

Abstract
Pharmacogenomics is a rapidly developing sector of human genetics research with arguably the highest potential for immediate benefit. There is a considerable body of evidence demonstrating that variability in drug-treatment response can be explained in part by genetic variation. Subsequently, much research has ensued and is ongoing to identify genetic variants associated with drug-response phenotypes. To reap the full benefits of the data we collect we must give careful consideration to the study population under investigation, the phenotype being examined and the statistical methodology used in data analysis. Here, we discuss principles of study design and optimizing statistical methods for pharmacogenomic studies when the outcome of interest is a continuous measure. We review traditional hypothesis testing procedures, as well as novel approaches that may be capable of accounting for more variance in a quantitative pharmacogenomic trait. We give examples of studies that have employed the analytical methodologies discussed here, as well as resources for acquiring software to run the analyses.
Keywords: data analysis, data mining, epistasis, genome-wide association, interaction, methodology, pharmacogenomics, statistics
Much evidence exists suggesting that individual variation in pharmacological traits, such as adverse drug response, stable dose of a drug with a narrow therapeutic range or treatment efficacy, can be attributed to genetic variation. Moreover, pharmacogenomics is a promising area of human genetic research with a high potential for generating immediate public health and economic benefits [1,2]. Technological advances in the last several years have allowed the pharmacogenomics field to progress from coarse genomic coverage with linkage maps and candidate gene association studies, to very-high-resolution association analyses using single nucleotide polymorphisms (SNPs) [3]. The completion and ongoing development of the International HapMap Project [4,5], a catalog of common human genetic variation at millions of poly morphic sites in several populations, allows for more powerful and strategic study design of both targeted and genome-wide scans. Several technologies are currently available that allow for rapid, highly accurate genotyping of over 1 million common SNPs at low cost per genotype. In addition to SNPs, the latest generation of GeneChips (Affymetrix) and BeadChips (Illumina) contain thousands of probes targeting known copy number variations (CNVs) based on the first-generation CNV maps available for the human genome [6–9].
Specifically for pharmacogenomic studies, several genomic tools are available that identify both rare and common variations associated with adverse drug reactions or dosing. To date, the most commonly applied modern genomic tool is the genome-wide association study (GWAS), which is generic and not specific to pharmacogenomics. GWAS has already been used in many pharmacogenomic studies to identify common variants that influence adverse drug reactions [10], drug dosing [11,12] and treatment efficacy [13]. While the GWAS approach has been successful in some pharmacogenomic studies, in many studies the GWAS platforms are insufficient because they do not represent some of the known rare variation associated with pharmacogenomic phenotypes [14]. Also the GWAS platforms do not contain variations that are difficult to assay such as CYP2D6 variants. To fill this niche, Affymetrix has developed a targeted genotyping platform that specifically covers over 200 genes known to be involved in absorption, distribution, metabolism and excretion (ADME) of pharmacological agents [15]. This platform is now being used extensively in pharmacogenomic research [16,17]. Finally, much research is being pursued in both the academic and private sector to develop methods for inexpensive whole-genome ‘next-generation’ sequencing [18–20], allowing for complete examination of all human genetic sequence variation, which will capture rare variation that is currently missed in GWAS studies.
In addition to benefitting from the technological advances in the field, pharmacogenomic studies can benefit from much of the recent improvements in study design and optimization of statistical methodology that has recently transformed disease gene-association studies. Here, we review the study designs and statistical methodology that are commonly used in pharmacogenomic studies of quantitative trait outcomes. We then discuss novel methodology research taking place in the human genetics community, and how these can be applied to optimize statistical analyses in pharmacogenomics research. A flowchart representation of the main topics covered in this review is shown in Figure 1.
Figure 1. Design and statistical workflow of a pharmacogenomic study.
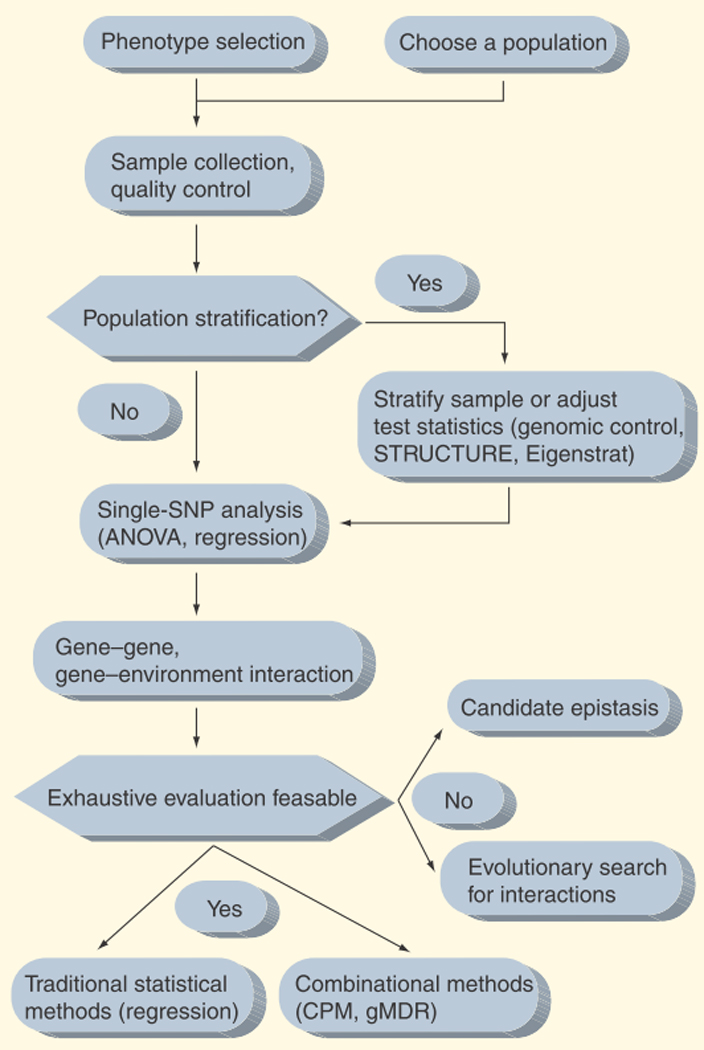
ANOVA: Analysis of variance; CPM: Combinatorial partitioning method; gMDR: Generalized multifactor dimensionality reduction; SNP: Single nucleotide polymorphism.
