

Abstract
Isoniazid (INH), a frontline antitubercular drug, inhibits InhA, the enoyl reductase from Mycobacterium tuberculosis, by forming a covalent adduct with the NAD cofactor. Here, we report that the INH-NAD adduct is a slow, tight-binding competitive inhibitor of InhA. Demonstration that the adduct binds to WT InhA by a two-step enzyme inhibition mechanism, with initial, weak binding (K–1 = 16 ± 11 nM) followed by slow conversion to a final inhibited complex (EI*) with overall Ki = 0.75 ± 0.08 nM, reconciles existing contradictory values for the inhibitory potency of INH-NAD for InhA. The first order rate constant for conversion of the initial EI complex to EI* (k2 = 0.13 ± 0.01 min–1) is similar to the maximum rate constant observed for InhA inhibition in reaction mixtures containing InhA, INH, NADH, and the INH-activating enzyme KatG (catalase/peroxidase from M. tuberculosis), consistent with an inhibition mechanism in which the adduct forms in solution rather than on the enzyme. Importantly, three mutations that correlate with INH resistance, I21V, I47T, and S94A, have little impact on the inhibition constants. Thus, drug resistance does not result simply from a reduction in affinity of INH-NAD for pure InhA. Instead, we hypothesize that protein–protein interactions within the FASII complex are critical to the mechanism of INH action. Finally, for M161V, an InhA mutation that correlates with resistance to the common biocide triclosan in Mycobacterium smegmatis, binding to form the initial EI complex is significantly weakened, explaining why this mutant inactivates more slowly than WT InhA when incubated with INH, NADH, and KatG.
Attempts to treat tuberculosis, a disease that kills more than two million people every year, are hindered by the spread of multidrug-resistant strains of Mycobacterium tuberculosis (MDRTB), the causative agent, and by the increased susceptibility of HIV-positive individuals to this disease (1–4). Although isoniazid (INH; Scheme 1) has been the most effective and widely used drug for the treatment of tuberculosis since the 1950s, the mode of action of this compound is still not completely understood. INH is a prodrug that is activated by the mycobacterial catalase-peroxidase enzyme KatG (Scheme 1) (5–8) and a substantial fraction of all clinical isolates that are resistant to INH result from KatG mutations (2, 9–11). Consequently, compounds that inhibit the ultimate molecular target(s) of INH, but that do not require activation by KatG, have tremendous promise as novel drugs for combating MDRTB.
Scheme 1.

Two enzymes, InhA and KasA, have been proposed as targets for INH. Both are members of the type II dissociated fatty acid biosynthesis pathway (FASII) in M. tuberculosis (Scheme 2), consistent with the observation that INH interferes with the biosynthesis of mycolic acids, very long chain fatty acid components of the mycobacterial cell wall. InhA, an enoyl reductase that catalyzes the NADH-dependent reduction of long chain trans-2-enoyl-acyl carrier proteins (ACPs), was first identified as a target by Jacobs and coworkers (6, 12) who observed mutations in the inhA gene in INH-resistant clinical isolates and identified a point mutant (S94A) that conferred resistance to INH and ethionamide in Mycobacterium smegmatis and in Mycobacterium bovis. Subsequently, Blanchard, Sacchettini, and coworkers (13–15) demonstrated that InhA was inhibited in vitro by a covalent adduct formed between activated INH and the nicotinamide head group of NAD (Scheme 1). InhA mutations observed in INH-resistant clinical isolates were found to be localized to the cofactor binding site and were shown to result in decreased affinity of the purified enzyme for NADH (12, 16), consistent with the hypothesis that binding of NADH to the enzyme precedes adduct formation. In addition, Vilcheze et al. (17) used a temperature-sensitive mutation in the inhA gene to show that the phenotypic response to InhA inactivation in M. smegmatis was identical to that caused by treatment with INH, thereby validating InhA as a target for drug discovery. However, although there is convincing evidence that InhA is inhibited by INH, Barry and coworkers (18) have also proposed that KasA, one of three ketoacyl synthases in the FASII pathway (Scheme 2), is a target for INH in vivo. Subsequent experiments involving the effect of drugs on gene (19, 20) and protein expression (21), and studying the effect of InhA and KasA expression on drug resistance (20, 22), have highlighted the apparent complexity in the mode of action of INH.
Scheme 2.

To better understand the molecular basis for INH resistance in clinical isolates carrying InhA mutations, we purified and isolated the INH-NAD adduct and quantified its affinity toward WT and drug-resistant mutants. Our studies show that INH-NAD is a slow, tight-binding competitive inhibitor of InhA that binds with an overall dissociation constant of Ki = 0.75 ± 0.08 nM. Importantly, the inhibition parameters for I21V and I47T, two InhA mutations detected in INH-resistant clinical isolates, and S94A, the mutant identified by Jacobs and coworkers, are similar to those for WT enzyme, indicating that resistance in these cases cannot be simply explained in terms of a decrease in the ability of InhA to be inhibited by INH.
