

Abstract
The mixed-lineage kinases (MLK) are serine/threonine protein kinases that regulate mitogen-activated protein (MAP) kinase signaling pathways in response to extracellular signals. Recent studies indicate that MLK activity may promote neuronal cell death through activation of the c-Jun NH2-terminal kinase (JNK) family of MAP kinases. Thus, inhibitors of MLK activity may be clinically useful for delaying the progression of neurodegenerative diseases, such as Parkinson's. In proliferating non-neuronal cells, MLK may have the opposite effect of promoting cell proliferation. In the current studies we examined the requirement for MLK proteins in regulating cell proliferation by examining MLK function during G2 and M-phase of the cell cycle. The MLK inhibitor CEP-11004 prevented HeLa cell proliferation by delaying mitotic progression. Closer examination revealed that HeLa cells treated with CEP-11004 during G2-phase entered mitosis similar to untreated G2-phase cells. However, CEP-11004 treated cells failed to properly exit mitosis and arrested in a pro-metaphase state. Partial reversal of the CEP-11004 induced mitotic arrest could be achieved by overexpression of exogenous MLK3. The effects of CEP-11004 treatment on mitotic events included the inhibition of histone H3 phosphorylation during prophase and prior to nuclear envelope breakdown and the formation of aberrant mitotic spindles. These data indicate that MLK3 might be a unique target to selectively inhibit transformed cell proliferation by disrupting mitotic spindle formation resulting in mitotic arrest.
Keywords: MAP kinase, Mixed-lineage kinase, Mitosis, Cell cycle, Phosphorylation
1. Introduction
Serine/threonine protein kinases belonging to the mixed-lineage kinase (MLK) family control the activity of mitogen-activated protein (MAP) kinases and are involved primarily regulating tissue development and apoptotic responses [1]. The MLK family includes MLK1–4, the dual leucine zipper-bearing kinase (DLK), and the leucine zipper kinase (LZK) [1,2]. MLK proteins each contain highly conserved structural motifs that are important for protein interactions and signal transduction including a Src homology 3 (SH3) domain, a catalytic domain, two leucine zipper (LeuZip) motifs, and a Cdc42/Rac interactive binding (CRIB) motif [1]. The regulation and function of the MLK1–4 subfamily has been best characterized for MLK2, which is localized to brain, skeletal muscle and testis, and MLK3, which is ubiquitously expressed [1]. Currently, only partial sequence information is known for MLK1 and the physiological functions for MLK4 have not been defined.
MLK proteins are activated by upstream G-proteins including Cdc42 and Rac1, which recruit cytoplasmic MLK proteins to the plasma membrane by interacting with the CRIB motif [3]. MLK protein activity may also be regulated through auto-inhibition involving intramolecular interactions between the SH3 domain and a single proline residue between the CRIB domain and the leucine zipper region [4]. Recently, the stability of MLK3 has been linked to the ability for MLK3 to interact with heat shock protein-90 (HSP90), where recent data suggests that geldanamycin inhibition of HSF90 results in decreased MLK3 stability and expression [5].
The functional role of MLK proteins in activating the stress-activated protein kinases and apoptotic pathways in neuronal cells has been well characterized [6–8]. MLK3 is a potent activator of the c-Jun N-terminal kinase (JNK) MAP kinases by directly phosphorylating and activating the JNK kinases, MKK4 and 7 [9,10]. MLK3 may also activate the p38 MAP kinases through direct phosphorylation of MKK3 and 6, which are the primary p38 MAP kinase activators [10]. MLK proteins have been proposed to play a critical role in the progression of neurodegenerative diseases such as Parkinson's, Huntington's, and Alzheimer's through a mechanism involving MLK induced JNK activation, cytochrome c release, and caspase activation of apoptotic pathways [6,8]. Thus, several small molecule inhibitors of MLK activity belonging to the indolocarbazole family, which include CEP-1347 (KT7515) and CEP-11004 (KT-8138), are being tested in clinical trials and may prove beneficial in preventing premature neuronal cell death [6–8,11,12].
In non-neuronal cells MLK proteins may also function in promoting cell proliferation. For example, overexpression of MLK3 has been reported to induce NIH 3T3 cell transformation and growth on soft agar through a mechanism involving MEK1 and ERK activation [13]. Similarly, growth factors may utilize MLK3 to activate B-Raf and the ERK pathway in proliferating tumor cells [14]. MLK3 activity may regulate cell proliferation by affecting centrosome organization and microtubule stability during mitosis through a mechanism that is JNK-independent [15]. Additional evidence suggests that MLK3 may promote cell transformation by mediating morphological changes in cells expressing an activated Rac1 mutant [16]. Recently, overexpression of a MLK-like protein, MLTK, was shown to be sufficient to induce cell transformation in a nude mouse model [17]. Thus, ample evidence links MLK proteins with promoting cell proliferation.
In the current studies, we sought to examine the requirement for MLK proteins in regulating cell proliferation specifically as cells progressed through G2-phase and mitosis. Our data suggest that inhibition of MLK3 causes a mitotic arrest through a mechanism involving disruption of microtubule formation and spindle pole assembly. These data suggest that inhibition of MLK3 may be a novel approach for inhibiting the proliferation of transformed cells.
2. Materials and methods
2.1. Cell culture and reagents
HeLa, NIH 3T3, HEK293, A549, or MRC-5 cells were grown in a DMEM supplemented with 10% fetal bovine serum (FBS) and penicillin (100 U/ml) and streptomycin (100 μg/ml). Estrogen receptor (ER) negative breast cancer cells, SUM-159, were obtained from the University of Michigan Human Breast Cancer Cell Lines (SUM-Lines) and grown in DMEM+10% FBS. In some experiments, cells were transfected with cDNA (1 μg) for pEGFP (BD Biosciences/Clontech, Palo Alto, CA), hemagglutinin (HA)-tagged MLK3, kinase dead mutant MLK3 (MLK3 K144R), GFP-tagged MLK2 (kindly provided by Dr. Donna Dorow), Flag-tagged DLK, or HA-tagged JNK1 using Lipofectamine® (Invitrogen, Carlsbad, CA). Transfected cells were harvested 16–24 h after transfection. Antibodies specific for MLK3 (C-20), JNK (C-17), phospho-JNK1/2 (G-7), Cyclin B1 (GNS1), MKK2 (C-16), and Cdc2 (#17) were purchased from Santa Cruz Biotech (Santa Cruz, CA). Antibodies for p62 nucleoporin (N43620) and phospho-specific histone H3 (serine 10) (Cat# 07-081) were purchased from BD Biosciences (Palo Alto, CA) and Upstate Biotechnology (Charlottesville, VA), respectively. CEP-11004 (kindly provided by Cephalon Inc. West Chester, PA) was reconstituted at a stock concentration of 0.4 mM in DMSO.
2.2. Cell synchronization
In some experiments, cells were synchronized by double thymidine block as we have previously described [18,19]. Briefly, cells were incubated for 16 h with 2 mM thymidine in 10% FBS containing DMEM. The excess thymidine was washed off with Hanks Buffered Saline Solution (HBSS) and cells were released back into the cell cycle with complete medium. After 8 h, cells were treated a second time with 2 mM thymidine in 10% FBS containing DMEM for another 16 h to arrest cells at the G1/S-phase boundary. Synchronized cells were then washed with HBSS, and released back into the cell cycle and harvested at various time points after G1/S release for analysis. Cell cycle analysis was done by fluorescence activated cell sorting (FACS) as described below. In some experiments, CEP-11004 was added to the synchronized cells 5 h after release from G1/S block, which corresponded to late S-phase or early G2-phase.