Phenotype selection & quantitative outcomes
The predominant study design for genetic association studies over the last decade has been the case–control design. In a disease gene association study, one would typically associate an allele or genotype frequency difference between affected and unaffected individuals [21]. The case–control design in disease gene studies has many advantages, namely the fact that relatively rare conditions can be ascertained after their onset, significantly reducing the costs of ascertainment and follow-up, typically incurred by large prospective studies [22]. The case–control design may be the most appropriate design in pharmacogenomics studies examining a purely discrete outcome, such as adverse drug reactions, where the phenotype neatly falls into one of two possible classes. However, analysis of a quantitative trait that varies continuously over a range of possible values may be more optimal in many cases. First, the outcome of interest in many pharmacogenomics investigations naturally varies on a continuous scale. Examples include determining the correct stable dosage for a drug with a narrow therapeutic range [23–25], predicting treatment efficacy [26–28] and predicting drug resistance [29]. Furthermore, even many adverse drug reactions that are typically thought of as discrete events can be measured on a continuous scale, rather than discretized based on an often arbitrary threshold. Examples include analyzing blood iron content as a continuously varying trait rather than anemic status as a binary variable, assaying the full spectrum of liver enzyme activity rather than the presence or absence of hepatotoxicity, or recording blood glucose concentration rather than hypoglycemia as a dichotomy. While artificially creating a discrete variable based on an arbitrary threshold in a naturally continuous trait can simplify analysis of the data, it can also be counterproductive as it comes with the cost of a dramatic decrease in statistical power. That is, the useful variance found in continuous outcome data is discarded in dichotomous outcome data. Therefore, creating categorical variables in such a way should be avoided as much as possible. Such genetic association study designs are common in other areas such as cardiovascular genetics [30–33], genetic analysis of gene expression levels (eQTLs) [34,35] and psychiatric genetics [36]. Finally, standard clinical chemistry and contemporary proteomic techniques have made collection of continuously varying biomarker traits relatively easy, accurate and inexpensive [37].
Regardless of whether the clinical outcome under investigation is a discrete or continuous end point, phenotype definition is crucial to optimizing statistical analysis for pharmacogenomic studies. This may be exceedingly difficult when a naturally continuous clinical outcome must be categorized into one class or another. However, even when a quantitative outcome is ascertained and recorded, care must be taken to select a continuously varying clinical feature that can be precisely defined and reliably measured. This may pose less of a problem with pharmacogenomic studies, where end points such as stable dosage of a drug or level of a particular circulating enzyme can easily be measured and recorded through medical record surveillance and by standard laboratory assays, respectively. However, one note of caution when using medical record mining to ascertain a phenotype is to consider the issue of compliance – measures should be taken to determine whether the patient actually took the prescribed dose before the analysis is performed. Finally, thoughtful consideration must be given to choosing which clinical features will be used to represent pharmacological end points. In addition to affecting the power of statistical analyses, the choice and specificity of phenotype definition have implications on the interpretation and reproducibility of one’s results. Standard measures should be used so that others may follow up and replicate the analysis of an identical outcome in another data set.
In summary, while the case–control design has dominated genetic association studies in recent years, continuous outcomes may be more readily ascertained in pharmacogenomics studies, and the methods designed to analyze this type of outcome (discussed later) tend to be statistically more powerful, taking into account the full range of phenotypic variability that may be influenced by genetic factors. It is important to choose an outcome that is reliably measured, as well as one that is a standard measure in the field so that others may easily follow-up results in future studies.
Choosing a study population & methods for addressing stratification
One of the largest sources of confounding in association studies using unrelated individuals stems from population stratification, which occurs when the study population contains multiple subgroups of individuals with distinct genetic backgrounds (usually the result of including multiple racial or ethnic subgroups into a single study population). This becomes problematic and leads to systematic type I and type II errors when two conditions apply [38]. The subgroups differ with respect to the frequency of an allele and the subgroups have different values for the quantitative outcome of interest (or differ in the frequency of the occurrence of an event if the outcome under investigation is categorical). There are great differences in allele frequency and linkage disequilibrium patterns between populations [4,5], and it is known that there are differences in drug response between ethnic groups [39]. Whether this difference is owing to other genetic factors or to environmental factors does not matter here. If a truly irrelevant polymorphism was more common in the subgroup that, for some other genetic or nongenetic reason, has a more favorable response to a drug, then the allele will appear associated with the favorable response in this data set. The association here would be purely artifactual, resulting from the failure to adjust for population stratification. Also, it is well known that admixture and unknown and/or unintentional population stratification leads to artificially increased linkage disequilibrium across the genome or genomic regions being studied. While this phenomenon is used as a tool in admixture mapping, it adversely affects genetic association studies employing a tagSNP or other LD-based approach to identifying the causal genetic variant regardless of whether or not the trait of interest varies between or across the subgroups [40]. Thus, in addition to causing excessive type I error inflation, population stratification could also obscure true genetic associations [38].
Selecting a population to minimize stratification
One solution to this problem is to use family-based designs, which are robust to population stratification. Rather than associating frequency of alleles across families, family-based designs typically link or associate outcomes to regions of genetic variation by following alleles through meioses within families, which inherently cannot contain population substructure. While this has been an important design for disease gene association studies and early pharmacogenetic studies of adverse drug reactions, it is typically not used by investigators in pharmacogenomics owing to the difficulty of finding sufficient numbers of families with uniform exposure to the drug or pharmacological treatment [41,42]. Therefore, discussions of statistical procedures used for family-based study designs will not be summarized in this review, but they are the subject of an extensive review available in [43]. A common strategy for avoiding bias induced by population stratification is to ensure that study samples are drawn from a genetically homogenous population. For example, the Framingham Heart Study original and offspring cohorts are mainly comprised of Americans of European descent, where the most common self-reported ancestry was Western European [44]. As expected, this geographically and racially homogenous population does not display any evidence of population substructure [45]. On the other hand, it is likely that findings in one genetically homogenous population may not replicate or explain disease susceptibility variance in other ethnic groups or in the broader general population [46]. In this case, population-based, diverse samples are desirable for genetic association studies focused on characterizing previous GWAS or candidate gene discoveries made in one population [47]. In addition to intentionally studying diverse populations, ascertainment of diverse samples may be unavoidable owing to economics, ease of recruitment and recruitment setting (e.g., an outpatient clinic in a diverse city). If sampling from a diverse population for a genetic association or pharmacogenomic study, care should be taken to record the racial and ethnic background of each individual in the study. In addition to self report, other extensive questionnaire tools have been developed to aid in collecting information regarding ancestry [48]. Analyses could then be carried out separately for each subgroup. However, self-reported race/ethnicity has been criticized for being an inaccurate assessment of genetic ancestry [49]. Since this inaccuracy can lead to population stratification, investigators have advocated the genotyping of ancestry informative markers (AIMs) to more accurately infer individual ancestry [50]. Some studies have suggested that self reporting can be equivalent to genetic ancestry determined by AIMs [51], but this is dependent on the specific markers genotyped as AIMs and the level of detectable substructure desired by the investigator.
