NS1 protein in NDV(F3aa)-NS1-infected Vero cells. Cells were infected at multiplicity of infection (MOI) 0.01, fixed at 18 hours postinfection, and stained with Dapi (blue), anti-NDV rabbit polyclonal antibody (green) and anti-NS1 mouse monoclonal antibody (red). NDV(B1) and NDV(F3aa)-infected cells were used as negative controls. (c) Time course of NS1 protein expression. Vero cells were infected with appropriate viruses at MOI 0.1 and collected at the indicated time points. Cells were lysed and analyzed by immunoblotting with antibodies to β-actin, NDV proteins, and influenza NS1. NDV, Newcastle disease virus.
NDV expressing influenza NS1 protein is effective in suppression of cellular innate immune responses
To confirm that the NS1 protein expressed within the context of the NDV genome antagonizes induction of the innate response in human cells, we performed an IFN bioassay for the time course of IFN induction (Supplementary Figure S1). To ensure that the recombinant virus is still capable of eliciting an antiviral response in noncancerous cells, the assay was performed in primary human foreskin fibroblasts (HFF-1) along with pancreatic cancer cell line Panc-1. Infection of HFF-1 cells with NDV(B1) and NDV(F3aa) viruses led to the induction of antiviral cytokines by 10 hours of infection (Supplementary Figure S1). By contrast, the infection with NDV(F3aa)-NS1 virus delayed the induction of antiviral cytokines by 6 hours, confirming the stronger ability of the NDV(F3aa)-NS1 virus to antagonize the induction of the innate immune response. Similar results were observed in Panc-1 cells (data not shown).
Recombinant NDV(F3aa)-NS1 efficiently replicates and lyses human and mouse melanoma cell lines
We proceeded to test the efficiency of the NDV(F3aa)-NS1 virus in lysis of various human tumor cell lines. The virus proved to be more effective than the NDV(F3aa) against the majority of cell lines tested (data not shown). We decided to focus on the melanoma lines as a model for our further studies.
To determine the extent of the NDV replication and cytotoxicity in the melanoma cell lines, we selected for our studies the human melanoma cell line SkMel-2, and the mouse melanoma cell line B16-F10. While all viruses exhibited significant cytolytic activity in both cell lines, NDV(F3aa), and NDV(F3aa)-NS1 were most effective (Figure 4a). At lower MOIs, NDV(F3aa)-NS1 proved to be the most effective cytolytic agent out of all viruses in both cell lines. Similar results were observed in the SkMel-119 and SkMel-197 cell lines (data not shown).
Figure 4.

NDV(F3aa)-NS1 virus replicates and induces oncolysis in human and mouse melanoma cell lines. (a) Cytotoxicity of the genetically engineered Newcastle disease virus (NDV) in B16-F10 and SkMel-2 cells. B16-F10 cells (left panel) and SkMel-2 cells (right panel) were infected with NDV(B1), NDV(F3aa), and NDV(F3aa)-NS1 viruses at the indicated MOIs. Cytotoxicity was assessed at 24, 48, and 72 hours by lactate dehydrogenase release assays. Lower MOIs were used in SkMel-2 cells due to higher susceptibility of the cells to NDV. (b) Efficiency of syncytia formation by the recombinant viruses. B16-F10 and SkMel-2 cells were infected with appropriate viruses at MOI 0.001 for 18 hours, and fixed and stained for NDV (green), NS1 (red), and Dapi (blue). (c) Replication of NDV in B16-F10 and SkMel-2 cell lines. Cells were infected at the indicated MOIs and the supernatants were collected at 24, 48, and 72 hours. (d) Interferon-β (IFN-β) induction in B16-F10 and SkMel-2 cells. Cells were infected with the indicated virus at MOI 1 and the supernatants were collected every 2 hours for 24 hours. Levels of IFN-β in the supernatant were assessed by murine and human IFN-β enzyme-linked immunosorbent assay.
Interestingly, both B16-F10 and SkMel-2 cells infected with the NDV(F3aa)-NS1 virus exhibited enhanced syncytia formation, when compared with the cells infected with NDV(F3aa) (Figure 4b). At MOIs of 0.1 and 1, the majority of NDV(F3aa)-NS1-infected cells were fused into syncytia by 24 hours of infection and detached from the plate by 48 hours. Formation of syncytia without significant early cytolysis suggested that expression of NS1 by NDV protected the cells from early lysis. This finding is consistent with the known antiapoptotic and anti-IFN properties of the influenza NS1 protein.26,27,31,32,33,34 We suspect that these functions of the NS1 protein allowed the fused cells to survive longer, which would in turn result in enhanced viral replication. Indeed, NDV(F3aa)-NS1 virus replicated to higher titers than the NDV(F3aa) virus (Figure 4c). Interestingly, when replication of the viruses was compared in the IFN-deficient cell line Vero and embryonated chick eggs, NDV(F3aa)-NS1 replicated to similar or even lower titers than the NDV(F3aa) virus (Supplementary Figure S2 and data not shown). These data support that the enhanced cytolytic effect and replication of NDV(F3aa)-NS1 virus are dependent on its ability to antagonize mammalian IFN response.
To confirm that the NDV(F3aa)-NS1 indeed antagonizes IFN induction in melanoma cell lines, we performed enzyme-linked immunosorbent assay (ELISA) for human and murine IFN-β secreted from the NDV-infected SkMel-2 and B16-F10 cells, respectively. As can be seen from Figure 4d, IFN-β induction was delayed in NDV(F3aa)-NS1-infected cells, when compared to the NDV(F3aa)-infected controls.