2.3. Cell proliferation assays
Following treatments cells were counted on a hemocy-tometer following trypsinization and staining with trypan blue (Sigma). Greater than 95% of the cells excluded trypan blue dye. Cell proliferation was also measured using colony formation assay. Growing cells were trypsinized and replated (1000 cells per 100 mm dish) in the presence or absence of various concentrations of CEP-11004. Following incubation for 8–14 days, cells were fixed for 10 min in 4% paraformaldehyde and stained with 0.2% crystal violet (in 20% methanol) for 1–2 min. Cells were washed several times with distilled water and colonies formed (at least 40 cells/per colony) were counted.
2.4. Immunoblotting and immunoprecipitation
For protein analysis, cells were washed twice with cold phosphate buffered saline (PBS), lysed with 300 μl of tissue lysis buffer (TLB) (20 mM Tris-base, pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, 25 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 10% glycerol, 1 mM sodium orthovanadate, 1 mM phenylmethy-sulfonyl fluoride (PMSF), and 1 mM benzamidine), and centrifuged at 20,000 × g to clarify lysates. Approximately 20 μg of total protein was separated by SDS-PAGE. Proteins were transferred to PVDF membrane (Perkin Elmer Life Sciences; Boston, MA), blocked for 1–2 h with 5% nonfat dry milk in Tris-buffered saline(TBS) (50 mM Tris-base, pH 7.4, 0.15 M NaCl, and 0.1% Tween-20), and incubated with the primary antibodies in TBS containing 1% BSA solution for 1 to 16 h. Membranes were washed several times in TBS-Tween solution and incubated with HRP conjugated anti-mouse or anti-rabbit antibodies (0.1 μg/ml). Immunoreactivity was detected by enhanced chemiluminescence (ECL; Amersham, Buckinghamshire, England).
In some experiments, proteins were immunoprecipitated using 1 μg/ml of primary antibodies incubated with cell lysate for 4–16 h at 4 °C. Protein A or G sepharose (Amersham/Pharmacia, Uppsala, Sweden)) was added for an additional 4 h and the immune complexes were washed twice with kinase buffer (KB, 25 mM Hepes, pH 7.4, 25 mM MgCl2, and 1 mM DTT). The proteins in the immune-complex were separated by SDS-PAGE and analyzed by immunoblotting.
2.5. Immunofluorescence
Cells grown on round glass coverslips in 6-cm plates were synchronized at the G1/S-phase boundary using excess thymidine as described in the previous section. At the indicated time after release back into the cell cycle, coverslips were fixed with 4% paraformaldehyde for 8 min and permeabilized with 0.1% Triton X-100 in PBS for 2 min. Cell staining patterns were confirmed in cells that were fixed and permeabilized with cold methanol for 10 min. Cells were stained with antibodies against MLK3, α-tubulin, cyclin B1, phospho-specific histone H3, or p62 nucleoporin followed by incubation with fluorescein or Texas Red conjugated secondary antibodies and counterstaining for cellular DNA with 4′,6-diamidino-2-phenylindole (DAPI, 0.2 μg/ml in PBS). Cells were identified using Nikon E800 Epi-fluorescence microscope (Image Systems, Columbia, MD) and captured with a Hamamatsu CCD camera. Cell images were processed using IPlab software (Scanalytics, Fairfax, VA).
2.6. MLK3 kinase assays
Wild type MLK3 or catalytically inactive MLK3KR (K144R) were immunoprecipitated from transfected cells treated with or without CEP-11004 and the immune-complex was washed with cold KB supplemented with 0.2 mM sodium orthovanadate. Immunoprecipitated MLK3 was incubated for 45 min at 30 °C with kinase buffer containing 10 μCi γ- 32P ATP, 20 μM cold ATP, and 0.5 μg myelin basic protein (MBP) as a substrate. The reaction was quenched with 2× SDS sample buffer, separated by SDS-PAGE and transferred to PVDF membranes (Perkin Elmer, Boston, MA). Radioactive phosphate incorporation into MBP and MLK3 were analyzed by phosphorimager (Amersham Biosciences/Molecular Dynamics) and immunoblotting, respectively.
2.7. FACS analysis
Synchronized cells were trypsinized, washed with PBS, fixed with 3 ml of cold (−20 °C) 70% ethanol, and stored at 4 °C overnight. Cells were then incubated with 100 μg/ml propidium iodide (Sigma) dissolved in 0.2 M Tris, pH 7.5, 20 mM EDTA, 1 mg/ml RNase A (Sigma) for 1 h at room temperature, and diluted with an equal volume of PBS. DNA content was measured by flow cytometry (FACScan Analyzer, BD Biosciences) and analyzed using the Sync Wizard Model, ModFit LT software (BD Biosciences). To determine the percentage of G1, S and G2/M-phase cells, the settings for 2N and 4N peaks were defined within each experiment from the G1/S arrested cells and applied to all samples within a given experiment.
2.8. Cdc2 kinase activity
Cdc2 was immunoprecipitated from cell lysates by incubating with Cdc2 antibody (0.5 μg) for 2 h on ice. The lysates were then incubated with protein G-sepharose with mixing at 4 °C for an additional 2 h. The immune-complex was washed with cold kinase buffer and incubated for 30 min at 30 °C with kinase buffer containing 10 μCi γ-32P ATP, 20 μM cold ATP, and 2.5 μg histone (Type III-SS, Sigma) as a substrate. The reactions were then stopped with SDS-PAGE sample buffer and the proteins were resolved by SDS-PAGE. Incorporation of 32P into histone was determined by phosphorimager analysis.
2.9. Mitotic index assay
Cells grown on coverslips were synchronized at the G1/ S-phase boundary as described above. In some cases, MLK3 was co-expressed with the pEGFP vector in order to identify transfected cells. At 5 h after release from thymidine-induced G1/S-phase block, cells were incubated in the presence or absence of varying concentrations of CEP-11004. At varying times after release, coverslips were fixed and cellular DNA was stained with DAPI. GFP-positive mitotic cells in prophase, pro-metaphase, metaphase, anaphase, or telophase were identified, counted, and expressed as a fraction of the total cells counted to determine the mitotic index. Within each experiment, 300 to 350 cells were counted for each condition and time point.
3. Results
3.1. Inhibition of MLK proteins blocks proliferation and causes G2 or M-phase arrest in HeLa cells
It was recently reported that inhibitors of MLK proteins prevented proliferation of Ras-transformed but had no effect on normal NIH 3T3 cells [20]. This prompted us to analyze the effects of MLK inhibition on cell cycle events in transformed cells. We first tested whether the addition of the MLK inhibitor CEP-11004 would inhibit proliferation of transformed HeLa cells. As shown, whereas HeLa cell proliferation was inhibited by CEP-11004, the proliferation of NIH 3T3 cells was unaffected (Fig. 1A). The proliferation of other transformed cells was examined using a colony formation assay. As shown in Fig. 1B, CEP-11004 caused a dose dependent inhibition of A549 airway epithelia carcinoma cell colony formation. A similar inhibition of colony formation was observed in HeLa cells and ER negative SUM159 breast cancer cells treated with CEP-11004 (data not shown).
Fig. 1.