Statistical methods for detecting & controlling for population stratification
Although the confounding effects of population stratification can be mitigated by carefully choosing a study population, it can never be completely eliminated. Furthermore, the confounding effects of population stratification becomes more severe as sample size increases [52,53]. Others have shown that even with a uniformly European sample of 3000 individuals, differences in genetic variation can be detected between populations centered in geographic areas as little as a few hundred kilometers apart [54]. Even slight differences in prevalence rates of the outcome of interest between these groups would cause spurious associations. To deal with these challenges, statistical methodology has been developed (and implemented into software) to aid in detecting and adjusting for population stratification in genetic association studies. One method, genomic control, aims to control for population stratification by first estimating an inflation factor, then adjusting all of the test statistics downward by this factor [53,55]. Several variations on genomic control have been developed, and a recent comprehensive review and critical evaluation of genomic control methods [56] recommended genomic control F (GCF) as the most appropriate variation [57]. GCF does not assume the inflation factor is measured without error, and refines this factor accordingly. Structured association [58], implemented in the STRUCTURE software, uses genotype data to infer population structure, then performs tests of association within each inferred subpopulation [201]. Investigators may also use STRUCTURE to identify individual samples that do not cluster with the majority of the samples. These samples can then be eliminated from the analysis. Since the risk of confounding by population stratification increases with sample size [38], and because large sample GWAS are becoming increasingly common, another method has been developed that utilizes large samples and thousands of markers throughout the genome to correct for population structure. Eigenstrat uses a principal components-based method to explicitly detect and correct for population stratification on a genome-wide scale in large sample sizes in a computationally efficient manner [59,60]. Eigenstrat was first described for case–control analysis but can also be used for quantitative trait outcomes. EIGENSOFT is a freely available open-source software for conducting Eigenstrat analyses, available online [202]. A recent report using large-scale simulation studies to compare methods for correcting for population stratification examined all of the aforementioned techniques, and found that principle components-based methods (as implemented in Eigensoft) outperformed both genomic control and structured association in terms of maximizing power, controlling type I error maintaining in computational efficiency [61].
Traditional statistical methods for pharmacogenomics analysis
Traditionally, the analysis of genetic factors that contribute to pharmacological or other quantitative traits involves testing each marker for association. When the outcome of interest is a categorical trait or event, traditional analytical methods test for allele or genotype frequency differences between cases and controls. When the trait or pharmacogenomic outcome is continuous such as stable dose or an efficacy measure, traditional approaches test for significant differences in the means of the outcome of interest across different genotypes at a given locus. Below, we outline several traditional statistical approaches for pharmacogenomic data analysis when the outcome of interest varies continuously. We will discuss their strengths and weaknesses, where to find software implementations that make them accessible and examples of how these techniques have been used in the analysis of pharmacogenomic data.
Analysis of variance
The analysis of variance (ANOVA) tests for significant differences in the mean value of a quantitative outcome (e.g., blood glucose level or stable warfarin dose) between individuals in groups based on genotype. The theoretical justification of ANOVA has been demonstrated in numerous statistical texts and its implementation is widely available in nearly all statistical computing software [62,63]. ANOVA has a clear interpretation, and when its assumptions are met, it is uniformly the most powerful statistical procedure for detecting differences in a continuous outcome between groups. ANOVA also allows very specific hypotheses to be tested, for example, testing the hypothesis that the homozygote for the minor allele has an increased responsiveness to a chemotherapeutic drug as measured by tumor shrinkage, when compared with individuals of both other genotypes.
Linear regression
Linear regression is a generalization of the analysis of variance, any analysis that can be performed in ANOVA can be performed equivalently in linear regression. Using the linear regression framework, a model can be fitted to test any specified mode of inheritance (dominant, additive or recessive). Linear regression also allows other clinical, genetic, or environmental components to be taken into account, or adjusted for, when testing for the unique effect of a genetic variant. Furthermore, linear regression allows very specific tests for both gene–gene and gene–environment interaction – a topic that will be discussed at length later in this review. While adding more predictor variables and inter action terms to a regression equation will always improve the model fit, care should be taken to choose a model that has the added advantage of parsimony. A commonly used measure for aiding in model selection is the Akaike Information Criterion (AIC), which gauges how closely the predicted values fit the actual values, with a penalty for each predictor variable added to the model [64]. Once a well-fitting model has been developed, the linear regression equation can be used as a prediction equation for the value of the quantitative trait of interest (i.e., the pharmacological event or trait) as a function of genetic variants or other variables present in the equation. It is important, however, that the predictive ability of a model be tested in an independent dataset. In addition to being standard with almost any statistical computing software, linear regression is also available in PLINK [65], an open-source software tailored specifically for the analysis of genetic data, freely available online [203].
Nonparametric or distribution-free methods
The analysis of variance and linear regression both have a very similar set of assumptions regarding the underlying distribution of data points that must hold in order for their estimates and standard errors to remain unbiased [62], namely that the outcome must follow a normal (Gaussian) distribution, and the variance the outcome must be equal across groups with different genotypes [62,63]. Many pharmacological or other clinical measures often do not adhere to these assumptions, often times being lognormal or exponentially distributed [32], or having a variance that differs dramatically across groups of subjects with different genotypes [66]. While the aforementioned methods are robust to small violations of these assumptions, substantial violations may warrant the use of nonparametric, or distribution-free methods. The Kruskal–Wallis procedure [63] is a nonparametric alternative to ANOVA for testing difference in group means. The Kruskal–Wallis procedure and methods similar to it usually rely on rank statistics. While the Kruskal–Wallis procedure is robust to violations of the assumptions of ANOVA, it is not as powerful as ANOVA when its assumptions hold. Although not currently available in PLINK, the Kruskal–Wallis procedure is available in most statistical computing software, including the freely available open-source R statistical computing language [204].
All of the aforementioned procedures share a feature in common; regardless of which of these is used, most analyses in pharmacogenomic data test for differences in the value of a quantitative outcome one variable at a time. Below we discuss the importance of gene–gene and gene–environment interactions, and recent developments for optimizing statistical analysis in pharmacogenomic studies that go beyond the traditional one-at-a-time approach that is most commonly used.