Overall, these findings suggested that the NDV(F3aa)-NS1 virus was an effective cytolytic agent for the melanoma cell lines in vitro. We proceeded to determine whether these properties of the virus would translate to being more effective as an antitumor agent in an animal tumor model.
NDV(F3aa)-NS1 effectively suppresses tumor growth and promotes mouse survival in a syngeneic murine melanoma model
To assess whether the observed in vitro cytolytic effect by NDV would translate to better antitumor efficacy in vivo, we used the syngeneic B16-F10 mouse footpad melanoma model. The B16-F10 cell line is known for its particularly aggressive tumor growth, early metastases, and very poor response to therapy.35,36,37,38,39 Because the naturally occurring NDV(B1) virus was previously demonstrated to be less effective in oncolysis than the fusogenic NDV(F3aa),24 and because the in vitro studies showed superior efficacy of the NDV(F3aa) viruses, NDV(B1) was not used in the mouse studies.
Toxicity studies were initially performed by inoculating 3 C57/BL6 mice subcutaneously and 3 C57/BL6 mice intravenously with 5 × 107 pfu of NDV(F3aa) and NDV(F3aa)-NS1 viruses. Over the next 2 weeks, none of the animals exhibited signs of distress and continued to gain weight (data not shown). To demonstrate the efficiency of NDV(F3aa)-NS1 virus in oncolytic therapy, we decided to use a low-dose treatment regimen (5 × 106 pfu) extended over 4–6 doses. For tumor studies, C57/BL6 mice from each group were inoculated into the right posterior footpad with 1 × 105 cultured B16-F10 cells. Tumors were allowed to develop for 7 days, at which point a pigmented tumor focus was visible in each of the animals. On day 7, the right posterior footpad of each animal was injected with 5 × 106 of NDV(F3aa), NDV(F3aa)-NS1, or phosphate-buffered saline (PBS) control. Eight mice were included in the control group, while 12–13 mice were used for each virus treatment group. The inoculation with 5 × 106 of each respective virus was repeated on days 9, 11, and 13 (four injections total). The most common side effect was the development of localized swelling at the injection site, which subsided over the next few days after the last inoculation.
None of the animals exhibited significant weight loss over the study period (data not shown). On day 25 after tumor implantation, 8/8 control mice developed tumors of significant size and were euthanized. In addition, six animals from the NDV(F3aa) group and five animals from the NDV(F3aa)-NS1 group were randomly selected, killed, and spleens were removed for analysis of tumor cellular immunity, while the rest of the animals continued to be followed for tumor growth (see below). As can be seen from Figure 5a, on day 25, only 2/13 animals in the NDV(F3aa) group and only 1/12 animals in the NDV(F3aa)-NS1 group exhibited significantly visible tumors, which were still smaller than the majority of tumors in the control group.
Figure 5.

NDV(F3aa)-NS1 suppresses tumor growth and promotes mouse survival in a syngeneic murine melanoma model. (a) Short-term tumor growth in B16-F10 melanoma-bearing mice treated with recombinant Newcastle disease viruses (NDVs) at 7 days. Mice were injected in the right posterior foot pad with 105 of cultured B16-F10 cells, and 7 days later were treated with 5 × 106 of the indicated viruses or phosphate-buffered saline (PBS) for a total of four injections. All eight mice from the control group and six randomly chosen mice from each virus group were killed on day 25 for immune studies (see Figure 6). (b) Short-term tumor growth in mice treated at 10 days. Starting on day 10 after tumor cell line injection, the mice were treated every other day with a total of six doses of 5 × 106 pfu of the indicated virus or PBS. When the largest tumors reached 8 mm in length, all of the animals were killed. (c) Long-term tumor growth follow-up in the treated mice. The remaining seven animals from each group in a were continued to be followed for 120 days, with tumor measurements being recorded every 2 days. (d) Summary of 120-day survival of the animals treated in a. Mice were killed when the tumors reached 8 mm in length. For experimental groups, only the mice included in the long-term study (n = 7 for each group) were included in the analysis (*P < 1 × 10−6).
To determine whether the viruses would be effective in clearing tumors at a later stage, the tumors were allowed to develop for 10 days and a total of six injections of each virus were used. As can be seen from Figure 5b, treatment with both NDV(F3aa) and NDV(F3aa)-NS1 viruses markedly suppressed tumor growth in all animals, with only minor tumors being detectable on the day of killing. These results indicate that increased number of treatments may be effective in clearing the tumors in later stages of development. The tumors were further processed for analysis for lymphocyte infiltration (see below).
The remaining animals from the early tumor treatment group continued to be followed to determine the long-term efficacy of each viral treatment. Over the next 120 days, 4/7 animals in the NDV(F3aa) group developed significant tumors and needed to be killed, while only 2/7 animals in the NDV(F3aa)-NS1 group developed tumors that required animal euthanasia (Figure 5c). Of note, these tumors took longer to develop than those in the animals from the NDV(F3aa) group. The remaining animals in each group either completely cleared the tumor (1/3 in the NDV(F3aa) group and 1/6 in the NDV(F3aa)-NS1 group), or had a persistent pigmented focus that remained stable. The overall survival for the animals in the long-term study was 0/8 for the control group, 3/7 for the NDV(F3aa) group, and 5/7 for the NDV(F3aa)-NS1 group (Figure 5d).