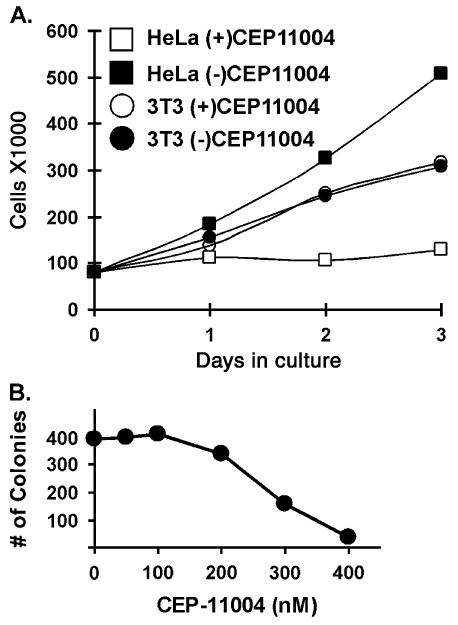
Inhibition of MLK activity blocks cell proliferation. (A) HeLa or NIH 3T3 cells were grown in the absence (closed squares or circles) or presence (open squares or circles) of CEP-11004 (500 nM) for up to 72 h. The number of cells was counted at time 0 and after 24, 48, or 72 h in each condition. (B) Trypsinized A549 cells (1000 per treatment) were plated in the presence of varying doses of CEP-11004. After 14 days, the resulting colonies were stained with crystal violet, and counted. Similar results were obtained with HeLa and SUM152 breast cancer cells.
To determine the nature of the CEP-11004 induced inhibition of cell proliferation, FACS analysis of DNA content was used to determine the effects of CEP-11004 on cell cycle progression. As shown, HeLa or HEK293 cells treated with CEP-11004 for 20 h accumulated 4N DNA indicative of arrest in G2 or M-phase of the cell cycle (Fig. 2A and B). In contrast, the DNA content profile in NIH 3T3 cells or normal diploid lung fibroblasts MRC-5 cells was similar in the absence or presence of CEP-11004 (Fig. 2C and D).
Fig. 2.
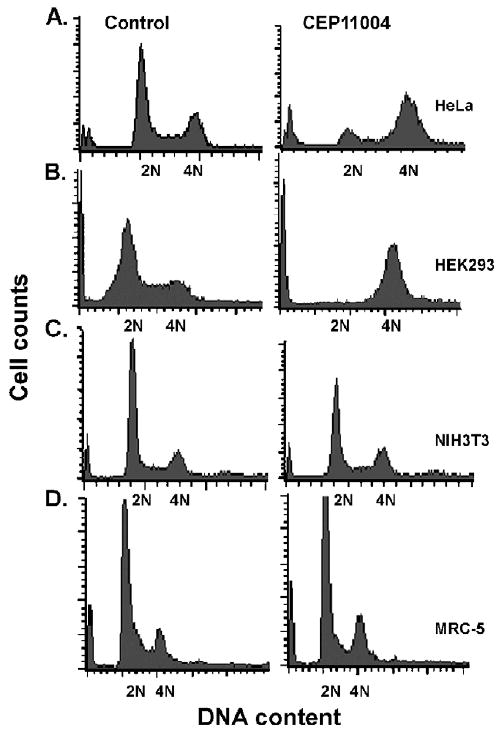
CEP-11004 causes G2/M-phase arrest in transformed cells. HeLa (A), HEK293 (B), NIH3T3 (C), or MRC5 (D) cells were treated in the absence (control) or presence of CEP-11004 (500 nM) for 15 h and 2N or 4N DNA content in cells was measured by FACS analysis following propidium iodide staining.
Based on previous data indicating that MLK3 is activated during G2-phase and mitosis [15], we examined the effects of CEP-11004 on cell cycle progression in synchronized HeLa cells. Cells synchronized at the G1/S-phase boundary were released back into the cell cycle for 5 h to allow completion of most of S-phase. The cells were treated with or without CEP-11004 for varying times and DNA content was determined by FACS. Between 7 and 9 h after release from G1/S-block, untreated cells are in G2 and M-phase as demonstrated by the increase in cells with 4N DNA (Fig. 3A). By 11 h after G1/S release, untreated cells have exited mitosis and re-entered G1-phase (Fig. 3A). In contrast, CEP-11004 treated cells arrest in G2 or M-phase as evident by the sustained accumulation of cells containing 4N DNA at 11 and 13 h after G1/S-phase release (Fig. 3B). In agreement, CEP-11004 induced arrest in G2 or M-phase was indicated by the sustained cyclin B1 expression (Fig. 3C) and elevated Cdc2 kinase activity at 11 and 13 h after G1/S-phase release as compared to untreated synchronized cells (Fig. 3D).
Fig. 3.
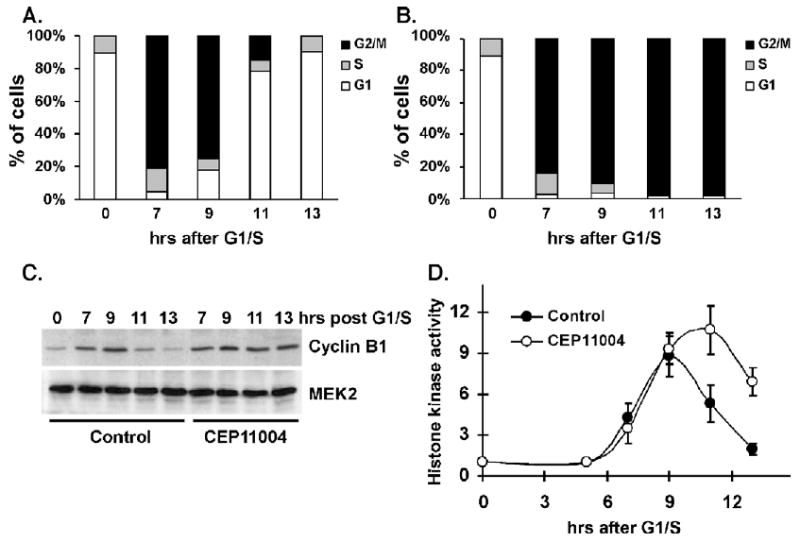
CEP-11004 treatment of synchronized cells during late S-phase/early G2-phase causes G2/M-phase arrest. HeLa cells were synchronized at the G1/S-phase boundary by double thymidine block, released back into the cell cycle, and treated 5 h after release in the absence (A) or presence (B) of CEP-11004 (500 nM). Cells were collected by trypsinization at indicated various times after G1/S release, the DNA was stained with propidium iodide, and the percentage of cells in G1, S, or G2/M-phase was determined by FACS. (C) HeLa cells were synchronized as in (A), released into the cell cycle, and treated in the absence or presence of CEP-11004 (500 nM). Lysates collected at various times were immunoblotted for cyclin B1 (top panel) or total MKK2 as a protein loading control (lower panel). (D) Histone kinase activity in synchronized cells was determined in untreated (closed circles) or CEP-11004 treated (open circles) cells following immunoprecipitation of Cdc2. Data shows the average and standard error from 3 independent experiments.