Experimental Procedures
Inactivation of WT InhA by INH or Benzoic Hydrazide and Isolation of the NAD Adducts. Inactivation reactions contained 0.3 μM InhA, 300 μM INH, 1 μM MnCl2, and 150 μM NADH in 100 ml of 100 mM Na2HPO4 (Pi) buffer, pH 7.5, at room temperature. Following InhA inhibition (2 h), the inactivation reactions were concentrated to 1 ml and the inhibited protein was purified by gel filtration using a 30 × 1-cm Sephadex G-25 column preequilibrated with Pi buffer. The fractions containing the InhA–inhibitor complex were pooled and concentrated. Urea was added to a final concentration of 6 M and the sample was applied to a mono Q HR5/5 FPLC column (Pharmacia). The adduct was eluted by using a linear gradient from 0 to 1 M KCl in 10 mM triethanolamine, pH 7.4, and was directly used for the inhibition experiments (23). Purified INH-NAD had a UV-visible absorbance spectrum similar to that reported earlier with λmax at 260 and 326 nm with an A260/A326 ratio of 3.8–3.9 (24). RP-HPLC analysis of the adduct was performed by using an analytical C18 Vydac column running a 0–20% gradient over 30 min with 20 mM NH4+ CH3COO–/1.75% CH3CN as buffer A and 95% CH3CN/5% H2O as buffer B. Adduct purified by ion exchange chromatography exhibited two peaks by HPLC that likely represent the two forms of INH-NAD assigned as the B and E species by Wilming and Johnsson (25). Reanalysis of these two fractions after HPLC demonstrated that they were unstable, as judged by changes in their UV-visible absorption as a function of time and the appearance of multiple peaks on further HPLC analysis, suggesting that the conditions used for HPLC may promote the interconversions described by Wilming and Johnsson. Importantly, adduct purified by ion exchange chromatography maintained a constant A260/A326 ratio and exact mass (electron spray ionization MS) for >24 h. A similar method was used to prepare the adduct derived from benzoic hydrazide, an analog of INH, and NADH (BH-NAD), which also had λmax at 260 and 326 nm and an A260/A326 ratio of 3.8–3.9. Like INH-NAD, the BH-NAD proved to be unstable after HPLC but was stable when purified by ion exchange chromatography. Adduct concentrations were determined by using ε260 = 27 mM–1·cm–1 or ε326 = 6.9 mM–1·cm–1 (24). Molecular weight was as follows: ESI-MS M– calculated for INH-NAD (C27H31N8O15P2) = 769.2 (found = 769.1); and ESI-MS M– calculated for BH-NAD (C28H32N7O15P2) = 768.2 (found = 768.1).
Slow-Binding Inhibition Kinetics. trans-2-Dodecenoyl-CoA (DD-CoA) as well as the WT and mutant InhA proteins were as used in a previous study (26). Kinetic assays were carried out on a Cary 100 Bio spectrophotometer (Varian) in Pi buffer at 25°C by following the oxidation of NADH to NAD+ at 340 nm. InhA activity was monitored by adding enzyme (0.1–1 nM) to assay mixtures containing DD-CoA (85 μM) and NADH (250 μM) plus inhibitor (INH-NAD 4–300 nM and BH-NAD 0.4–200 nM). Assays at lower enzyme concentrations were stabilized by the addition of 8% (vol/vol) glycerol and BSA (0.1 mg/ml). Reactions were allowed to proceed until the progress curves became linear, indicating that the steady-state velocity had been attained. The resulting progress curves were fitted to the integrated rate Eq. 1 for slow binding inhibition (27, 28) by nonlinear regression analysis.
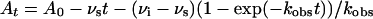 |
[1] |
In Eq. 1, At and A0 are the absorbances at time t and time 0, kobs is the pseudo-first order rate constant for approach to the steady state, whereas νi and νs correspond to the initial and final slopes of the progress curve. Values for νi, νs, and kobs were obtained at each inhibitor concentration. Low enzyme concentrations were used so that [E] « [I] and also to ensure that only a small fraction of the DD-CoA and NADH were consumed during the course of the measurement. This ensured that progress curves were approximately linear in the absence of added inhibitor. Reactions at the lowest inhibitor concentrations ([I]/[E] = 4–30) approached the steady state only after a significant fraction (10–20%) of the DD-CoA had been consumed. Consequently, data were truncated at an A340 value where the rate of DD-CoA reduction in the absence of inhibitor had decreased 10%, ensuring that substrate depletion did not significantly contribute to curvature in the progress curves.
Results were analyzed in terms of a two-step inhibition mechanism in which the initial rapid binding of the inhibitor to enzyme is followed by a second slow step that results in the final enzyme–inhibitor complex (Scheme 3). The following equations (29) were used to obtain the equilibrium and kinetic constants for the inhibition.