Tumors treated with NDV exhibit a high degree of CD4+ and CD8+ cell infiltration
We proceeded to determine whether the virus-treated tumors demonstrated a higher level of immune cell infiltration. As the tumors from the early treatment group were either too small or undetectable in size for cellular fractionation, tumors from the later treatment group were used in the analysis. Tumors from the killed mice described above were collected on day 22, dissected and filtered, and stained for CD4 and CD8 antigen expression. As can be seen from Figure 6a, tumors from the animals treated with NDV(F3aa) and NDV(F3aa)-NS1 viruses exhibited a high degree of both CD4 and CD8 cell infiltration, suggesting the development of an immune response to the infection and/or tumor. These results also indicated that suppression of the innate immune response by the NDV(F3aa)-NS1 virus had no negative effect on the adaptive immune response to the tumor and the infection.
Figure 6.

NDV treatment leads to tumor lymphocyte infiltration and generation of antimelanoma immune responses. (a) Tumor infiltration with CD4+ and CD8+ cells. Tumors collected from the animals described in 6D were dissociated into single-cell suspensions and analyzed by flow cytometry for presence of CD4 and CD8 cells. (b) Interferon-γ (IFN-γ) release from stimulated splenocytes. Splenocytes collected from the killed animals in 6a were cocultured with mitomycin-inactivated B16-F10 cells and IFN-γ was measured in the supernatants on day 3 of coculture (*P < 0.003, **P < 0.00006). (c) Melanoma-specific cytotoxicity of the stimulated splenocytes. Stimulated splenocytes described in b were cocultured with fresh B16-F10 cells for 4 hours at the indicated ratios and specific cytotoxic activity was determined by measurements of lactate dehydrogenase release (*P < 0.015, **P < 0.0007).
NDV-treated mice develop immune response against melanoma cells
To determine whether the treated mice develop an adaptive immune response to melanoma cells, animals killed on day 25 were assessed for the development of cytotoxic T lymphocyte (CTL) responses against B16-F10 cells. Splenocytes from the animals were cocultured with mitomycin C–inactivated B16-F10 cells for 5 days and assessed for IFN-γ release on day 3 and for B16-F10-specific CTL activity on day 5. As can be seen from Figure 6b,c, treatment with both NDV(F3aa) and NDV(F3aa)-NS1 viruses resulted in enhanced IFN-γ release and enhanced CTL activity, when compared to the control. These results suggested that tumor treatment with NDV resulted in generation of tumor-specific CTL responses, which may have contributed to the long-lasting antitumor effect of the virus.
Discussion
Sensitivity of NDV to the antiviral effects of IFN has been previously proposed to underlie its selective oncolytic properties.20,21 Based on these findings, the use of IFN-sensitive viruses has been suggested for oncolytic virus therapy, as many tumors have been demonstrated to be deficient in type I IFN response. Indeed, infection with viruses which are poor antagonists of type I IFN response results in enhanced apoptosis and release of cytokines, which stimulate immune responses to the virus and tumor.32,40 Despite these findings, the use of naturally occurring strains of NDV in human clinical trials suggested that many of the human tumors still demonstrate resistance to NDV infection.12,14 Studies of human tumor cell lines showed that tumor cells are still capable of mounting antiviral responses that could limit NDV replication and its oncolytic efficacy.24,41,42 In fact, sensitivity of NDV to the antiviral effects of human IFN was shown to play a major role in its restriction to the avian host.22 It then follows that, if the virus is too attenuated, the extent of the infection might not be significant enough to infect a wide field of cancer cells and to generate a potent adaptive immune response, especially because most of the tumors possess a high degree of vascular compromise and are limited in recruitment of immune cells. Thus, an oncolytic virus must possess an ability to replicate in cancer cells enough to cause effective oncolysis and allow for efficient antigen presentation. We felt that naturally occurring NDV strains, while effectively infecting tumor cells in vitro, might fail to spread in vivo to cause significant tumor regression or to allow for access by immune cells. Supporting our hypothesis, engineered measles virus with strong anti-IFN activity has been recently shown to be a more effective oncolytic agent than its IFN-sensitive counterpart.41 We used the reverse-genetics system for the lentogenic (avirulent) NDV Hitchner B1 strain (NDV(B1)) to engineer a virus with two alterations to improve the viral oncolytic properties: modification of the viral fusion protein to allow a more efficient spread between the infected cells, and introduction of an IFN-antagonist protein to attenuate the innate immune response to the infection.
The cleavage site of the NDV F protein has been postulated to be a major determinant of virulence.19,43,44 Modification of the cleavage site of the NDV F protein to a polybasic amino acid sequence allows the protein to be cleaved by intracellular proteases, making the virus more effective in entering cells and forming syncytia.19,43,44 In fact, introduction of the fusogenic NDV F into the VSV genome increased its syncytia-forming ability, which enhanced its oncolytic potential in head and neck squamous carcinomas.45 Importantly, while the fusogenic NDV appears to be slightly more virulent in avian species,44 it appeared to have no adverse effects in mice (ref. 24 and this study). As expected, in our study, generation of the virus with modified fusion protein (NDV(F3aa)) allowed for more efficient virus spread among tumor cells through formation of syncytia, resulted in increased viral replication, and showed enhanced oncolysis in various tumor cell lines, when compared to the wild-type NDV.