3.2. CEP-11004 inhibits MLK3 but not other related kinases
We next examined the specificity of CEP-11004 for inhibition of MLK proteins and not other related kinases. These experiments focused on the MLK3 isoform for the following reasons. First, MLK3 is ubiquitously expressed and the best characterized of the MLK proteins. Second, although MLK2 shares more than 70% sequence identify in the kinase catalytic domain and other regions with MLK3, MLK2 expression is restricted to brain, skeletal muscle, and testis [21]. Third, the full sequence of MLK1 has yet to be identified. Lastly, MLK3 has recently been implicated to be involved in G2/M-phase transitions [15]. Therefore, we first tested the ability for CEP-11004 to block MLK3 activity. HA-tagged MLK3 wild type was overexpressed in HeLa cells and treated with or without CEP-11004 for 4 h before harvesting. As shown in Fig. 4A, CEP-11004 treatment significantly inhibited expressed MLK3 activity compared to untreated cells. Next, the effect of CEP-11004 on a related mixed-lineage kinase, dual leucine zipper kinase (DLK), activity was measured in transfected CHO cells. Using JNK MAP kinase phosphorylation as a measure of DLK or MLK3 activity, CEP-11004 inhibited MLK3 but not DLK induced JNK phosphorylation (Fig. 4B). In agreement with others [15], we have demonstrated that MLK3 has higher activity (∼4 fold) during G2 and M-phase transitions as compared to G1/S phase and this activity is blocked by CEP-11004 (data not shown).
Fig. 4.
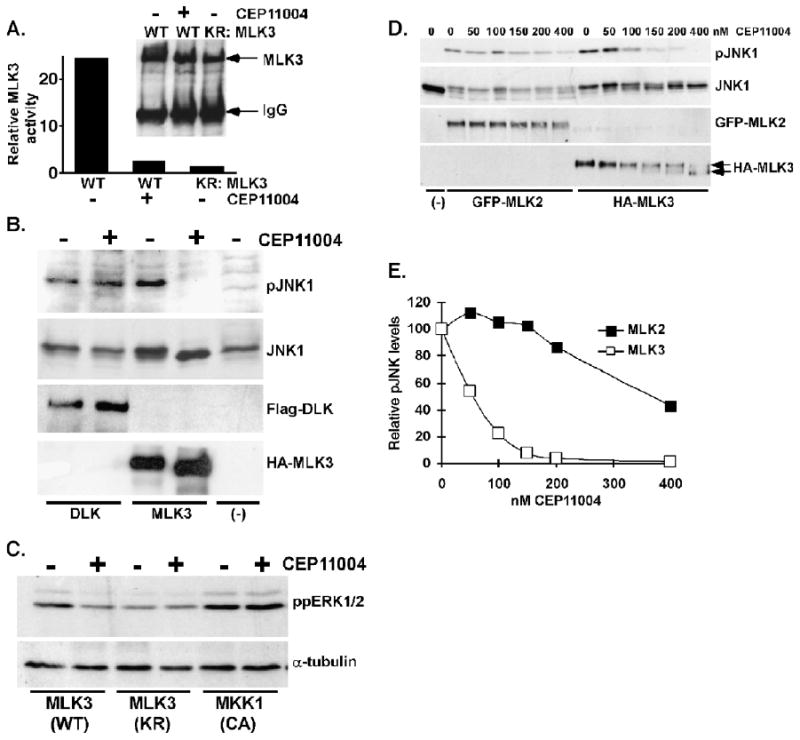
CEP-11004 targets MLK3 but not DLK or MKK1 activity. (A) HeLa cells were transfected with HA-tagged MLK3 wild type (WT) or a catalytically inactive MLK3 (KR) mutant. The MLK3 wild type transfected cells were incubated with or without CEP-11004 (500 nM) for 4 h prior to harvesting. Wild type and inactive MLK3 were immunoprecipitated with a MLK3 antibody and incubated with MBP as a substrate in an in vitro kinase assay. The graph shows the relative amount of phosphate incorporation into MBP under each condition and the immunoblot shows the amount of the MLK3 proteins in the immunoprecipitates. (B) HeLa cells were transfected with HA-tagged JNK1 plus MLK3 wild type or Flag-tagged DLK and then treated in the absence or presence of CEP-11004 (500 nM) for 4 h prior to harvesting. Immunoblots for active JNK (pJNK), HA-JNK1, Flag-DLK, or HA-MLK3 are shown in the top to bottom panels, respectively. (C) HeLa cells were transfected with wild type (WT) MLK3, inactive (KR) MLK3, or a constitutively active MKK1 mutant (CA) and then treated with or without CEP-11004 (500 nM). The relative level of active ERK was determined by immunoblotting with a phospho-specific ERK1/2 (ppERK) antibody (upper panel). Protein loading was monitored by immunoblotting for α-tubulin (lower panel). (D) HeLa cells were co-transfected with HA-JNK1 and GFP tagged MLK2 or HA-MLK3. Transfected cells were then treated for 1 h with various concentrations of CEP-11004 and JNK activity was determined by immunoblotting with a phospho-specific JNK1/2 antibody (pJNK, upper panel). The expression of HA-JNK1, GFP-MLK2, or HA-MLK3 is shown in the lower panels. The upper and lower arrows in the HA-MLK3 blot indicate phosphorylated and de-phosphorylated forms of MLK3, respectively. (E) Graph of the relative JNK activity as quantified by measuring the ratio of pJNK to total JNK by densitometry in cells expressing MLK2 (closed squares) or MLK3 (open squares).
Overexpression of MLK3 has been shown to induce ERK activation through MEK1 [13]. Therefore, the effect of CEP-11004 on ERK activation following overexpression of MLK3 wild type or a constitutively active MKK1 mutant was examined. Although both MLK3 and active MKK1 stimulate ERK activity, only MLK3-induced ERK activation was inhibited by CEP-11004 (Fig. 4C). Lastly, we compared the ability for CEP-11004 to inhibit MLK2 and MLK3 proteins. GFP-tagged MLK2 or HA-tagged MLK3 were co-expressed with HA-tagged JNK1 for 24 h and then treated with or without CEP-11004 for an additional 3 h before harvesting. Whereas MLK3 induced JNK1 phosphorylation was inhibited by 80% with 100 nM CEP-11004, significant inhibition of MLK2 activation of JNK1 required greater than 400 nM CEP-11004 (Fig. 4D and E). Interestingly, CEP-11004This is consistent with CEP-11004 having approximately 3 fold higher specificity towards MLK3 as compared to MLK2 [11]. Thus, these data support MLK3 as the major MLK isoform targeted by CEP-11004.
3.3. CEP-11004 induces mitotic arrest in pro-metaphase
Since it did not appear that CEP-11004 affected progression into mitosis (Fig. 3), the nature of CEP-11004 induced M-phase arrest was characterized by examining the various stages of mitosis in synchronized cells treated in the presence or absence of CEP-11004. G1/S-phase cells grown on coverslips were released back into the cell cycle for 5 h, treated with or without 400 nM CEP-11004 for an additional 4 or 6 h incubation (9 and 11 h, respectively, after release from G1/S block), and the DNA was stained with DAPI. Chromosome morphology was examined by fluorescence microscopy and the percentage of cells in prophase, pro-metaphase, metaphase, or anaphase/telophase was determined. At 9 h after G1/S, CEP-11004 treated cells began to show accumulation of pro-metaphase cells compared to untreated cells (Fig. 5A, left graph). Furthermore, cells in metaphase or anaphase/telophase were not evident in CEP-11004 treated cells (Fig. 5A, left graph). This difference became even more apparent at 11 h after G1/S release as CEP-11004 treated cells primarily accumulated in pro-metaphase unlike control cells, which showed an increased number of anaphase/telophase cells (Fig. 5A, right graph). The number of cells accumulating in pro-metaphase in response to CEP-11004 was dose-dependent. G1/S-phase synchronized cells that were released back into the cell cycle for 5 h and then treated with various concentrations of CEP-11004 for an additional 8 h (13 h after G1/S release), showed a dose-dependent increase in the percentage of pro-metaphase cells (Fig. 5B).
Fig. 5.