Rare variation & epistasis
Despite the dizzying pace of advances in genotyping technologies that have made GWAS accessible, we have not been able to fully take advantage of the wealth of data generated by these studies because our analytical strategies have not kept pace. As mentioned previously, the most commonly used analytical procedures for analyzing pharmacogenomic data are tests of association at a single genetic variant at a time. This approach has been arguably successful in identifying genetic variants associated with complex traits, but these variants collectively explain little of the genetic component expected based on family and twin studies [68].
One potential explanation for this is the fact that current GWAS techniques are based largely on the ‘common disease common variant’ (CDCV) hypothesis, which is largely unsupported by empirical evidence [69]. This is because most of our richest resource on human genetic variation is primarily limited to common variants, and because current GWAS technology is focused on providing assays for polymorphisms with high frequency [4,5]. An alternative to the CDCV hypothesis is that the missing genetic component to pharmacogenomic traits may lie in rare variation, which is by and large overlooked by current GWAS techniques. Whole-genome sequencing technologies [18–20] are currently being developed that will allow for examination of rare variation within the next 2 years.
In addition to rare variation, many investigators have speculated that the missing genetic component lies in gene–gene and gene–environment interactions. Indeed, it is generally accepted that common traits are complex and are influenced by an intricate interplay of multiple genetic and environmental exposure [70–73]. This belief has been shared by biologists for over 70 years, when it was first emphasized by Sewall Wright that any biological or evolutionary end point is dependent on complex interactions between genes and environmental factors [74]. It is still thought that gene–gene and gene–environment interactions are ubiquitous given the complex biomolecular interactions that are essential for regulation of gene expression and complex metabolic networks [75], and are likely to play a role in influencing human traits [76]. Furthermore, while recent perspectives have emphasized the fact that most true genetic associations to complex traits carry a vanishingly small effect size [77–80], others have shown experimentally that gene–gene interaction is pervasive and often carries surprisingly large effects [81]. Although there is little empirical evidence of gene–gene interaction associations in pharmacogenomic outcomes, owing to the complexity of drug metabolism and transport, it is probable that many drug-treatment outcomes are explained by combinations of genetic variation in the context of gene–gene interactions.
Interactions & quantitative outcomes: new approaches
Compelling evidence makes it clear that epistasis exists in humans and model organisms and influences human traits, yet there is no consensus on how to best optimize statistical analysis for investigating interactions in pharmacogenomic studies. One approach is to evaluate multimarker combinations for potential inter active effects based on biological criteria [82]. This may include, for instance, testing for interactions between genes that share a similar structure or function, or genes in the same pathway or biological process, such as a receptor and its ligand. Using this strategy would bias the analysis in favor of models with an established biological foundation in the literature, and novel interactions between SNPs would be missed. Furthermore, the entire analysis is conditional upon the quality of the biological information used. Another approach is to select SNPs based on the strength of their independent main effects, evaluating interactions only between SNPs that meet a certain effect size or significance threshold. This strategy assumes that relevant interactions occur only between markers that independently have some major effect on the phenotype alone. This assumption is neither biologically nor statistically well grounded. Biologically, compensatory mechanisms and redundancy at other loci can mitigate the effects of a devastating mutation or polymorphism at another locus, thus rendering its effect undetectable. This is evident in the many gene-knockout mouse lines that show no apparent phenotype [83–90]. Statistically, the main effect components and interactions between them are mathematically independent effects [62]. Furthermore, theoretical studies have shown that traits can be influenced exclusively through the interaction of two or more genetic variants [91,92], and that filtering based on significant main effects would miss these types of discoveries.
Exhaustive evaluation
A strategy to search for a gene–gene or gene–environment interaction that influences a pharmacogenomic outcome without preconditioning on single SNP main effects is to exhaustively evaluate the relationship between the pharmacogenomic outcome of interest and every possible combination of genetic and environmental exposures. While one may wish to fit ANOVA or linear regression models to every possible 2-, 3-, or n-way combination SNPs, this approach becomes problematic for several reasons. First, when interactions among multiple genetic and/or environmental components are considered, there are many combinations that are present in only a few individuals or perhaps none at all. This is known as the curse of dimensionality [93], and results in unstable estimates of population parameters from large sample-based methods such as ANOVA and linear regression. Furthermore, while the interpretation of the statistical significance of models fit using traditional methods is fairly straightforward, correction must be made for multiple testing. Tests of interactions are large in number, and are not independent, making multiple testing correction difficult. Also, as mentioned previously, these methods are uniformly the most powerful technique for detecting differences in the mean value of an outcome, but this only holds when all assumptions are met. For many pharmacogenomic studies, however, these assumptions are typically violated to some degree. Finally, these methods are typically the most efficient only when a mode of inheritance is specified (e.g., dominant, recessive and additive). One of the first methods proposed that would obviate some of these issues is the combinatorial partitioning method (CPM) [94]. CPM works by expanding multilocus genotype combinations and then partitioning these genotypes into groups that explain the largest proportion of variance in the quantitative trait outcome. A later improvement on this method was the restricted partitioning method (RPM) [95], which does not spend valuable computing resources evaluating multilocus genotype partitions that explain little variance. A third similar approach is generalized multifactor dimensionality reduction (gMDR) [96], a variation on the widely used MDR case–control framework [97]. An advantage of exhaustive approaches, such as CPM, RPM and gMDR, is that they will search through every possible multivariable model for a given dimensionality to find the optimal set of genes and environmental factors to most accurately model a quantitative outcome. These methods will report an optimal set of genes found and the amount of variance in the outcome explained by partitioning multilocus genotypes at these genes. As with any data mining technique, however, care must be taken to avoid overfitting [98], or ‘memorizing’ each data point, rather than discovering the true underlying model. Cross-validation is a widely employed and easily implemented technique for mitigating the risk of overfitting a model to a particular dataset [99]. Furthermore, while the theoretical sampling distribution of test statistics for these methods is unknown, statistical significance of models discovered with these combinatorial procedures may be empirically estimated using permutation testing [100]. Here, the null hypothesis will be empirically generated by shuffling the outcome variable values among individuals in the dataset, and running the modeling procedure many times, generating a null sampling distribution of the statistic reported by these methods (e.g., R2). The statistic from the nonpermuted analysis is compared against this null sampling distribution to estimate statistical significance. Since permutation testing requires running the modeling procedure usually thousands of times on permuted data, this can be computationally intensive even in small datasets. However, one group has recently reported a way to approximate permutation testing that requires as little as 1/50th of the time a full permutation test would require [101].