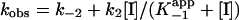 |
[2] |
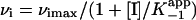 |
[3] |
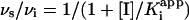 |
[4] |
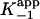 and
and 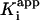 are the apparent dissociation constants for the initial enzyme–inhibitor complex (EI) and the final enzyme–inhibitor complex (EI*), respectively. Because inhibition is competitive with respect to both DD-CoA and NADH, the true values of K–1 and Ki are obtained by dividing the apparent values by 1 + [S1]/Km1 + [S2]/Km2, where [S1] and [S2] are the concentrations of the two substrates and Km1 and Km2 are their respective Michaelis constants (29). Kinetic parameters for WT and mutant InhA enzymes obtained through regular steady-state kinetics were similar to the values reported earlier (20, 27); the KmDD-CoA values used were 46, 70, 36, 104, and 307 μM for WT, I21V, I47T, S94A, and M161V, respectively, and the KmNADH values were 66, 120, 152, and 250 μM and 1.3 mM, respectively.
are the apparent dissociation constants for the initial enzyme–inhibitor complex (EI) and the final enzyme–inhibitor complex (EI*), respectively. Because inhibition is competitive with respect to both DD-CoA and NADH, the true values of K–1 and Ki are obtained by dividing the apparent values by 1 + [S1]/Km1 + [S2]/Km2, where [S1] and [S2] are the concentrations of the two substrates and Km1 and Km2 are their respective Michaelis constants (29). Kinetic parameters for WT and mutant InhA enzymes obtained through regular steady-state kinetics were similar to the values reported earlier (20, 27); the KmDD-CoA values used were 46, 70, 36, 104, and 307 μM for WT, I21V, I47T, S94A, and M161V, respectively, and the KmNADH values were 66, 120, 152, and 250 μM and 1.3 mM, respectively.
Scheme 3.

Measurement of koff. The dissociation rate constants of INH-NAD from WT InhA and the I47T mutant were determined in dialysis experiments with radiolabeled adduct. The enzyme was inhibited by INH and 32P-NAD (1 Ci/μmol; 1 Ci = 37 GBq), concentrated and purified by gel-filtration chromatography. An excess of BH-NAD was added to the dialysis buffer (final concentration 25 nM in 22 ml) to displace the INH-32P-NAD adduct from the inhibited InhA (0.5 nM, 500 μl), which was placed in the dialysis bag. The dialysate was sampled at various times to measure the displaced INH-32P-NAD. The data were fitted to Eq. 5, where koff is the dissociation rate constant, Nt are the radioactive counts (cpm) at time t, and N0 are the final radioactive counts (cpm).
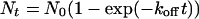 |
[5] |
Results
Adduct Synthesis. The INH-NAD adduct was purified by isolating inhibited enzyme from inactivation reactions containing INH, NADH and Mn2+, followed by release of the adduct by denaturation and purification by ion exchange chromatography (23). INH-NAD adduct purified by using aqueous buffers proved sufficiently stable (>24 h) for the inhibition studies. While we were investigating the stability of the INH-NAD adduct, discussions with W. van der Donk (personal communication) led to the hypothesis that an adduct formed from benzoic hydrazide, thereby lacking the pyridine nitrogen, might be more stable than the INH-NAD adduct. The BH-NAD adduct was synthesized by using the procedure described for INH-NAD except that complete inhibition of 0.3 μM InhA occurred with t1/2 = 24 h. The BH-NAD absorption spectrum remained constant for >48 h, supporting the hypothesis that this adduct is more stable than INH-NAD.
Progress Curves for the Inhibition of WT and Mutant InhAs by the INH-NAD Adduct. Progress curves for the reaction of WT InhA (Fig. 1) and the mutant InhAs (data not shown) with DD-CoA and NADH in the presence of varying [INH-NAD] showed that the turnover velocity decreased exponentially with time, from an initial velocity νi to a final, steady-state velocity νs (Fig. 1 A). Higher concentrations of INH-NAD cause the steady state to be reached more quickly but give a lower νs. This behavior indicates that INH-NAD is a slow, tight-binding reversible inhibitor (27) that interacts rapidly with the enzyme to form an initial complex, EI, that then slowly converts to a more stable (i.e., more strongly bound) final inhibited complex, EI* (Scheme 3). Individual progress curves for the reaction of WT InhA (Fig. 1) and mutant InhAs (data not shown) fitted well to Eq. 1, which describes this inhibition mechanism, allowing values for νi, νs, and kobs to be determined for each inhibitor concentration.
Fig. 1.

(A) Time-dependent inactivation of WT InhA by 0–80 nM INH-NAD. The solid curves represent the best fit of the data to Eq. 1 for slow binding inhibition. (B) kobs from A plotted as a function of [I]. Data are fitted by using Eq. 2 to give values for k2, k–2, and 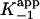 . (C) Initial velocity (νi) from A plotted as a function of [I]. Data are fitted by using Eq. 3 to give a value for
. (C) Initial velocity (νi) from A plotted as a function of [I]. Data are fitted by using Eq. 3 to give a value for 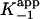 . (D) Ratio of νs and νi from A plotted as a function of [I]. Data are fitted by using Eq. 4 to give a value for
. (D) Ratio of νs and νi from A plotted as a function of [I]. Data are fitted by using Eq. 4 to give a value for 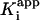 .
.