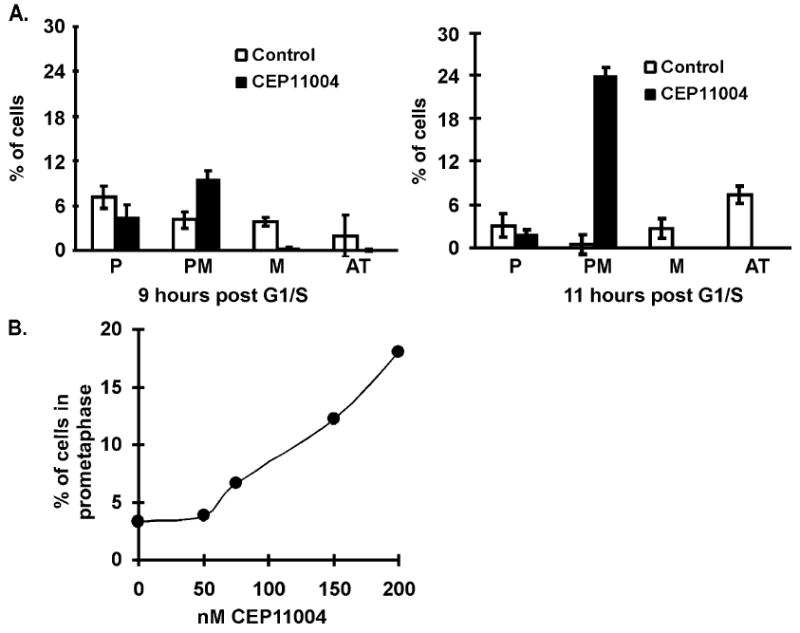
CEP-11004 causes cell cycle arrest in pro-metaphase. HeLa cells grown on coverslips were synchronized at the G1/S boundary with excess thymidine and released back into the cell cycle. At 5 h after release, cells were treated in the absence or presence of CEP-11004 (500 nM) for an additional 4 or 6 h (9 or 11 h cumulative time after G1/S release). (A) The percentage of cells in prophase (P), pro-metaphase (PM), metaphase (M), or anaphase/telophase (AT) was determined by DAPI staining of chromosomes in control (open bars) or CEP-11004 treated (closed bars) cells. The left and right graphs represent the percentage of mitotic cells 9 and 11 h, respectively, after G1/S-phase release. Data represent the average and standard error from 3 independent experiments. (B) The number of cells in pro-metaphase were counted at 11 h after G1/S-phase release following treatment with increasing concentrations of CEP-11004 (added at 5 h after G1/S-phase release). 300–350 cells were counted at each concentration.
3.4. MLK3 overexpression reverses the effect of CEP-11004 on mitotic arrest arrest
Given that the previous data indicated that inhibition of MLK proteins caused mitotic arrest in pro-metaphase and that MLK3 was a major target of CEP-11004, we tested whether MLK3 overexpression could reverse mitotic arrest induced by CEP-11004. HeLa cells were co-transfected with pEGFP, to identify transfected cells, in the presence of control vector or HA-MLK3 and then synchronized at the G1/S boundary using excess thymidine. At 5 h after G1/S release, cells were incubated with various concentrations of CEP-11004 for an additional 8 h. Mitotic progression was examined by immunoblotting for cyclin B1, determining the mitotic index, and assaying Cdc2 kinase activity. Cyclin B1 levels were elevated in mock transfected cells treated with 100 or 200 nM CEP-11004, which is consistent with a mitotic arrest (Fig. 6A). In contrast, MLK3 transfected cells had lower cyclin B1 levels after CEP-1104 treatment consistent with the ability for exogenous MLK3 to prevent the mitotic arrest induced by these concentrations of CEP-11004 (Fig. 6A).
Fig. 6.
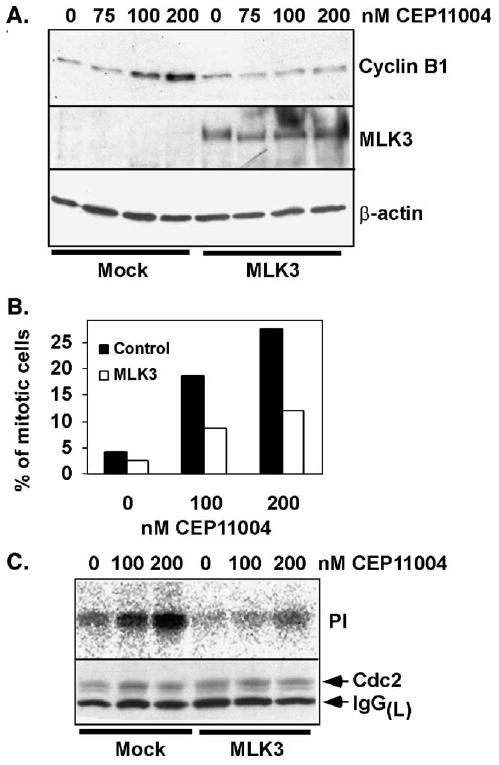
Exogenous MLK3 inhibits CEP-11004-induced mitotic arrest. HeLa cells were co-transfected with pEGFP in the presence or absence (mock) of HA-tagged MLK3 wild type and then synchronized by double thymidine block. At 5 h after G1/S release, the cells were treated with various concentrations of CEP-11004 (75, 100, 200 nM) for 8 h and then harvested (13 h post G1/S release). (A) Immunoblots for cyclin B1 (top panel), MLK3 (middle panel), and β-actin (lower panel) as a protein loading control. (B) Synchronized HeLa cells transfected with pEGFP plus or minus MLK3 were grown on coverslips, treated with or without various concentrations of CEP-11004, fixed at 13 h after release from G1/S, and stained with DAPI. The number of mitotic cells (prophase, pro-metaphase, metaphase, and anaphase/telophase cells) was determined by DAPI staining in GFP positive cells (closed bars) or GFP positive cells that co-express MLK3 (open bars). (C) Relative Cdc2 kinase activity in lysates collected 13 h after G1/S release in cells that were transfected with pEGFP alone or pEGFP and MLK3. Radiolabeled phosphate incorporation into histone substrate (upper panel) and the immunoblot for Cdc2 in the immunoprecipitates (lower panel) are shown. IgG(L) indicates the antibody light chain.
Next, the mitotic index was determined by DAPI staining in mock or MLK3 transfected cells treated with varying concentrations of CEP-11004. As expected, mock transfected cells showed higher levels of mitotic cells following CEP-11004 treatment consistent with mitotic arrest (Fig. 6B). In contrast, MLK3 transfected cells showed a reduced mitotic index following CEP-11004 treatment (Fig. 6B). Lastly, Cdc2 kinase activities in mock or MLK3 transfected cells treated with 100 or 200 nM CEP-11004 were measured. Similar to the cyclin B1 expression, mock transfected cells treated with CEP-11004 exhibited higher Cdc2 activity compared to MLK3 transfected cells (Fig. 6C). These data suggest that exogenous MLK3 is able to overcome the CEP-11004 induced mitotic arrest.