Despite the enhanced replication and spread, the NDV(F3aa) virus still induced significant antiviral signaling in the infected tumor cells (Figure 2). We felt that this effect would impose a limitation on the oncolytic efficacy of NDV in vivo. We thus sought to generate a virus that would attenuate the antiviral response to the infection, yet would still be sensitive enough to remain nonpathogenic in animal models. We were able to achieve this effect by introducing into NDV(F3aa) the NS1 protein of influenza A virus, which was previously shown to block the induction of antiviral signaling in influenza virus-infected cells.25,26,27 Infection of human primary fibroblasts with NDV(F3aa)-NS1 virus showed that the virus was still capable of inducing a strong antiviral response in noncancerous cells, though the induction was delayed when compared to the NDV(B1) and NDV(F3aa) (Supplementary Figure S1). These findings directly translated into increased viral oncolytic efficacy: When compared to the NDV(F3aa) virus, NDV(F3aa)-NS1 replicated more efficiently and resulted in enhanced formation of syncytia between tumor cells (Figure 4). This efficacy was further demonstrated in the syngeneic B16-F10 murine melanoma model. Intratumoral treatment with NDV(F3aa)-NS1 virus led to an effective tumor arrest or regression and high percentage of animal survival (Figure 5). Moreover, suppression of innate responses by the NS1 protein had no major effect on the generation of adaptive immune responses to the infected tumor cells, as was demonstrated by tumor lymphocyte infiltration, and generation of tumor-specific CTL responses (Figure 6).
Of note, none of the animals developed side effects to the virus, suggesting that the virus is still sufficiently attenuated not to cause disease. Several factors play a role in maintenance of the observed therapeutic safety margin. First of all, while the NS1 protein enhances the ability of NDV to replicate more efficiently in mammalian cells, the loss of viral species specificity for avian cells is not absolute.22 In particular, viral receptor specificity for avian cells as a result of binding of the viral HN to α2,3-linked sialoglycoproteins limits its infectivity in mammalian cells, as was shown for influenza hemagglutinin protein.46,47 Second, the Hitchner B1 strain used in our studies is a lentogenic (nonpathogenic) avian strain, possessing other attenuating mutations, which likely also limit its replication in mammalian cells. Third, as we show in Supplementary Figure S1, while the NDV(F3aa)-NS1 virus delayed the induction of IFN response in the primary human cells HFF-1, it did not completely abolish it. In fact, by 16 hours it resulted in induction of enough IFN to suppress further NDV replication, as shown by the bioassay (Supplementary Figure S1).
In our studies we demonstrated that intravenous or subcutaneous injection of at least 5 × 107 pfu of NDV(F3aa) or NDV(F3aa)-NS1 resulted in no significant side effects. For the study of viral oncolytic efficacy, we used a tenfold lower dose (5 × 106 pfu) per injection for a total of four treatments, primarily due to limitation of a solution volume that could be injected into the footpad. This dose is lower than the doses used in the majority of the previous studies using naturally occurring NDV strains.24,48,49 We speculate that the use of higher doses and administration of longer treatment regimens would result in an even more significant oncolytic effect and survival. Indeed, we show that treatment of B16-F10 tumors at later stages with six doses of NDV was effective in induction of tumor regressions, when compared to the untreated controls. In further support of these conclusions, previous studies showed that intratumoral and systemic administration of NDV were effective in treatment of human neuroblastoma xenografts49 and that multiple NDV doses were more effective than a single dose.
While our study used the syngeneic mouse melanoma model as the primary assessment tool of the viral oncolytic efficacy, we also show that the NDV(F3aa)-NS1 virus is a more effective oncolytic agent in a variety of human tumor cell lines. In particular, the virus proved to be cytotoxic to all of the human malignant melanoma cell lines tested, where it replicated to significantly higher titers and formed larger syncytia than its NDV(F3aa) counterpart. We speculated that these findings will be translated into the oncolytic efficacy of the virus in the in vivo models of human melanomas and other tumors. Studies are currently underway to determine the oncolytic effect of the NDV(F3aa)-NS1 virus against other human tumor types.
To our knowledge, this is the first report of the use of reverse genetics to enhance the oncolytic efficacy of NDV through modulation of the innate immune responses. Influenza virus NS1 protein exerts its anti-IFN effect through inhibition of RIG-I-mediated activation of type I IFN promoter.25,26 Similar to influenza NS1, proteins of other viruses have been previously shown to block innate antiviral signaling, both at the level of type I IFN induction and at the level of the response to circulating IFN. We are currently investigating the therapeutic efficacy and safety of recombinant NDV expressing these proteins against a variety of tumor types. The use of this strategy may represent an attractive means of maximizing the oncolytic efficacy of NDV, while maintaining the safety margin of the naturally occurring NDV strains.