Consistent with the mechanism shown in Scheme 3, kobs displayed a hyperbolic dependence on [INH-NAD] (Fig. 1B), and nonlinear curve fitting by using Eq. 2 gave values for k2, k–2, and 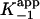 (Table 1). To accurately fit the data for I47T, the value of k–2, given by the intercept on the y axis, was constrained to the value of koff (0.013 min–1) obtained in the radioactive dialysis experiments (see below). Because the rapid, reversible binding of E+I to form EI is also manifest as an instantaneous effect of [I] on the initial reaction velocity νi,
(Table 1). To accurately fit the data for I47T, the value of k–2, given by the intercept on the y axis, was constrained to the value of koff (0.013 min–1) obtained in the radioactive dialysis experiments (see below). Because the rapid, reversible binding of E+I to form EI is also manifest as an instantaneous effect of [I] on the initial reaction velocity νi, 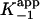 can also be determined by fitting the dependence of νi on [I] with Eq. 3 (Fig. 1C). Finally,
can also be determined by fitting the dependence of νi on [I] with Eq. 3 (Fig. 1C). Finally, 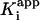 , the overall apparent dissociation constant for the dissociation of EI* to free E and free I was obtained by using Eq. 4 to fit the dependence of νs/νi on [I] (Fig. 1D). As expected, the values of
, the overall apparent dissociation constant for the dissociation of EI* to free E and free I was obtained by using Eq. 4 to fit the dependence of νs/νi on [I] (Fig. 1D). As expected, the values of 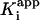 for each enzyme are smaller than the corresponding values of
for each enzyme are smaller than the corresponding values of 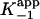 , indicating that the interaction of the adduct with the enzyme is substantially stronger in EI* than in EI (Table 1).
, indicating that the interaction of the adduct with the enzyme is substantially stronger in EI* than in EI (Table 1).
Table 1. Kinetic constants for the inhibition of WT and mutant InhA by INH-NAD.
| Enzyme |
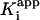 , nM , nM |
K-1, nM | k2*, min-1 | k-2, min-1 | k2/K-1*, min-1·nM-1 |
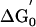 †, nM †, nM |
Ki‡, nM |
|---|---|---|---|---|---|---|---|
| WT | 100 ± 75* | 16 ± 11§ | 0.13 ± 0.01 | 0.016 ± 0.007* | 0.0088 ± 0.0066 | 5.0 ± 0.5 | 0.75 ± 0.08 |
| 150 ± 42¶ | 23 ± 6∥ | 0.017 ± 0.001** | |||||
| I21V | 13 ± 6* | 3.0 ± 1.3§ | 0.095 ± 0.008 | <0.02†† | 0.045 ± 0.004 | 9.1 ± 3.4 | 2.1 ± 0.8 |
| 89 ± 14¶ | 21 ± 3∥ | ||||||
| I47T | 17 ± 6* | 3.4 ± 1.2§ | 0.093 ± 0.012 | 0.013 ± 0.005** | 0.027 ± 0.013 | 7.0 ± 0.9 | 1.4 ± 0.2 |
| 74 ± 22¶ | 15 ± 4∥ | ||||||
| S94A | 280 ± 80* | 99 ± 28§ | 0.44 ± 0.33 | 0.004 ± 0.001* | 0.0045 ± 0.0045 | 15.0 ± 3.0 | 5.3 ± 1.1 |
| 230 ± 70¶ | 81 ± 23∥ | ||||||
| M161V | 1,100 ± 340* | 770 ± 230§ | 0.15 ± 0.36 | 0.020 ± 0.003* | 0.0002 ± 0.0005 | 5.0 ± 0.7 | 3.4 ± 0.5 |
| 1,400 ± 1,100¶ | 940 ± 720∥ |
From plot of kobs versus [I]
From plot of vs/vi versus [I].
Corrected value of Ki.
Corrected value of K-1 from plot of kobs versus [I].
From plot of vi versus [I].
Corrected value of K-1 from plot of vi versus [I].
koff determined by monitoring the rate of release of INH-32P-NAD.
Upper limit of k-2 estimated from plot of kobs versus [I].
As expected (15), kobs was observed to decrease with an increase in the concentration of either substrate (data not shown), showing that inhibition is competitive toward both substrates in agreement with previous studies (30). To correct for the concentration of substrates in the inhibition assays, a factor of 1 + [S1]/Km1 + [S2]/Km2 was used to calculate the true inhibition constants, K–1 and Ki, from the apparent inhibition constants, 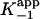 and
and 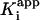 (29). The corrected values of K–1 and Ki are given in Table 1, together with the value of k2/K–1, the second order rate constant for the reaction of E+I to form EI*.
(29). The corrected values of K–1 and Ki are given in Table 1, together with the value of k2/K–1, the second order rate constant for the reaction of E+I to form EI*.
Direct Determination of koff. Time-dependent inactivation of WT InhA and the mutant proteins by using varying concentrations of BH-NAD adduct showed that this compound is also a slow, tight-binding inhibitor. These preliminary data indicated that BH-NAD was a suitable competing ligand for use in the radioactive dialysis experiments. Subsequently, the dissociation rate constant (koff) of INH-NAD from WT InhA and the I47T mutant were independently determined by using radiolabeled ligand (Table 1).