3.5. CEP-11004 delays histone H3 phosphorylation in early mitosis
The mechanisms of CEP-11004 induced mitotic arrest were further examined by monitoring mitosis-specific phosphorylation events. Phosphorylation of histone H3 at serine 10 is commonly used as a marker of mitotic progression [22,23]. HeLa cells were treated with or without CEP-11004 at 5 h after release from G1/S-phase thymidine block and allowed to incubate for an additional 4 h. Cells were then fixed and processed for immunofluorescence of cyclin B1 and phospho-histone H3 (pH3) to identify the mitotic cells. During prophase in control cells, cyclin B1 and pH3 staining were observed in the nucleus prior to nuclear envelope breakdown (Fig. 7A). In contrast, cells treated with CEP-11004 did not contain nuclear pH3 staining during prophase, even though these cells showed strong nuclear reactivity with cyclin B1 (Fig. 7A). This effect on inhibited pH3 staining in early mitotic cells was observed with CEP-11004 doses as low as 200 nM (data not shown). These data suggest that MLK protein activity directly or indirectly is involved in histone H3 phosphorylation during mitosis. This is consistent with the localization of MLK3 in prophase cells, which suggested MLK3 targeting to the nucleus and centrosomes in prophase cells (Fig. 7B).
Fig. 7.
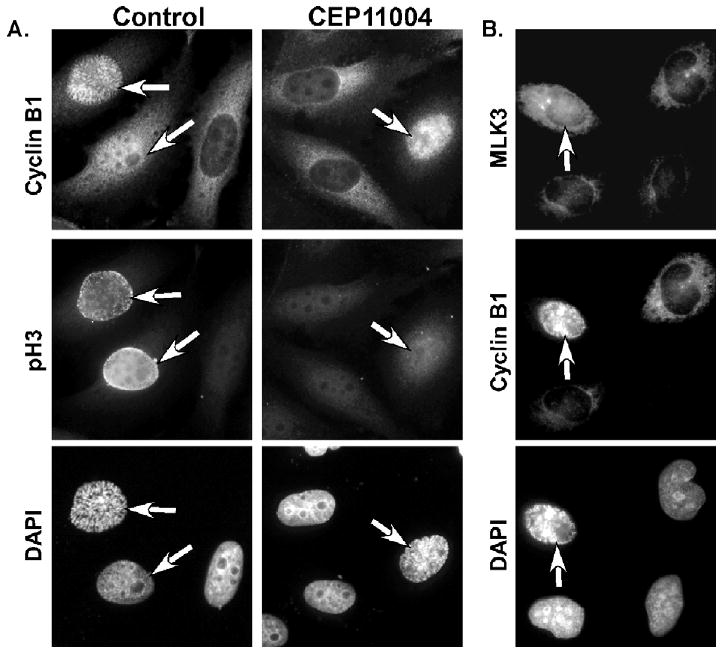
CEP-11004 delays nuclear histone H3 phosphorylation in early prophase. HeLa cells grown on coverslips were synchronized at G1/S with excess thymidine and released back into the cell cycle. At 5 h after release, cells were incubated in the absence or presence of CEP-11004 (500 nM). After an additional 4 h incubation (9 h after G1/S release), cells were fixed and co-immunostained for cyclin B1 or phosphorylated histone H3 (pH3) in the top and middle panels, respectively, of (A). MLK3 and cyclin B1 expression are shown in the top and middle panels, respectively, of (B). Mitotic cells (indicated by arrows) were determined by the presence of nuclear cyclin B1 and/or pH3 staining. Cells in (A) and (B) were counterstained with DAPI (lower panels).
We next compared pH3 staining before and after nuclear envelope breakdown (NEB). As expected, in untreated cells, pH3 staining was observed in early mitosis before NEB and later stages of mitosis after NEB (Fig. 8A and B). The integrity of the nuclear envelope was determined by staining for the p62 nucleoporin protein (Fig. 8). Similar to Fig. 7, pH3 staining in early mitosis before NEB was not observed in CEP-11004 treated cells (Fig. 8A). However, pH3 staining was readily apparent after NEB in pro-metaphase of both untreated and CEP-11004 treated cells (Fig. 8B). Together, these data indicate that MLK activity is important for histone H3 phosphorylation during early mitosis through a Cdc2/cyclin B1 independent mechanism.
Fig. 8.
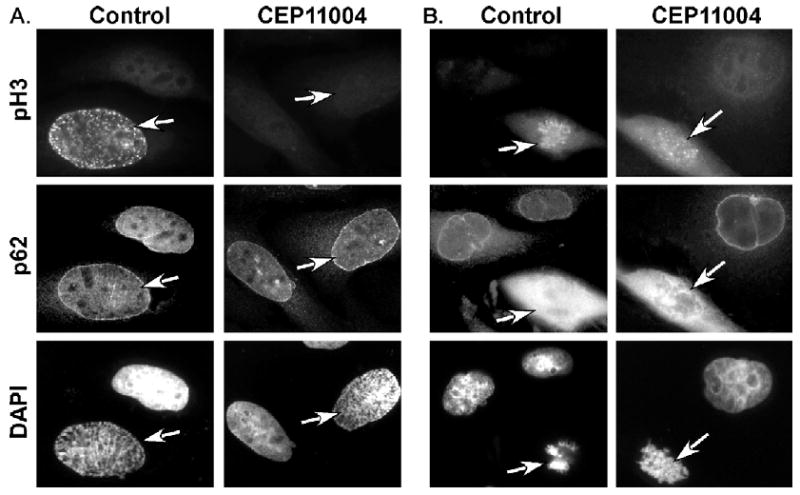
CEP-11004 inhibits histone H3 phosphorylation prior to nuclear envelope breakdown. HeLa cells were prepared for immunostaining as described in Fig. 5. Cells were stained for pH3 (top panels) and nucleoporin (p62, middle panels) as a marker of the nuclear envelope. Cells in prophase (A; pre nuclear envelope breakdown) or pro-metaphase (B; post nuclear envelope breakdown) that had been treated in the absence or presence of CEP-11004 were identified. Mitotic cells, as indicated by the arrows, were identified by DAPI staining (bottom panels) of condensed chromosomes. Adjacent interphase cells are shown for comparison.
3.6. CEP-11004 causes aberrant spindle pole formation during mitosis
Although overexpression of MLK3 has recently been suggested to promote microtubule instability [15], it is not clear what effects inhibition of MLK3 activity have on microtubule organization in mitotic cells. Thus, we next examined the effects of CEP-11004 on spindle pole assembly in mitotic cells. Synchronized cells were treated at the end of S-phase with or without CEP-11004 for 4 h followed by immunostaining for α-tubulin as a marker of the microtubules and γ-tubulin as a marker of the spindle pole. As shown, CEP-11004 treatment caused significant disruption of the microtubule organization (α-tubulin, Fig. 9A) in mitotic cells but had no effect on the number of spindle poles (γ-tubulin, Fig. 9A). In addition, the chromosome organization as determined by DAPI staining in CEP-11004 was highly disorganized (Fig. 9A). Greater than 60% of the mitotic cells contained the aberrant microtubule organization (Fig. 9B). Examination of interphase cells treated for 4 h with CEP-11004 showed no apparent effects on microtubules as compared to untreated cells (data not shown). Thus, the inability for CEP-11004 treated cells to progress through metaphase is likely due to defects in microtubule organization and aberrant spindle formation.
Fig. 9.