Materials and Methods
Cell lines, antibodies, and other reagents. Human melanoma cell lines SkMel-2, SkMel-119, and SkMel-197 cells were maintained in Roswell Park Memorial Institute medium supplemented with penicillin, streptomycin, and 10% fetal calf serum. Hep-2, A549, B16-F10, and Panc-1 cells were maintained in high-glucose DMEM medium supplemented with 10% fetal calf serum, penicillin, and streptomycin. HFF-1, SCC-15, SCC-25, and Vero cells were maintained in MEM supplemented with 10% fetal calf serum, penicillin, and streptomycin. Rabbit polyclonal serum to NDV and mouse monoclonal anti-NS1 antibody were described previously.22,26 Antibody to β-actin was from Sigma (St Louis, MO). Fluorochrome-conjugated secondary antimouse and antirabbit antibodies for microscopy were from Molecular Probes (Eugene, OR). Conjugated anti-CD4 and anti-CD8 antibodies for flow cytometry were purchased from BD Pharmingen (San Jose, CA). Cytotox lactate dehydrogenase (LDH) release assay kits were purchased from Promega (Madison, WI). IFN-β ELISA kits were purchased from PBL (Piscataway, NJ).
Virus cloning and rescue. The NDV mutant viruses with modified F cleavage site (NDV(F3aa)) were previously described.43 To generate NDV(F3aa) virus expressing NS1, a DNA fragment encoding the influenza A/PR/8/34 NS1 protein flanked by the appropriate NDV-specific RNA transcriptional signals was inserted into the XbaI site created between the P and M genes of pT7NDV/F3aa. Viruses were rescued from cDNA using methods described previously18 and sequenced by reverse transcription PCR for insert fidelity.
IFN induction bioassay and ELISA. To determine the amount of IFN produced in cells infected with the different recombinant NDVs, we modified a bioassay described previously.50 Briefly, HFF-1 or Panc-1 cells were infected in six-well dishes with the viruses of interest at MOI 0.1. Infection supernatants were collected at different time points postinfection. The virus present in supernatants was inactivated in Stratalinker 1800 (Stratagene, Cedar Creek, TX) with six pulses of 300 mJ/cm2 UV light. Inactivated supernatants were then serially diluted and used to treat Vero cells in 96-well plates for 6 hours. Supernatants from uninfected cells and human IFN-β (R&D Systems, Minneapolis, MN) were used as negative and positive controls, respectively. Vero cells were subsequently washed and infected with NDV(B1)-GFP virus at MOI 0.1 for 20 hours. At 20 hours postinfection, the cells were examined for GFP expression under fluorescent microscope. Presence of antiviral cytokines in the supernatant induces an antiviral state in Vero cells, which prevents subsequent infection with NDV(B1)-GFP. In this assay, the amount of IFN is inversely proportional to the amount of GFP expression. IFN-β secretion was further measured by ELISA, according to the manufacturer's instructions (PBL).
LDH release assays. Cells were infected in 12-well plates for 24, 48, and 72 hours in triplicate for each condition. At each time point, the media was aspirated and the cells were washed with 1 ml of PBS. The cells were subsequently incubated with 1% Triton X-100 at 37 °C for 30 minutes. LDH activity in the lysates was determined using the Promega CytoTox 96 assay kit, according to the manufacturer's instructions.
Infections and virus titers. Cells of interest were incubated at room temperature with the virus in 12-well culture dishes at the indicated MOIs in a total volume of 100 µl. One hour after the incubation, the infection media was aspirated and the cells were incubated at 37 °C in 1 ml of DMEM with 0.3% bovine serum albumin. To the cells infected with wild-type NDV(B1) virus 10% chick allantoic fluid was added to the medium to allow for fusion protein activation. After 24, 48, and 72 hours, the supernatants were collected and the virus titers were determined by serial dilution and immunofluorescence in Vero cells.
Fluorescence microscopy. Cells were cultured on 10 mm cover slips and infected with viruses of interest at an MOI of 0.001. Twenty hours later, the cells were fixed with 5% formaldehyde in PBS and permeabilized with 1% Triton X-100. Proteins of interest were visualized by indirect immunofluorescence. Cells were probed with specific primary antibody for 2 hours at room temperature, washed, and labeled with secondary antibody conjugated to a specific fluorophore. DAPI staining was used to visualize cell nuclei. Labeled cells were visualized by laser scanning confocal microscopy (Leica TCS-SP, Bannockburn, IL) with TCS-SP software for image capture.
Mouse experiments. Cultured B16-F10 cells (1 × 105) were inoculated into the right posterior footpad of 6–8-week old C57/BL6J mice in a total volume of 50 µl. On day 7 or on day 10 postinoculation, the mice were treated by intratumoral injection of 5 × 106 NDV of interest or PBS, in a total volume of 50 µl. The treatments were repeated every other day for a total of four or six treatments, respectively.
Tumor sizes and mouse weights were recorded every other day. According to the institutional protocols, the animals were euthanized when the tumors reached 8 mm in length. On day 25, all eight animals from the control group and five animals from each treatment group were euthanized, and their spleens, popliteal lymph nodes, and tumors were collected. The remaining mice in each treatment group were observed for 120 days with measurement of tumor sizes every other day.
Splenocyte collection, IFN-γ release, and CTL assays. Spleens were removed from the euthanized animals, and splenocytes were isolated by passing the spleens through 80 µm nylon mesh filters. Cultured B16-F10 cells (5 × 105) were treated with 50 µg/ml of mitomycin C for 2 hours at 37 °C to induce cell cycle arrest. After the treatment, the cells were washed with PBS and incubated with 1 × 107 splenocytes in Roswell Park Memorial Institute medium with 10% fetal calf serum for 5 days. On day 3, the supernatants were collected and tested for IFN-γ release by ELISA using Quantikine M kit (R&D Systems). On day 5, the splenocytes were collected, washed, counted, and cocultured for 4 hours with 1 × 103 B16-F10 cells at the stimulator:effector ratios of 1:1.25, 1:2.5, 1:5, 1:10, 1:20, and 1:40. Specific CTL activity was determined by LDH release from the target cells utilizing the CytoTox 96 LDH kit from Promega according to the manufacturer's instructions.