Exhaustive approaches such as the mentioned earlier are ideally suited for exploring interactions in small pharmacogenomic datasets comprised of only a few variables, such as in a candidate gene study. However, the computational resources required to exhaustively search for interactions in GWAS scale data is often prohibitive. For example, the number of two-way interactions that can be evaluated in a GWAS with 500 k SNPs is 1.2 × 1011. Memory issues aside, it would take many years on a desktop computer to run this analysis. This limitation is the motivation for developing techniques that still utilize the full dimensionality of the data without exhaustively searching all possible combinations of variables with the goal of discovering a well fitting model that explains variance in a pharmacogenomic trait. Below we will discuss three computational strategies for discovering gene–gene interactions in genome-wide scale pharmacogenomic data where an exhaustive approach would most likely be computationally prohibitive.
Evolutionary computing
Sharing many similarities with Darwinian evolution of biological organisms, evolutionary computing has been proposed as a way to discover gene–gene and gene–environment interactions that contribute to a human phenotype [102]. Individuals in biological populations can be thought of as candidate solutions to a problem, where the problem in nature is to survive and reproduce. Individuals that are more fit will be selected, and their genes will be propagated in future generations in the population. By analogy, evolutionary computing commences by defining a population of candidate solutions to a problem, where the problem is to find a model containing influential genes that can explain a large proportion of variance in the outcome. ‘Individuals’ are candidate solutions, which are mathematical models containing genetic and environmental variables attempting to explain variance in the outcome of interest in the study. The candidate solutions that explain more variance in the outcome are the models that contain combinations of variables that truly influence the phenotype, and these models are selected and reproduced in subsequent generations of evolutionary computing. In addition, after this phase of selecting the ‘most fit’ individuals, models may be ‘mutated’ (switching one genetic variant out for another in the dataset), or undergo ‘recombination’ with another well-fitting model. Evolutionary computing can be thought of as a pattern-recognition or machine-learning approach for discovering complex genetic models that influence a trait. Evolutionary computing has been used extensively in other disciplines to model complex processes [103–113]. In addition to using evolutionary computing for genetic association studies [114–121], evolutionary computing has been used in other biological applications including microarray analysis [122], cancer classification [123], molecular docking [124] and protein folding [125]. A team of leaders in the field have recently prepared a book [126], giving an overview of genetic programming (a widely used type of evolutionary computing) [205]. Weka [206] is a freely available, open-source, extensible software for evolutionary computing and other data mining methods, available at [129].
Candidate epistasis
Another recently described strategy, the Biofilter, combines a bioinformatics approach with traditional statistical hypothesis testing [130]. Several years ago, when the genome was too large to fully interrogate with genotyping, pharmacogenomics and other disease gene-mapping investigators relied on the candidate gene study design. Here, candidates were selected for genotyping based on their hypothesized biological function, and statistical tests were carried out on a SNP-by-SNP basis. Now, with the advent of GWAS and the impending arrival of inexpensive whole-genome sequencing, assaying human variation across the entire genome is no longer the issue it was in the past. While millions of SNPs can be tested one-by-one for association to a trait, the interactome is too large for us to fully investigate. The approach used reduces the interaction search space by assessing specific combinations of genetic variants based on prior statistical and biological knowledge [130]. The method creates multi-SNP models based on information from publicly available bioinformatics data sources that can then be straightforwardly tested using logistic or linear regression.
Expert knowledge-guided evolutionary computation
Finally, there is much interest in the computer science community to develop strategies for incorporating expert knowledge into evolutionary computation [131]. As mentioned previously, it is theoretically possible and probable in some cases that a trait is influenced exclusively by the interaction of two or more genetic variants, with neither genetic variant having a main effect by itself. This represents the worst-case scenario for an evolutionary method. In fact, it has been shown that evolutionary methods perform little better than randomly testing models for association with the outcome when the underlying model is purely epistatic [132]. However, supplementing an evolutionary procedure with expert knowledge has been shown to increase the statistical sensitivity of evolutionary methods for finding these difficult-to-model interactions [133]. In these reports, the authors used a data-driven approach, relying upon prior statistical expert knowledge as a result of preprocessing the data. The notion presented in [127] suggests a different approach, where expert knowledge is gleaned extrinsically, without any data analysis or preprocessing [130]. Here, multigene groupings were created based on representation in publicly accessible biological databases, such as the Gene Ontology [134] or Kyoto Encyclopedia of Genes and Genomes (KEGG) [135]. Multi-SNP models from these gene groupings were then prioritized in analysis. A very promising approach involves combining a bioinformatics approach such as this with evolutionary computation, allowing investigators to take advantage of the many decades of biomedical research to guide a machine learning procedure. Furthermore, while the aforementioned bioinformatics approach is gene-centric, incorporation of these principles into a stochastic evolutionary procedure would allow for discovery of gene–gene interactions between genetic variants that may not be in gene regions (e.g., an interaction between variants in a micro-RNA and its target).
Expert commentary
In this review, we have presented study design strategies and statistical methodologies for optimizing statistical analysis in pharmacogenomic studies. Careful consideration of the phenotype under study, the population in which the study is carried out and the procedures used to model genetic influences are all equally important for achieving maximum statistical power and breadth of interpretation. Irrespective of the aforementioned considerations, one of the most important aspects of pharmacogenomics and any other genetic association study is replication in an independent sample and/or functional studies. The NCI-NHGRI Working Group on Replication in Association Studies recently established recommendations for bona fide replication of GWAS results [136]. Basic conditions for a successful replication include a sufficient sample size to replicate the genetic effect size estimated in the discovery data set, an independent replication set, the same outcome phenotype for both data sets, a similar study population, similar direction of effect from the same SNP or a SNP in near perfect LD, a consistent genetic model and adequate reporting of replication study design and analysis. Replication of a multi-SNP model presents new challenges and how to effectively test for replication of higher-order models remains an open question in the field of human genetic epidemiology.
Replication of pharmacogenomic studies may post a significantly greater challenge than many other genetic association studies of complex human traits and diseases. Many pharmacogenomic traits such as adverse drug reaction are, by US FDA design, rare. Investigators in the field struggle to amass sufficient numbers of samples for the initial GWAS, leaving them stranded with no replication set. For example, statin-induced myopathy occurs in approximately one out of 10,000 patients prescribed standard doses. A recent GWAS of this adverse event for a commonly prescribed medication had only 85 cases for the initial GWAS and no independent replication cohort [137]. This highlights the important role that large drug-exposed cohorts or populations, such as biobanks linked to electronic medical records [138,139], will play in fulfilling the need to identify sufficient samples for discovery and replication datasets.