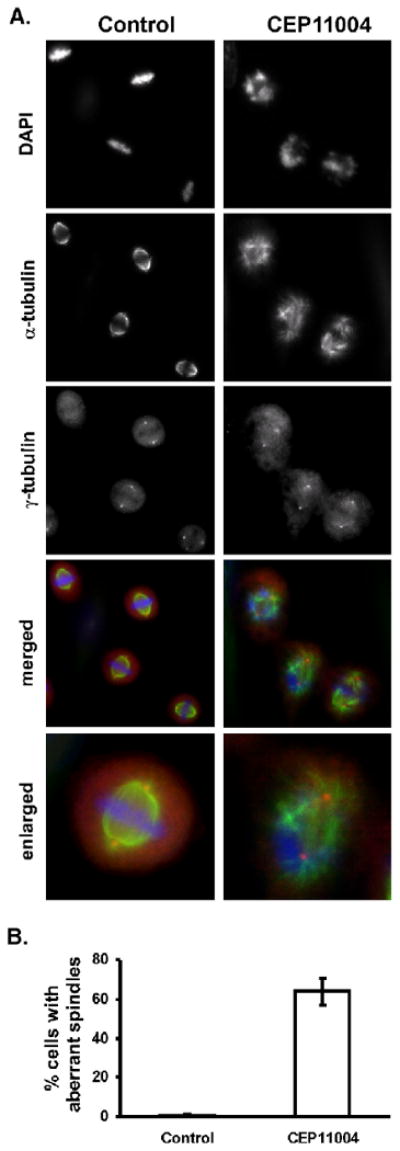
CEP-11004 induces aberrant mitotic spindles. HeLa cells grown on coverslips were treated in the absence or presence of CEP-11004 as described in Fig. 5. (A) Cells were collected at 9 h after release from G1/S block and the organization of the mitotic spindle was evaluated by immunostaining for α or γ-tubulin to identify microtubules and the spindle poles. Cells were counterstained with DAPI to identify mitotic chromosomes. The merged Images shows α-tubulin in green, γ-tubulin in red, and DAPI in blue pseudocolor. (B) The percentage of the mitotic cells with aberrant spindles in control or CEP-11004 treated cells was quantified in 3 separate experiments. At least 300 mitotic cells were counted in each experiment under each condition.
4. Discussion
The findings of the present study indicate that MLK proteins, and in particular MLK3, may be an important regulator of microtubule organization during mitosis in transformed HeLa cells. As such, inhibition of MLK activity with CEP-11004 blocked mitotic progression and caused cells to arrest in pro-metaphase. Previous studies in transfected Cos7 cells suggested that MLK2 will also associate with microtubules [24]. However, because we show that MLK3, and not MLK2, is more sensitive to inhibition by CEP-11004 we propose that the microtubule disruption induced by CEP-11004 is primarily due to the loss of MLK3 activity. In addition, we demonstrate that CEP-11004 does not affect the activity of other MLK-related (DLK) or non-MLK related protein kinases (MKK1). Nevertheless, others have suggested that the effects of MLK inhibitors on Ras-induced proliferation of NIH 3T3 cells may be due to simultaneous inhibition of the p21-activated kinase-1 (Pak1) [20]. In the present studies, Pak1 activity was not tested in the presence or absence CEP-11004 because the doses that were used to initiate cell cycle arrest in the current study were approximately 10 fold less than the reported IC50 concentrations needed for the MLK inhibitor to block Pak1 activity [20]. Future analysis of potential inhibition of other kinases by CEP-11004 and other MLK inhibitors will nonetheless need to be considered.
Although we observed a profound effect on the mitotic microtubule organization within a few hours exposure to CEP-11004, it is unclear what MLK-regulated microtubule associated proteins are being affected. Expression of ectopic MLK3 has been shown to destabilize microtubules in HEK293 cells through a mechanism that does not involve JNK or p38 MAP kinase activation [15]. However, it is not clear whether this observation is unique to MLK3 or an over-expression artifact. MLK2 and active JNK have also been shown to co-localize along microtubules and interact with motor proteins [24]. Whether these interactions were occurring in a cell cycle-dependent manner was not determined. We have recently described a novel role for direct MLK3 phosphorylation of golgin-160, a Golgi complex protein that may mediate cellular responses to apoptotic stimuli [25]. Thus, MLK3 may have additional substrates that may help explain MLK3 functions during the cell cycle that are independent of downstream MAP kinase activation.
MLK proteins, and especially MLK3, appear to be multifunctional kinases involved in regulating cell death or proliferation depending on the cell type and stimuli. MLK3 function in cell death has been largely attributed to its role in activating the stress activated protein kinases, which include JNK and p38 [1]. However, recent studies also show that MLK3 can mediate cell proliferation through activation of the B-Raf and ERK pathway [14]. To identify mechanisms by which MLK proteins may affect cell proliferation, we examined the effects of MLK inhibition during G2-phase on subsequent progression through mitosis. One objective of this approach was to avoid long term exposures to pharmacological agents in order to observe changes to specific cell cycle events in highly synchronized cells. From our data, we propose that MLK3 inhibition by CEP-11004 prevents cell proliferation by disrupting microtubule events necessary for mitotic progression. This may at first appear to contrast with a previous study that cited unpublished data indicating that down-regulation of MLK3 using RNA interference (RNAi) does not affect cell proliferation or DNA content by flow cytometry [15]. However, it is difficult to directly compare the two studies and several reasons could explain the discrepancy between the data by Swenson et al. and the data in the current paper. First, the studies by Swenson et al. down-regulated MLK3 in the human osteosarcoma SAOS2 cell line, which reportedly has a functional G2-phase checkpoint in response to colcemid treatment as compared to HeLa cells [26]. Second, it is unlikely that RNAi treatment is sufficient to completely deplete MLK3 and the remaining MLK3 may be sufficient to promote mitotic progression in these cells. Third, the MLK3-depleted SAOS2 cells were generated by incubation with RNAi for 2–3 days, which could allow other compensatory mechanisms that would substitute for MLK3 during mitotic progression.
Interestingly, another MLK inhibitor, CEP-1347, which has similar biological properties to CEP-11004 has been reported to activate the ERK pathway [12]. However, these experiments were done in neuronal cells and we have found no evidence that CEP-11004 causes ERK activation in non-neuronal cells. In contrast, CEP-11004 treatment inhibited MLK3 induced ERK activation (Fig. 4C), which is consistent with a role for MLK3 in activating the ERK pathway in tumor cells [14]. This further supports the opposing roles for MLK3 in promoting proliferation of non-neuronal tumor cells but cell death of neuronal cells. In addition, these data suggest a dual function for MLK3 characterized by negative regulation of the ERK pathway in neuronal cells but positive regulation of ERK activity in non-neuronal tumor cells.
Although CEP-11004 clearly inhibited phosphorylation of histone H3 at S10 in early mitosis, it is not clear whether MLK3 is directly or indirectly responsible for the phosphorylation. Histone H3 is phosphorylated throughout the cell cycle by a number of kinases including PKA, Msk1, ERK, and p38 MAP kinase [27]. Although all of these kinases can phosphorylate S10 on histone H3, the functional role of phosphorylation is not clear and may depend on the phase of the cell cycle that it occurs. Our immunofluorescence data suggests that MLK3 might localize to the nucleus of mitotic cells (Fig. 7B) and support a direct phosphorylation of histone H3 by MLK3. Consistent with this is the finding that MLK3 is related to NIMA, a mitotic kinase that phosphorylates S10 on histone H3 in vitro [28]. Even though MLK3 can activate ERK and p38 signaling pathways, and histone H3 phosphorylation at serine 10 may be mediated by ERK and p38 MAP kinases in response to stress stimuli [29], future studies will need to determine whether these kinases contribute to histone phosphorylation during early mitosis prior to NEB. Consistent with this is the presence of active ERK in the nucleus of early mitotic cells prior to NEB [30,31]. Other potential candidate kinases that regulate phosphorylation of histone H3 at S10 during mitosis are the aurora A and B kinases, with aurora B being the most likely physiological histone H3 kinase since aurora A localizes primarily to the centrosome [32]. Nonetheless, aurora B depletion does not eliminate S10 phosphorylation on histone H3 indicating the involvement of other mitotic kinases [27]. Although aurora kinases are regulated by phosphorylation, there is no evidence to link MLK proteins with regulation of the aurora kinases.