Discussion
In this work, we have purified and isolated the INH-NAD adduct and quantified its affinity toward WT and drug-resistant mutants of InhA. I21V and I47T are clinically observed drug-resistant mutants (16, 31), while the S94A mutation has been shown to confer resistance to INH in M. smegmatis, M. bovis, and M. tuberculosis (6). The M161V InhA mutant was identified in a triclosan-resistant strain of M. smegmatis (32) and was shown to be inhibited significantly less rapidly by activated INH than the WT enzyme. M161V InhA is therefore of interest because the latter observation implies that exposure to triclosan, an antibacterial additive in many consumer products, could stimulate the emergence of INH-resistant InhA in M. tuberculosis (33, 34).
Interaction of INH-NAD with WT InhA. Our data show that INH-NAD acts as a slow, tight-binding competitive inhibitor of WT InhA. The results indicate a mechanism of at least two steps, in which an initial enzyme–inhibitor complex EI is formed rapidly and then slowly converts to a final inhibited complex EI*. The inhibition constant for the formation of EI is K–1 = 16 ± 11 nM, whereas for the overall inhibition of the enzyme Ki = 0.75 ± 0.08 nM, indicating that conversion of EI to EI* is accompanied by a significant [–RTln(K–1/Ki) = –1.8 kcal/mol] increase in the strength of interaction with the adduct (Fig. 2). Importantly, the K–1 values obtained by analysis of kobs versus [I] (16 ± 11 nM) and νi versus [I] (23 ± 6 nM) are in good agreement with each other, supporting the robustness of the data and the validity of the method used for data analysis. In addition, the rate of dissociation of the adduct from the enzyme determined by analysis of the kobs versus [I] data (k–2 = 0.016 ± 0.007 min–1) is identical within experimental error to the value of koff determined directly by monitoring release of radiolabeled adduct from the enzyme (0.017 ± 0.001 min–1). The close agreement between k–2 and koff supports the notion that the compound isolated from the inhibited enzyme and used in the inhibition experiments is identical to the species that initially inhibits the enzyme. The slow rate of adduct dissociation from the enzyme, characterized by t1/2 = 43 min, may explain why inhibition by INH had previously been considered irreversible (7, 35).
Fig. 2.

Free-energy profile for the interaction of INH-NAD with InhA. 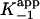 was calculated for the EI and EI* states by using K–1 and Ki and applying a standard state of 1 μM. The free energy of the transition state for the conversion of EI to EI* (‡2) is determined from k2.(A) Free-energy profile for WT InhA. (B) Free-energy profiles for WT enzyme (heavy line) and M161V (light line, ⋄), and data points for I21V (▿), I47T (○), and S94A (□).
was calculated for the EI and EI* states by using K–1 and Ki and applying a standard state of 1 μM. The free energy of the transition state for the conversion of EI to EI* (‡2) is determined from k2.(A) Free-energy profile for WT InhA. (B) Free-energy profiles for WT enzyme (heavy line) and M161V (light line, ⋄), and data points for I21V (▿), I47T (○), and S94A (□).
Two previous estimates of the affinity for INH-NAD binding to InhA led to conflicting conclusions. Wilming and Johnsson (25) reported that the INH-NAD adduct was a competitive inhibitor of InhA with a KI of 100 ± 50 nM, determined by initial rate assays. KI determined in this way is actually an IC50 for the initial interaction of adduct and enzyme and is therefore related to K–1 determined in the present work (16 ± 11 nM) by the expression IC50 = K–1(1 + [S1]/Km1 + [S2]/Km2), where [S1], Km1, [S2], and Km2 refer to the concentrations and Km values for octenoyl-CoA and NADH used in their study. In a separate study, Lei et al. (24) estimated a Kd of <0.4 nM based on the observation that no significant dissociation of the adduct could be detected from 0.36 μM inhibited complex. This approach measures the dissociation of I from EI*, and, consistent with this notion, their estimate for Kd compares well with our value for Ki (0.75 ± 0.08 nM), which directly corresponds to the dissociation constant for EI*. Consequently, K–1 and Ki determined in the present work provide an explanation for the two apparently conflicting estimates for the binding of the INH-NAD adduct to WT InhA published previously.
Interaction of INH-NAD with Mutant InhAs. Extension of the inhibition analysis to the mutant enzymes enabled us to directly probe the molecular basis for INH resistance. Our studies reveal that INH-NAD is also a slow, tight-binding inhibitor of the mutant enzymes. For I21V and I47T, the Ki values are very close to the value obtained for WT InhA. In addition, the values of K–1 for I21V and I47T are, if anything, slightly lower than that observed for the WT enzyme, while values of the other rate constants for enzyme inhibition, notably k2, k2/K–1 and k–2, as well as koff for I47T determined by dialysis, are also very similar. The similarities in the interaction of INH-NAD with WT, I21V, and I47T InhA (Table 1) can clearly be seen in Fig. 2, in which free energy profiles have been constructed for the binding of INH-NAD to each enzyme. These results show that no significant differences exist between the inhibition of WT, I21V, and I47T InhA by INH in vitro that could account for the correlation between these InhA mutations and drug resistance in vivo.