Five-year view
Several years have elapsed since the advent of genome-wide association studies, bringing several success stories as well as many disappointments. We must ensure that our strength in study design and optimal statistical methodology keeps pace with the relentless progression of genotyping and sequencing technology so that we may reap the benefits of the wealth of data we will soon face. The characteristic that most bleeding-edge statistical methodologies have in common is that they abandon the simple approach of considering one genetic variant in isolation when modeling the etiology of complex phenotypes. Methods exploiting existing domain knowledge are likely one of many solutions required for the challenging task of properly mining large genomic data sets to identify all variation, alone or in combination, that has an impact on human health.
Key issues
Pharmacogenomics is a promising area of human genetic research with a high potential for generating immediate public health and economic benefits.
Many pharmacological outcomes are naturally continuous variables and the full distribution should be considered rather than relying on artificial categories for ease of analysis.
The pharmacological phenotype should be precisely defined and reliably measured to optimize statistical power and potential reproducibility by others.
Careful consideration should be given to choosing a study population and/or using available statistical methods to minimize the effects of population stratification.
Traditional statistical hypothesis testing techniques for quantitative outcomes (analysis of variance, linear regression) are widely available in nearly any statistical computing software. Investigators typically use these methods to test genetic markers one-by-one for association with the trait of interest.
Gene–gene and gene–environment interactions are pervasive and ubiquitous, and exist even in the absence of main effects. Optimal statistical methods will account for interactions between multiple variables.
Exhaustive searches for interaction (combinatorial partitioning method, restricted partitioning method and generalized multifactor dimensionality reduction) are reasonable approaches for small datasets but quickly become unwieldy with GWAS-scale data.
As advances in genotyping and sequencing technology progress, novel computational methods that take advantage of the wealth of domain knowledge will become increasingly necessary.
Replication is key: regardless of the phenotype studied, population used or methods applied, it is essentially a requirement to provide either statistical replication in an independent sample, or evidence of a functional role for any pharmacogenomic association discovered. Large cohorts or populations will be indispensable for ascertaining enough individuals for discovery and replication datasets.
Footnotes
Financial & competing interests disclosure
This work by Stephen D Turner, Dana C Crawford and Marylyn D Ritchie was funded by the NIH Pharmacogenetics Research Network (PGRN) Pharmacogenomics of Arrhythmia Therapy U01 (HL65962) and the training program on genetic variation and human phenotypes training grant (5T32GM080178-02). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Stephen D Turner, Center for Human Genetics Research, Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville TN, 37232, USA, Tel.: +1 615 343 6549, Fax: +1 615 322 6974, stephen@chgr.mc.vanderbilt.edu.
Dana C Crawford, Center for Human Genetics Research, Assistant Professor, Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville TN, 37232, USA, Tel.: +1 615 343 7852, Fax: +1 615 322 6974, crawford@chgr.mc.vanderbilt.edu.
Marylyn D Ritchie, Center for Human Genetics Research, Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville TN, 37232, USA, Tel.: +1 615 343 5851, Fax: +1 615 322 6974, ritchie@chgr.mc.vanderbilt.edu.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Roses AD. Pharmacogenetics. Hum. Mol. Genet. 2001;10(20):2261–2267. doi: 10.1093/hmg/10.20.2261. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein DB, Tate SK, Sisodiya SM. Pharmacogenetics goes genomic. Nat. Rev. Genet. 2003;4(12):937–947. doi: 10.1038/nrg1229. [DOI] [PubMed] [Google Scholar]
- 3.Risch N, Merikangas K. The future of genetic studies of complex human disorders. Science. 1996;273(5281):1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 4.International hapmap consortium. The International HapMap Project. Nature. 2003;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 5.International hapmap consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itsara A, Cooper GM, Baker C, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84(2):148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40(10):1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 8.Jakobsson M, Scholz SW, Scheet P, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451(7181):998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 9.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson MR, Bacanu SA, Mosteller M, et al. Genomewide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J. 2009;9(1):23–33. doi: 10.1038/tpj.2008.4. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi F, McGinnis R, Bourgeois S, et al. A genomewide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5(3):e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper GM, Johnson JA, Langaee TY, et al. A genomewide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood. 2008;112(4):1022–1027. doi: 10.1182/blood-2008-01-134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu C, Batliwalla F, Li W, et al. Genomewide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol. Med. 2008;14(9–10):575–581. doi: 10.2119/2008-00056.Liu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koska MT. CEOs make the most of trustees’ business acumen. Hospitals. 1990;64(11):28–33. [PubMed] [Google Scholar]
- 15.Dumaual C, Miao X, Daly TM, et al. Comprehensive assessment of metabolic enzyme and transporter genes using the Affymetrix Targeted Genotyping System. Pharmacogenomics. 2007;8(3):293–305. doi: 10.2217/14622416.8.3.293. [DOI] [PubMed] [Google Scholar]
- 16.Caldwell MD, Awad T, Johnson JA, et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008;111(8):4106–4112. doi: 10.1182/blood-2007-11-122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mega JL, Close SL, Wiviott SD, et al. Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med. 2009;360(4):354–362. doi: 10.1056/NEJMoa0809171. [DOI] [PubMed] [Google Scholar]
- 18.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24(3):133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Schuster SC. Next-generation sequencing transforms today’s biology. Nat. Methods. 2008;5(1):16–18. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 20.Von Bubnoff A. Next-generation sequencing: the race is on. Cell. 2008;132(5):721–723. doi: 10.1016/j.cell.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 21.Haines JL, Pericak-Vance MA. Approaches to gene mapping in complex human diseases. John Wiley and Sons; 1998. pp. 323–333. [Google Scholar]
- 22.Leon Gordis. Epidemiology. Saunders; 2008. pp. 177–199. [Google Scholar]
- 23.Klein TE, Altman RB, Eriksson N, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med. 2009;360(8):753–764. doi: 10.1056/NEJMoa0809329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sills GJ. Pharmacogenetics of epilepsy: one step forward? Epilepsy Curr. 2005;5(6):236–238. doi: 10.1111/j.1535-7511.2005.00074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arranz MJ, Munro J, Birkett J, et al. Pharmacogenetic prediction of clozapine response. Lancet. 2000;355(9215):1615–1616. doi: 10.1016/s0140-6736(00)02221-2. [DOI] [PubMed] [Google Scholar]