Flow cytometry. The tumors of killed animals were dissected and manually dissociated with scissors. Dissociated tissue was then collected and incubated at 37 °C in 3 ml of Roswell Park Memorial Institute medium and 50 µl of Liberase Blendzyme 3 (Roche Diagnostics, Indianapolis, IN). After 30 minutes of incubation, 120 µl of 0.5 M EDTA were added to the cell homogenates and mixed for 5 minutes. Cells were then filtered using a cell strainer and stained with anti-CD4, and anti-CD8 antibodies (GK1.5, and 53–6.7, respectively; BD Pharmingen) and flow cytomety was done in a Cytomics FC500 machine (Beckman Coulter, Miami, FL) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Supplementary MaterialFigure S1. Genetically engineered NDV(F3aa)-NS1 suppresses the induction of antiviral state in the infected cells. IFN induction assay was performed in HFF-1 cells, as described in figure 2. HFF-1 were infected with NDV(B1), NDV(F3aa), NDV(F3aa)-NS1 viruses, or were mock-infected. Infection supernatants were collected post-infection at 2 hour intervals for 14 hours and were UV-inactivated. The supernatants were then used to treat Vero cells for 6 hours, which were subsequently infected with NDV-GFP at MOI 0.1 for 20 hours.Figure S2. Replication of NDV(F3aa) and NDV(F3aa)-NS1 viruses in the interferon-deficient cell line Vero. Cells were infected at the indicated MOIs in triplicate and the virus production in the supernatant was assessed at 24, 48, and 72 hours by immunofluorescence.
Supplementary Material
Genetically engineered NDV(F3aa)-NS1 suppresses the induction of antiviral state in the infected cells. IFN induction assay was performed in HFF-1 cells, as described in figure 2. HFF-1 were infected with NDV(B1), NDV(F3aa), NDV(F3aa)-NS1 viruses, or were mock-infected. Infection supernatants were collected post-infection at 2 hour intervals for 14 hours and were UV-inactivated. The supernatants were then used to treat Vero cells for 6 hours, which were subsequently infected with NDV-GFP at MOI 0.1 for 20 hours.
Replication of NDV(F3aa) and NDV(F3aa)-NS1 viruses in the interferon-deficient cell line Vero. Cells were infected at the indicated MOIs in triplicate and the virus production in the supernatant was assessed at 24, 48, and 72 hours by immunofluorescence.
Acknowledgments
This work was partially supported from the Northeast Biodefense grant U54 AI057158 (P.P.), NIH training grant T32AI07647 (D.Z.), Bill and Melinda Gates Foundation grant 38648 (P.P.), and Award from the Flight Attendant Medical Research Institute (Y.F.). Mount Sinai School of Medicine owns patent positions for reverse genetics of Newcastle disease viruses.
REFERENCES
- Alexander DJ. Newcastle Disease, Newcastle Disease Virus—An Avian Paramyxovirus. Kluwer Academic: Dordrecht, the Netherlands; 1988. pp. 1–22. [Google Scholar]
- Sinkovics JG., and , Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16:1–15. doi: 10.1016/s1386-6532(99)00072-4. [DOI] [PubMed] [Google Scholar]
- Karcher J, Dyckhoff G, Beckhove P, Reisser C, Brysch M, Ziouta Y, et al. Antitumor vaccination in patients with head and neck squamous cell carcinomas with autologous virus-modified tumor cells. Cancer Res. 2004;64:8057–8061. doi: 10.1158/0008-5472.CAN-04-1545. [DOI] [PubMed] [Google Scholar]
- Liang W, Wang H, Sun TM, Yao WQ, Chen LL, Jin Y, et al. Application of autologous tumor cell vaccine and NDV vaccine in treatment of tumors of digestive tract. World J Gastroenterol. 2003;9:495–498. doi: 10.3748/wjg.v9.i3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Gerhards R, Kirches E., and , Firsching R. Preliminary results of active specific immunization with modified tumor cell vaccine in glioblastoma multiforme. J Neurooncol. 2001;53:39–46. doi: 10.1023/a:1011856406683. [DOI] [PubMed] [Google Scholar]
- Steiner HH, Bonsanto MM, Beckhove P, Brysch M, Geletneky K, Ahmadi R, et al. Antitumor vaccination of patients with glioblastoma multiforme: a pilot study to assess feasibility, safety, and clinical benefit. J Clin Oncol. 2004;22:4272–4281. doi: 10.1200/JCO.2004.09.038. [DOI] [PubMed] [Google Scholar]
- Cassel WA., and , Murray DR. Treatment of stage II malignant melanoma patients with a Newcastle disease virus oncolysate. Nat Immun Cell Growth Regul. 1988;7:351–352. [PubMed] [Google Scholar]
- Batliwalla FM, Bateman BA, Serrano D, Murray D, Macphail S, Maino VC, et al. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med. 1998;4:783–794. [PMC free article] [PubMed] [Google Scholar]
- Wallack MK, Sivanandham M, Balch CM, Urist MM, Bland KI, Murray D, et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from a phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial J Am Coll Surg 199818769, 77–77.discussion [DOI] [PubMed] [Google Scholar]
- Ockert D, Schirrmacher V, Beck N, Stoelben E, Ahlert T, Flechtenmacher J, et al. Newcastle disease virus-infected intact autologous tumor cell vaccine for adjuvant active specific immunotherapy of resected colorectal carcinoma. Clin Cancer Res. 1996;2:21–28. [PubMed] [Google Scholar]