For S94A, Ki and K–1 are ≈5-fold higher than for WT InhA. However, k2, the first order rate constant for the conversion of EI to EI*, and k2/K–1, the second order rate constant for formation of EI from E+I, are identical within experimental error to the values for WT InhA. Comparison of INH-NAD binding to S94A and WT InhA is shown graphically in Fig. 2, which highlights the similarities in the interaction of adduct with both enzymes. Based on these data, it seems unlikely that the small differences in Ki and K–1 are sufficient to account for the 80-fold increase in MIC observed in strains of M. smegmatis containing the S94A InhA mutation (21). Indeed, because the concentration of adduct within the cell is presumably at least equivalent to that of the enzyme (1–10 μM), small changes in nM inhibition constants are unlikely to have any significant effect on the fraction of uninhibited enzyme.
Whereas Ki for M161V is similar to that observed for WT InhA, k2/K–1, the second order rate constant for the reaction of E+I to form EI*, is substantially reduced. This effect is entirely due to an increase in K–1; k2, the rate constant for conversion of EI to EI*, is unchanged. Thus, whereas the final inhibited complexes (EI*) for WT and M161V InhA have similar stabilities, the rate of EI* formation for M161V InhA is significantly slower because of the weaker interaction of INH-NAD in the initial EI complex. A graphical representation of the M161V binding data is shown in Fig. 2, illustrating that the only significant difference in the interaction of INH-NAD with all four enzymes is the increase in free energy of the EI state for M161V and the corresponding increase in the free energy of the transition state for conversion of EI to EI* for this mutant. Although we do not know the structural change that accompanies conversion of EI to EI*, it is clear that the M161V mutation causes a 10-fold reduction in the affinity of the INH-NAD adduct in the EI complex but not in EI*. This destabilization persists in the transition state for conversion to EI* but is largely relieved in EI* itself. This observation suggests that the M161 side chain makes important contacts with the adduct when it first binds to the enzyme but that formation of EI* reduces the importance of this interaction, possibly due to movement of M161 away from the inhibitor.
Mechanism of Drug Resistance. Previous studies, in which it was shown that the InhA mutations resulted in a decreased affinity of the enzyme for NADH, supported an attractive hypothesis that binding of NADH to the enzyme was a prerequisite for formation of the INH-NAD adduct and that drug resistance arose from decreased occupancy of the enzyme by NADH at physiologically relevant NADH concentrations (10 μM). Indeed, Kd for the interaction of NADH with InhA is raised 23-, 60-, and 141-fold for I21V, S94A, and I47T, respectively, compared with WT enzyme (16). However, these changes in NADH affinity do not carry over into significant alterations in affinity of INH-NAD for either enzyme, and the present data support a model in which the INH-NAD adduct is formed initially in solution and then binds to the enzyme. This conclusion is supported by the observation that the pseudo-first order rate constants for the inactivation of I21V, I47T, and S94A by INH, in the presence of 10 μM NADH and the INH activator KatG or Mn2+, are only 1.2- to 1.5-fold slower than the rate of inactivation of WT InhA (16, 34). Furthermore, the maximum rate we have observed for InhA inactivation in the presence of activated INH when INH, KatG, and NADH were preincubated for 5 h before InhA addition (t1/2 ≈ 5 min; data not shown) is close to k2 determined in the present work (0.13 ± 0.01 min–1), again supporting the notion that the INH-NAD adduct does not form on the enzyme. Finally, WT InhA is inactivated at similar rates when NADH (70 μM) is replaced by NAD+ (75 μM) in the inactivation reaction (ref. 7 and data not shown), and because the affinity of NAD+ for InhA is very low (Kd > 250 μM) (13), this result also argues against the notion that cofactor binding precedes adduct formation in the enzyme's active site (25).
In the present study, only the M161V mutation has a significant impact on the interaction of INH-NAD with the enzyme. The alteration in the stability of the intermediate EI complex can completely account for the significant reduction (56-fold) reported in the rate of inactivation of this mutant by activated INH (34). However, even for this mutant, the overall potency of inhibition by INH-NAD is decreased only ≈5-fold compared with WT InhA.
Our results clearly establish that the INH-NAD adduct is an extremely potent inhibitor of InhA, with Ki ≈1 nM. However, the similarity in kinetic and thermodynamic parameters for the interaction of INH-NAD with WT, I21V, I47T, and S94A InhA indicates that INH-resistance observed in bacterial strains harboring the latter mutations does not arise from an alteration in the affinity of the adduct for these enzymes. There are at least two distinct hypotheses for how these apparently contradictory observations might be reconciled. Firstly, it is possible that InhA is inhibited by INH in vivo, but that this inhibition is unrelated to the antimycobacterial activity of INH and, thus, mutations in InhA are inconsequential to the mechanism of drug resistance. This explanation is consistent with the body of work that points to KasA as the target of INH action but suffers from the major weakness that it requires both the high potency with which INH-NAD inhibits InhA and also the multiple examples of mutations in InhA found in INH-resistant strains to be essentially irrelevant to INH activity. An alternative explanation is suggested by the results of Bloch et al. (36), who have described the purification of a high MW FASII complex from M. smegmatis that catalyzes the acyl carrier protein (ACP)-dependent synthesis of long chain fatty acids and that contains both InhA and MabA (FabG1) (37, 38). This result suggests that InhA interacts directly with other components of the FASII pathway. If these interactions serve to modulate the catalytic properties of InhA, then it is possible that the resistance-associated mutations in InhA affect the sensitivity of the enzyme to INH inhibition only in the context of the multienzyme complex, and not when InhA is tested in isolation as is the case in our in vitro assays. In addition, the presence of both InhA and KasA within the same noncovalent complex might ultimately account for the two apparently conflicting proposals concerning the in vivo target of INH.