- 26.Nagasubramanian R, Innocenti F, Ratain MJ. Pharmacogenetics in cancer treatment. Annu. Rev. Med. 2003;54:437–452. doi: 10.1146/annurev.med.54.101601.152352. [DOI] [PubMed] [Google Scholar]
- 27.Wessels JA, van der Kooij SM, le CS, et al. A clinical pharmacogenetic model to predict the efficacy of methotrexate monotherapy in recent-onset rheumatoid arthritis. Arthritis Rheum. 2007;56(6):1765–1775. doi: 10.1002/art.22640. [DOI] [PubMed] [Google Scholar]
- 28.Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drugs safety, and confirmation of a pathogenetic hypothesis (Alzheimer’s disease) Pharmacogenomics J. 2007;7(1):10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- 29.Siddiqui A, Kerb R, Weale ME, et al. Association of multidrug resistance in epilepsy with apolymorphism in the drug-transporter gene ABCB1. N. Engl. J. Med. 2003;348(15):1442–1448. doi: 10.1056/NEJMoa021986. [DOI] [PubMed] [Google Scholar]
- 30.Aulchenko YS, Ripatti S, Lindqvist I, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009;41(1):47–55. doi: 10.1038/ng.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabatti C, Service SK, Hartikainen AL, et al. Genomewide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 2009;41(1):35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat. Genet. 2009;41(1):56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newton-Cheh C, Johnson T, Gateva V, et al. Genomewide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009 doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 2009;10(3):184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon AL, Liang L, Moffatt MF, et al. A genomewide association study of global gene expression. Nat. Genet. 2007;39(10):1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 36.Meyer-Lindenberg A, Weinberger DR. Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat. Rev. Neurosci. 2006;7(10):818–827. doi: 10.1038/nrn1993. [DOI] [PubMed] [Google Scholar]
- 37.Cristea IM, Gaskell SJ, Whetton AD. Proteomics techniques and their application to hematology. Blood. 2004;103(10):3624–3634. doi: 10.1182/blood-2003-09-3295. [DOI] [PubMed] [Google Scholar]
- 38.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004;36(5):512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 39. Need AC, Motulsky AG, Goldstein DB. Priorities and standards in pharmacogenetic research. Nat. Genet. 2005;37(7):671–681. doi: 10.1038/ng1593. • Contains an extensive list of US FDA-approved drugs withdrawn from major markets between 1990–2005 with genes implicated in variable drug response.
- 40.Zhu X, Tang H, Risch N. Admixture mapping and the role of population structure for localizing disease genes. Adv. Genet. 2008;60:547–569. doi: 10.1016/S0065-2660(07)00419-1. [DOI] [PubMed] [Google Scholar]
- 41.Britt BA, Locher WG, Kalow W. Hereditary aspects of malignant hyperthermia. Can. Anaesth. Soc. J. 1969;16(2):89–98. doi: 10.1007/BF03005788. [DOI] [PubMed] [Google Scholar]
- 42.Denborough MA, Forster JF, Lovell RR, Maplestone PA, Villiers JD. Anaesthetic deaths in a family. Br. J. Anaesth. 1962;34:395–396. doi: 10.1093/bja/34.6.395. [DOI] [PubMed] [Google Scholar]
- 43.Laird NM, Lange C. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 2006;7(5):385–394. doi: 10.1038/nrg1839. [DOI] [PubMed] [Google Scholar]
- 44.Govindaraju DR, Cupples LA, Kannel WB, et al. Genetics of the Framingham Heart Study population. Adv. Genet. 2008;62:33–65. doi: 10.1016/S0065-2660(08)00602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilk JB, Manning AK, Dupuis J, et al. No evidence of major population substructure in the Framingham Heart Study. Genet. Epidemiol. 2005;29:234–292. [Google Scholar]
- 46.Ioannidis JP, Thomas G, Daly MJ. Validating, augmenting and refining genomewide association signals. Nat. Rev. Genet. 2009;10(5):318–329. doi: 10.1038/nrg2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manolio TA. Collaborative genomewide association studies of diverse diseases: programs of the NHGRI’s office of population genomics. Pharmacogenomics. 2009;10(2):235–241. doi: 10.2217/14622416.10.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin SS, Kelsey JL. Use of race and ethnicity in epidemiologic research: concepts, methodological issues, and suggestions for research. Epidemiol. Rev. 2000;22(2):187–202. doi: 10.1093/oxfordjournals.epirev.a018032. [DOI] [PubMed] [Google Scholar]
- 49.Race ethnicity and genetics working group: the use of racial, ethnic, and ancestral categories in human genetics research. Am. J. Hum. Genet. 2005;77(4):519–532. doi: 10.1086/491747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seldin MF, Price AL. Application of ancestry informative markers to association studies in European Americans. PLoS Genet. 2008;4(1):E5. doi: 10.1371/journal.pgen.0040005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yaeger R, vila-Bront A, Abdul K, et al. Comparing genetic ancestry and self-described race in african americans born in the United States and in Africa. Cancer Epidemiol. Biomarkers Prev. 2008;17(5):1329–1338. doi: 10.1158/1055-9965.EPI-07-2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pritchard JK, Rosenberg NA. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 1999;65(1):220–228. doi: 10.1086/302449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reich DE, Goldstein DB. Detecting association in a case–control study while correcting for population stratification. Genet. Epidemiol. 2001;20(1):4–16. doi: 10.1002/1098-2272(200101)20:1<4::AID-GEPI2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 54.Novembre J, Johnson T, Bryc K, et al. Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 56.Dadd T, Weale ME, Lewis CM. A critical evaluation of genomic control methods for genetic association studies. Genet. Epidemiol. 2009;33(4):290–298. doi: 10.1002/gepi.20379. [DOI] [PubMed] [Google Scholar]
- 57.Devlin B, Bacanu SA, Roeder K. Genomic Control to the extreme. Nat. Genet. 2004;36(11):1129–1130. doi: 10.1038/ng1104-1129. [DOI] [PubMed] [Google Scholar]
- 58.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genomewide association studies. Nat. Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 60.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):E190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang F, Wang Y, Deng HW. Comparison of population-based association study methods correcting for population stratification. PLoS ONE. 2008;3(10):e3392. doi: 10.1371/journal.pone.0003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maxwell SE, Delaney HD. Designing Experiments and Analyzing Data. Lawrence Erlbaum Associates; 2004. [Google Scholar]
- 63.Sokal RR, Rohlf FJ. In: Biometry. Freeman, editor. 1995. pp. 179–450. [Google Scholar]
- 64.Agresti A. Categorical Data Analysis. San Francisco, CA, USA: John Wiley & Sons; 1990. pp. 141–142. [Google Scholar]
- 65.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mushiroda T, Ohnishi Y, Saito S, et al. Association of VKORC1 and CYP2C9 polymorphisms with warfarin dose requirements in Japanese patients. J. Hum. Genet. 2006;51(3):249–253. doi: 10.1007/s10038-005-0354-5. [DOI] [PubMed] [Google Scholar]
- 67.Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456(7218):18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 68.Iles MM. What can genomewide association studies tell us about the genetics of common disease? PLoS Genet. 2008;4(2):E33. doi: 10.1371/journal.pgen.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265(5181):2037–2048. doi: 10.1126/science.8091226. •• Overview on using traditional linkage and association methods in the genetic analysis of complex phenotypes.