- Schlag P, Manasterski M, Gerneth T, Hohenberger P, Dueck M, Herfarth C, et al. Active specific immunotherapy with Newcastle-disease-virus-modified autologous tumor cells following resection of liver metastases in colorectal cancer. First evaluation of clinical response of a phase II-trial. Cancer Immunol Immunother. 1992;35:325–330. doi: 10.1007/BF01741145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorence RM, Roberts MS, O'Neil JD, Groene WS, Miller JA, Mueller SN, et al. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr Cancer Drug Targets. 2007;7:157–167. doi: 10.2174/156800907780058853. [DOI] [PubMed] [Google Scholar]
- Hotte SJ, Lorence RM, Hirte HW, Polawski SR, Bamat MK, O'Neil JD, et al. An optimized clinical regimen for the oncolytic virus PV701. Clin Cancer Res. 2007;13:977–985. doi: 10.1158/1078-0432.CCR-06-1817. [DOI] [PubMed] [Google Scholar]
- Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221–228. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL, et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol. 2002;20:2251–2266. doi: 10.1200/JCO.2002.08.042. [DOI] [PubMed] [Google Scholar]
- Csatary LK, Gosztonyi G, Szeberenyi J, Fabian Z, Liszka V, Bodey B, et al. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- Romer-Oberdorfer A, Mundt E, Mebatsion T, Buchholz UJ., and , Mettenleiter TC. Generation of recombinant lentogenic Newcastle disease virus from cDNA. J Gen Virol. 1999;80 Pt 11:2987–2995. doi: 10.1099/0022-1317-80-11-2987. [DOI] [PubMed] [Google Scholar]
- Nakaya T, Cros J, Park MS, Nakaya Y, Zheng H, Sagrera A, et al. Recombinant Newcastle disease virus as a vaccine vector. J Virol. 2001;75:11868–11873. doi: 10.1128/JVI.75.23.11868-11873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters BP, de Leeuw OS, Koch G., and , Gielkens AL. Rescue of Newcastle disease virus from cloned cDNA: evidence that cleavability of the fusion protein is a major determinant for virulence. J Virol. 1999;73:5001–5009. doi: 10.1128/jvi.73.6.5001-5009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S, Takimoto T, Scroggs RA., and , Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol. 2006;80:5145–5155. doi: 10.1128/JVI.02618-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiola C, Peeters B, Fournier P, Arnold A, Bucur M., and , Schirrmacher V. Tumor selective replication of Newcastle disease virus: association with defects of tumor cells in antiviral defence. Int J Cancer. 2006;119:328–338. doi: 10.1002/ijc.21821. [DOI] [PubMed] [Google Scholar]
- Park MS, Garcia-Sastre A, Cros JF, Basler CF., and , Palese P. Newcastle disease virus V protein is a determinant of host range restriction. J Virol. 2003;77:9522–9532. doi: 10.1128/JVI.77.17.9522-9532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Krishnamurthy S, Panda A., and , Samal SK. Newcastle disease virus V protein is associated with viral pathogenesis and functions as an α interferon antagonist. J Virol. 2003;77:8676–8685. doi: 10.1128/JVI.77.16.8676-8685.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A, Park MS, Martinez O, Chua MA, Xiao S, Cros JF, et al. Use of reverse genetics to enhance the oncolytic properties of Newcastle disease virus. Cancer Res. 2007;67:8285–8292. doi: 10.1158/0008-5472.CAN-07-1025. [DOI] [PubMed] [Google Scholar]
- Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M., Jr, and , Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of β interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, et al. Influenza A virus NS1 protein prevents activation of NF-κB and induction of α/β interferon. J Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sastre A, Egorov A, Matassov D, Brandt S, Levy DE, Durbin JE, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- Palese P., and , Shaw M. Fields BN, KD, Howley PM. edsOrthomyxoviridae: the virus and their replication. Lippincott Williams & Wilkins: Philadelphia; Field's Virology. 2007;2:1647–1689. [Google Scholar]
- Noah DL, Twu KY., and , Krug RM. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAS. Virology. 2003;307:386–395. doi: 10.1016/s0042-6822(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Vigil A, Martinez O, Chua MA., and , Garcia-Sastre A. Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol Ther. 2008;16:1883–1890. doi: 10.1038/mt.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, et al. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol. 2000;74:7989–7996. doi: 10.1128/jvi.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann M, Romirer I, Sachet M, Fleischhacker R, Garcia-Sastre A, Palese P, et al. A genetically engineered influenza A virus with ras-dependent oncolytic properties. Cancer Res. 2001;61:8188–8193. [PubMed] [Google Scholar]