The significance of the delayed histone H3 phosphorylation that we observe following inhibition of MLK proteins during mitotic progression is not known. The requirement for phosphorylation of histone H3 on S10 during mitosis remains controversial. For example, although phosphorylation of histone H3 at S10 has been suggested to be required for proper chromosome condensation and mitotic progression in tetrahymena [33,34], a yeast mutant strain containing an alanine mutation at S10 of histone H3 proliferates as well as the wild type strain [35].
Of particular interest is the observation that inhibition of MLK proteins may preferentially inhibit mitotic transitions and cell proliferation of transformed cells, such as HeLa, A549, or HEK293, but not in non-transformed NIH 3T3 or normal diploid human MRC-5 lung fibroblasts. The inherent abnormalities of cell cycle regulation in the transformed cells may make these cells more sensitive to undergo cell cycle arrest and apoptosis in response to MLK inhibitors. If this turns out to be true, then inhibition of MLK proteins may be a useful approach for preventing cancer cell proliferation without affecting normal cell function. A recent report indicating that MLK3 is up regulated in breast cancer cell lines supports this area of study for the development of new cancer therapies [5].
Acknowledgments
We thank Dr. Donna Dorow (University of Dundee, Scotland, UK) for providing the GFP-MLK2 construct.
Abbreviations
- MLK
Mixed-lineage kinase
- MAP
Mitogen-Activated Protein
- ERK
Extracellular signal-Regulated Kinase
- JNK
c-Jun NH2-terminal Kinase
- DLK
Dual Leucine Zipper-Bearing Kinase
- CRIB
Cdc42/Rac Interactive Binding
References
- 1.Gallo KA, Johnson GL. Nat Rev Mol Cell Biol. 2002;3:663. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 2.Mata M, Merritt SE, Fan G, Yu GG, Holzman LB. J Biol Chem. 1996;271:16888. doi: 10.1074/jbc.271.28.16888. [DOI] [PubMed] [Google Scholar]
- 3.Burbelo PD, Drechsel D, Hall A. J Biol Chem. 1995;270:29071. doi: 10.1074/jbc.270.49.29071. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Gallo KA. J Biol Chem. 2001;276:45598. doi: 10.1074/jbc.M107176200. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Wu W, Du Y, Santos SJ, Conrad SE, Watson JT, Grammatikakis N, Gallo KA. J Biol Chem. 2004;279:19457. doi: 10.1074/jbc.M311377200. [DOI] [PubMed] [Google Scholar]
- 6.Mota M, Reeder M, Chernoff J, Bazenet CE. J Neurosci. 2001;21:4949. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang LH, Besirli CG, Johnson EM., Jr Annu Rev Pharmacol Toxicol. 2004;44:451. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- 8.Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. Mol Cell Biol. 2001;21:4713. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. J Biol Chem. 1999;274:10195. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- 10.Tibbles LA, Ing YL, Kiefer F, Chan J, Iscove N, Woodgett JR, Lassam NJ. EMBO J. 1996;15:7026. [PMC free article] [PubMed] [Google Scholar]
- 11.Murakata C, Kaneko M, Gessner G, Angeles TS, Ator MA, O'Kane TM, McKenna BA, Thomas BA, Mathiasen JR, Saporito MS, Bozyczko-Coyne D, Hudkins RL. Bioorg Med Chem Lett. 2002;12:147. doi: 10.1016/s0960-894x(01)00690-4. [DOI] [PubMed] [Google Scholar]
- 12.Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA. J Biol Chem. 2002;277:49473. doi: 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- 13.Hartkamp J, Troppmair J, Rapp UR. Cancer Res. 1999;59:2195. [PubMed] [Google Scholar]
- 14.Chadee DN, Kyriakis JM. Nat Cell Biol. 2004;6:770. doi: 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- 15.Swenson KI, Winkler KE, Means AR. Mol Biol Cell. 2003;14:156. doi: 10.1091/mbc.E02-02-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert JM, Karnoub AE, Graves LM, Campbell SL, Der CJ. J Biol Chem. 2002;277:4770. doi: 10.1074/jbc.M109379200. [DOI] [PubMed] [Google Scholar]
- 17.Cho YY, Bode AM, Mizuno H, Choi BY, Choi HS, Dong Z. Cancer Res. 2004;64:3855. doi: 10.1158/0008-5472.CAN-04-0201. [DOI] [PubMed] [Google Scholar]
- 18.Dangi S, Cha H, Shapiro P. Cell Signal. 2003;15:667. doi: 10.1016/s0898-6568(03)00002-0. [DOI] [PubMed] [Google Scholar]
- 19.Cha H, Hancock C, Dangi S, Maiguel D, Carrier F, Shapiro P. Biochem J. 2004;378:857. doi: 10.1042/BJ20031173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nheu TV, He H, Hirokawa Y, Tamaki K, Florin L, Schmitz ML, Suzuki-Takahashi I, Jorissen RN, Burgess AW, Nishimura S, Wood J, Maruta H. Cancer J. 2002;8:328. doi: 10.1097/00130404-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 21.Katoh M, Hirai M, Sugimura T, Terada M. Oncogene. 1995;10:1447. [PubMed] [Google Scholar]
- 22.Hendzel MJ, Wei Y, Mancini MA, VanHooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Chromosoma. 1997;106:348. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 23.Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. Proc Natl Acad Sci U S A. 1998;95:7480. doi: 10.1073/pnas.95.13.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagata K, Puls A, Futter C, Aspenstrom P, Schaefer E, Nakata T, Hirokawa N, Hall A. EMBO J. 1998;17:149. doi: 10.1093/emboj/17.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cha H, Smith BL, Gallo K, Machamer CE, Shapiro P. J Cell Sci. 2004;117:751. doi: 10.1242/jcs.00897. [DOI] [PubMed] [Google Scholar]
- 26.Jha MN, Bamburg JR, Bedford JS. Cancer Res. 1994;54:5011. [PubMed] [Google Scholar]
- 27.Prigent C, Dimitrov S. J Cell Sci. 2003;116:3677. doi: 10.1242/jcs.00735. [DOI] [PubMed] [Google Scholar]
- 28.DeSouza CP, Osmani AH, Wu LP, Spotts JL, Osmani SA. Cell. 2000;102:293. doi: 10.1016/s0092-8674(00)00035-0. [DOI] [PubMed] [Google Scholar]
- 29.Zhong SP, Ma WY, Dong Z. J Biol Chem. 2000 doi: 10.1074/jbc.M909934199. [DOI] [PubMed] [Google Scholar]
- 30.Zecevic M, Catling AD, Eblen ST, Renzi L, Hittle JC, Yen TJ, Gorbsky GJ, Weber MJ. J Cell Biol. 1998;142:1547. doi: 10.1083/jcb.142.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, Ahn NG. J Cell Biol. 1998;142:1533. doi: 10.1083/jcb.142.6.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascreau G, Arlot-Bonnemains Y, Prigent C. Prog Cell Cycle Res. 2003;5:369. [PubMed] [Google Scholar]
- 33.Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Cell. 1999;97:99. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- 34.Van Hooser A, Goodrich DW, Allis CD, Brinkley BR, Mancini MA. J Cell Sci. 1998;111(Pt 23):3497. doi: 10.1242/jcs.111.23.3497. [DOI] [PubMed] [Google Scholar]
- 35.Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD. Cell. 2000;102:279. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]