In support of the hypothesis that heterotypic associations might modulate the sensitivity of InhA to inhibition, there is evidence for allostery in ligand binding to InhA. Results from cross-linking studies and analytical ultracentrifugation indicate that adduct binding to InhA modulates contacts at the dimer–dimer interface in the InhA tetramer (data not shown). In addition, Basso et al. (16) observed that InhA mutations occurring in INH-resistant strains not only decreased the affinity of NADH for the enzyme but also resulted in the appearance of cooperativity in cofactor binding. These data indicate that homotypic interactions within the InhA tetramer can modulate, or be modulated by, ligand binding and that this allosteric response to ligand binding is affected by InhA mutations that occur in INH-resistant bacterial strains. If InhA does participate in heterotypic protein–protein interactions in vivo, it follows that these interactions could indeed modulate InhA activity and sensitivity toward INH.
Acknowledgments
We thank Dr. Robert A. Rieger for electron spray ionization MS data collection and Dr. Melanie Nilsson for performing initial HPLC analysis of the INH-NAD adduct. This work was supported by National Institutes of Health Grant AI44639 (to P.J.T.). P.J.T. is an Alfred P. Sloan Fellow.
Abbreviations: DD-CoA, trans-2-dodecenoyl-CoA; INH, isoniazid; KatG, catalase/peroxidase from Mycobacterium tuberculosis.
References
- 1.Bloom, B. R. & Murray, C. J. (1992) Science 257, 1055–1064. [DOI] [PubMed] [Google Scholar]
- 2.Heym, B., Honore, N., Truffot-Pernot, C., Banerjee, A., Schurra, C., Jacobs WR, Jr., van Embden, J. D., Grosset, J. H. & Cole, S. T. (1994) Lancet 344, 293–298. [DOI] [PubMed] [Google Scholar]
- 3.Perlman, D. C., ElSadr, W. M., Heifets, L. B., Nelson, E. T., Matts, J. P., Chirgwin, K., Salomon, N., Telzak, E. E., Klein, O., Kreiswirth, B. N., et al. (1997) AIDS 11, 1473–1478. [DOI] [PubMed] [Google Scholar]
- 4.Rattan, A., Kalia, A. & Ahmad, N. (1998) Emerg. Infect. Dis. 4, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang, Y., Heym, B., Allen, B., Young, D. & Cole, S. (1992) Nature 358, 591–593. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee, A., Dubnau, E., Quemard, A., Balasubramanian, V., Um, K. S., Wilson, T., Collins, D., de Lisle, G. & Jacobs, W. R., Jr. (1994) Science 263, 227–230. [DOI] [PubMed] [Google Scholar]
- 7.Johnsson, K., King, D. S. & Schultz, P. G. (1995) J. Am. Chem. Soc. 117, 5009–5010. [Google Scholar]
- 8.Basso, L. A., Zheng, R. J. & Blanchard, J. S. (1996) J. Am. Chem. Soc. 118, 11301–11302. [Google Scholar]
- 9.Stoeckle, M. Y., Guan, L., Riegler, N., Weitzman, I., Kreiswirth, B., Kornblum, J., Laraque, F. & Riley, L. W. (1993) J. Infect. Dis. 168, 1063–1065. [DOI] [PubMed] [Google Scholar]
- 10.Musser, J. M., Kapur, V., Williams, D. L., Kreiswirth, B. N., van Soolingen, D. & van Embden, J. D. (1996) J. Infect. Dis. 173, 196–202. [DOI] [PubMed] [Google Scholar]
- 11.Ramaswamy, S. V., Reich, R., Dou, S. J., Jasperse, L., Pan, X., Wanger, A., Quitugua, T. & Graviss, E. A. (2003) Antimicrob. Agents Chemother. 47, 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dessen, A., Quemard, A., Blanchard, J. S., Jacobs, W. R., Jr., & Sacchettini, J. C. (1995) Science 267, 1638–1641. [DOI] [PubMed] [Google Scholar]
- 13.Quemard, A., Sacchettini, J. C., Dessen, A., Vilcheze, C., Bittman, R., Jacobs, W. R., Jr., & Blanchard, J. S. (1995) Biochemistry 34, 8235–8241. [DOI] [PubMed] [Google Scholar]
- 14.Quemard, A., Dessen, A., Sugantino, M., Jacobs, W. R., Jr., Sacchettini, J. C. & Blanchard, J. S. (1996) J. Am. Chem. Soc. 118, 1561–1562. [Google Scholar]