- Zhirnov OP, Konakova TE, Wolff T., and , Klenk HD. NS1 protein of influenza A virus down-regulates apoptosis. J Virol. 2002;76:1617–1625. doi: 10.1128/JVI.76.4.1617-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasakova J, Ferko B, Kittel C, Sereinig S, Romanova J, Katinger H, et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J Gen Virol. 2005;86:185–195. doi: 10.1099/vir.0.80422-0. [DOI] [PubMed] [Google Scholar]
- Poste G, Doll J, Hart IR., and , Fidler IJ. In vitro selection of murine B16 melanoma variants with enhanced tissue-invasive properties. Cancer Res. 1980;40:1636–1644. [PubMed] [Google Scholar]
- Lee YS, Kim JH, Choi KJ, Choi IK, Kim H, Cho S, et al. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7–1 in an immunocompetent murine model. Clin Cancer Res. 2006;12:5859–5868. doi: 10.1158/1078-0432.CCR-06-0935. [DOI] [PubMed] [Google Scholar]
- Entin I, Plotnikov A, Korenstein R., and , Keisari Y. Tumor growth retardation, cure, and induction of antitumor immunity in B16 melanoma-bearing mice by low electric field-enhanced chemotherapy. Clin Cancer Res. 2003;9:3190–3197. [PubMed] [Google Scholar]
- Rochlitz C, Dreno B, Jantscheff P, Cavalli F, Squiban P, Acres B, et al. Immunotherapy of metastatic melanoma by intratumoral injections of Vero cells producing human IL-2: phase II randomized study comparing two dose levels. Cancer Gene Ther. 2002;9:289–295. doi: 10.1038/sj.cgt.7700441. [DOI] [PubMed] [Google Scholar]
- Seliger B, Wollscheid U, Momburg F, Blankenstein T., and , Huber C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001;61:1095–1099. [PubMed] [Google Scholar]
- Muster T, Rajtarova J, Sachet M, Unger H, Fleischhacker R, Romirer I, et al. Interferon resistance promotes oncolysis by influenza virus NS1-deletion mutants. Int J Cancer. 2004;110:15–21. doi: 10.1002/ijc.20078. [DOI] [PubMed] [Google Scholar]
- Haralambieva I, Iankov I, Hasegawa K, Harvey M, Russell SJ., and , Peng KW. Engineering oncolytic measles virus to circumvent the intracellular innate immune response. Mol Ther. 2007;15:588–597. doi: 10.1038/sj.mt.6300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, et al. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci USA. 2002;99:10736–10741. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, Steel J, Garcia-Sastre A, Swayne D., and , Palese P. Engineered viral vaccine constructs with dual specificity: avian influenza and Newcastle disease. Proc Natl Acad Sci USA. 2006;103:8203–8208. doi: 10.1073/pnas.0602566103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw OS, Koch G, Hartog L, Ravenshorst N., and , Peeters BP. Virulence of Newcastle disease virus is determined by the cleavage site of the fusion protein and by both the stem region and globular head of the haemagglutinin-neuraminidase protein. J Gen Virol. 2005;86:1759–1769. doi: 10.1099/vir.0.80822-0. [DOI] [PubMed] [Google Scholar]
- Shin EJ, Chang JI, Choi B, Wanna G, Ebert O, Genden EM, et al. Fusogenic vesicular stomatitis virus for the treatment of head and neck squamous carcinomas. Otolaryngol Head Neck Surg. 2007;136:811–817. doi: 10.1016/j.otohns.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Rivetz B., and , Lipkind M. Interaction of Newcastle disease virus strains differing in virulence with chicken red blood cell receptors. Arch Virol. 1985;85:231–255. doi: 10.1007/BF01314234. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. Sialobiology of influenza: molecular mechanism of host range variation of influenza viruses. Biol Pharm Bull. 2005;28:399–408. doi: 10.1248/bpb.28.399. [DOI] [PubMed] [Google Scholar]
- Schirrmacher V, Griesbach A., and , Ahlert T. Antitumor effects of Newcastle disease virus in vivo: local versus systemic effects. Int J Oncol. 2001;18:945–952. doi: 10.3892/ijo.18.5.945. [DOI] [PubMed] [Google Scholar]
- Phuangsab A, Lorence RM, Reichard KW, Peeples ME., and , Walter RJ. Newcastle disease virus therapy of human tumor xenografts: antitumor effects of local or systemic administration. Cancer Lett. 2001;172:27–36. doi: 10.1016/s0304-3835(01)00617-6. [DOI] [PubMed] [Google Scholar]
- Quinlivan M, Zamarin D, Garcia-Sastre A, Cullinane A, Chambers T., and , Palese P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J Virol. 2005;79:8431–8439. doi: 10.1128/JVI.79.13.8431-8439.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetically engineered NDV(F3aa)-NS1 suppresses the induction of antiviral state in the infected cells. IFN induction assay was performed in HFF-1 cells, as described in figure 2. HFF-1 were infected with NDV(B1), NDV(F3aa), NDV(F3aa)-NS1 viruses, or were mock-infected. Infection supernatants were collected post-infection at 2 hour intervals for 14 hours and were UV-inactivated. The supernatants were then used to treat Vero cells for 6 hours, which were subsequently infected with NDV-GFP at MOI 0.1 for 20 hours.
Replication of NDV(F3aa) and NDV(F3aa)-NS1 viruses in the interferon-deficient cell line Vero. Cells were infected at the indicated MOIs in triplicate and the virus production in the supernatant was assessed at 24, 48, and 72 hours by immunofluorescence.