- 15.Rozwarski, D. A., Grant, G. A., Barton, D. H. R., Jacobs, W. R., Jr., & Sacchettini, J. C. (1998) Science 279, 98–102. [DOI] [PubMed] [Google Scholar]
- 16.Basso, L. A., Zheng, R., Musser, J. M., Jacobs, W. R., Jr., & Blanchard, J. S. (1998) J. Infect. Dis. 178, 769–775. [DOI] [PubMed] [Google Scholar]
- 17.Vilcheze, C., Morbidoni, H. R., Weisbrod, T. R., Iwamoto, H., Kuo, M., Sacchettini, J. C. & Jacobs, W. R., Jr. (2000) J. Bacteriol. 182, 4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mdluli, K., Slayden, R. A., Zhu, Y., Ramaswamy, S., Pan, X., Mead, D., Crane, D. D., Musser, J. M. & Barry, C. E., III (1998) Science 280, 1607–1610. [DOI] [PubMed] [Google Scholar]
- 19.Wilson, M., DeRisi, J., Kristensen, H. H., Imboden, P., Rane, S., Brown, P. O. & Schoolnik, G. K. (1999) Proc. Natl. Acad. Sci. USA 96, 12833–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slayden, R. A., Lee, R. E. & Barry, C. E., III (2000) Mol. Microbiol. 38, 514–525. [DOI] [PubMed] [Google Scholar]
- 21.Kremer, L., Dover, L. G., Morbidoni, H. R., Vilcheze, C., Maughan, W. N., Baulard, A., Tu, S. C., Honore, N., Deretic, V., Sacchettini, J. C., et al. (2003) J. Biol. Chem. 278, 20547–20554. [DOI] [PubMed] [Google Scholar]
- 22.Larsen, M. H., Vilcheze, C., Kremer, L., Besra, G. S., Parsons, L., Salfinger, M., Heifets, L., Hazbon, M. H., Alland, D., Sacchettini, J. C. & Jacobs, W. R., Jr. (2002) Mol. Microbiol. 46, 453–466. [DOI] [PubMed] [Google Scholar]
- 23.Orr, G. A. & Blanchard, J. S. (1984) Anal. Biochem. 142, 232–234. [DOI] [PubMed] [Google Scholar]
- 24.Lei, B., Wei, C. J. & Tu, S. C. (2000) J. Biol. Chem. 275, 2520–2526. [DOI] [PubMed] [Google Scholar]
- 25.Wilming, M. & Johnsson, K. (1999) Angew. Chem. Int. Ed. Engl. 38, 2588–2590. [DOI] [PubMed] [Google Scholar]
- 26.Parikh, S., Moynihan, D. P., Xiao, G. & Tonge, P. J. (1999) Biochemistry 38, 13623–13634. [DOI] [PubMed] [Google Scholar]
- 27.Morrison, J. F. & Walsh, C. T. (1988) Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301. [DOI] [PubMed] [Google Scholar]
- 28.Stone, S. R. & Hermans, J. M. (1995) Biochemistry 34, 5164–5172. [DOI] [PubMed] [Google Scholar]
- 29.Hood, D. B., Huntington, J. A. & Gettins, P. G. (1994) Biochemistry 33, 8538–8547. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen, M., Quemard, A., Broussy, S., Bernadou, J. & Meunier, B. (2002) Antimicrob. Agents Chemother. 46, 2137–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musser, J. M. (1995) Clin. Microbiol. Rev. 8, 496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMurry, L. M., McDermott, P. F. & Levy, S. B. (1999) Antimicrob. Agents Chemother. 43, 711–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMurry, L. M., Oethinger, M. & Levy, S. B. (1998) Nature 394, 531–532. [DOI] [PubMed] [Google Scholar]
- 34.Parikh, S. L., Xiao, G. & Tonge, P. J. (2000) Biochemistry 39, 7645–7650. [DOI] [PubMed] [Google Scholar]
- 35.Johnsson, K. & Schultz, P. G. (1994) J. Am. Chem. Soc. 116, 7425–7426. [Google Scholar]
- 36.Odriozola, J. M. & Bloch, K. (1977) Biochim. Biophys. Acta 488, 198–206. [DOI] [PubMed] [Google Scholar]
- 37.Marrakchi, H., Laneelle, G. & Quemard, A. (2000) Microbiology 146, 289–296. [DOI] [PubMed] [Google Scholar]
- 38.Marrakchi, H., Ducasse, S., Labesse, G., Montrozier, H., Margeat, E., Emorine, L., Charpentier, X., Daffe, M. & Quemard, A. (2002) Microbiology 148, 951–960. [DOI] [PubMed] [Google Scholar]
- 39.Rozwarski, D. A., Vilcheze, C., Sugantino, M., Bittman, R. & Sacchettini, J. C. (1999) J. Biol. Chem. 274, 15582–15589. [DOI] [PubMed] [Google Scholar]