- 70. Moore JH, Williams SM. New strategies for identifying gene–gene interactions in hypertension. Ann. Med. 2002;34(2):88–95. doi: 10.1080/07853890252953473. •• Essay on statistical and biological interaction.
- 71.Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. Bioessays. 2005;27(6):637–646. doi: 10.1002/bies.20236. [DOI] [PubMed] [Google Scholar]
- 72.Ritchie MD, Hahn LW, Roodi N, et al. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001;69(1):138–147. doi: 10.1086/321276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright S. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. Proc. 6th Intl. Congress of Genetics. 1932;1:356–366. [Google Scholar]
- 74.Gibson G. Epistasis and pleiotropy as natural properties of transcriptional regulation. Theor. Popul. Biol. 1996;49(1):58–89. doi: 10.1006/tpbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 75. Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum. Hered. 2003;56(1–3):73–82. doi: 10.1159/000073735. • The importance of gene–gene interaction in complex traits.
- 76.Hirschhorn JN. Genomewide association studies – illuminating biologic pathways. N. Engl. J. Med. 2009;360(17):1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 77.Goldstein DB. Common genetic variation and human traits. N. Engl. J. Med. 2009;360(17):1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- 78.Hardy J, Singleton A. Genomewide association studies and human disease. N. Engl. J. Med. 2009;360(17):1759–1768. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kraft P, Hunter DJ. Genetic risk prediction – are we there yet? N. Engl. J. Med. 2009;360(17):1701–1703. doi: 10.1056/NEJMp0810107. [DOI] [PubMed] [Google Scholar]
- 80.Shao H, Burrage LC, Sinasac DS, et al. Genetic architecture of complex traits: large phenotypic effects and pervasive epistasis. Proc. Natl Acad. Sci. USA. 2008;105(50):19910–19914. doi: 10.1073/pnas.0810388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carlson CS, Eberle MA, Kruglyak L, Nickerson DA. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429(6990):446–452. doi: 10.1038/nature02623. [DOI] [PubMed] [Google Scholar]
- 82.Baba T, Azuma S, Kashiwabara S, Toyoda Y. Sperm from mice carrying a targeted mutation of the acrosin gene can penetrate the oocyte zona pellucida and effect fertilization. J. Biol. Chem. 1994;269(50):31845–31849. [PubMed] [Google Scholar]
- 83.Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79(4):679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- 84.Gorry P, Lufkin T, Dierich A, et al. The cellular retinoic acid binding protein I is dispensable. Proc. Natl Acad. Sci. USA. 1994;91(19):9032–9036. doi: 10.1073/pnas.91.19.9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gruda MC, van AJ, Rizzo CA, Durham SK, Lira S, Bravo R. Expression of FosB during mouse development: normal development of FosB knockout mice. Oncogene. 1996;12(10):2177–2185. [PubMed] [Google Scholar]
- 86.Itohara S, Mombaerts P, Lafaille J, et al. T cell receptor δ gene mutant mice: independent generation of α β T cells and programmed rearrangements of γδ TCR genes. Cell. 1993;72(3):337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- 87.Killeen N, Stuart SG, Littman DR. Development and function of T cells in mice with a disrupted CD2 gene. EMBO J. 1992;11(12):4329–4336. doi: 10.1002/j.1460-2075.1992.tb05532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kneitz B, Herrmann T, Yonehara S, Schimpl A. Normal clonal expansion but impaired Fas-mediated cell death and anergy induction in interleukin-2-deficient mice. Eur. J. Immunol. 1995;25(9):2572–2577. doi: 10.1002/eji.1830250925. [DOI] [PubMed] [Google Scholar]
- 89.Zheng H, Jiang M, Trumbauer ME, et al. Mice deficient for the amyloid precursor protein gene. Ann. NY Acad. Sci. 1996;777:421–426. doi: 10.1111/j.1749-6632.1996.tb34456.x. [DOI] [PubMed] [Google Scholar]
- 90. Culverhouse R, Suarez BK, Lin J, Reich T. A perspective on epistasis: limits of models displaying no main effect. Am. J. Hum. Genet. 2002;70(2):461–471. doi: 10.1086/338759. • Theoretical justiication for the possibility of interaction with little or no main effect component.
- 91.Moore J, Hahn L, Ritchie M, Thornton T, White B. Application of genetic algorithms to the discovery of complex models for simulation studies in human genetics; Proceedings of the Genetic and Evolutionary Algorithm Conference; 2002. pp. 1150–1155. [PMC free article] [PubMed] [Google Scholar]
- 92.Bellman R. Adaptive control processes. Princeton University Press; 1961. [Google Scholar]
- 93.Nelson MR, Kardia SLR, Sing CF. The combinatorial partitioning method. lecture notes in computer. Science. 2000;1848:293–304. [Google Scholar]
- 94.Culverhouse R, Klein T, Shannon W. Detecting epistatic interactions contributing to quantitative traits. Genet. Epidemiol. 2004;27(2):141–152. doi: 10.1002/gepi.20006. [DOI] [PubMed] [Google Scholar]
- 95.Lou XY, Chen GB, Yan L, et al. A generalized combinatorial approach for detecting gene-by-gene and gene-by-environment interactions with application to nicotine dependence. Am. J. Hum. Genet. 2007;80(6):1125–1137. doi: 10.1086/518312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ritchie MD, Motsinger AA. Multifactor dimensionality reduction for detecting gene–gene and gene–environment interactions in pharmacogenomics studies. Pharmacogenomics. 2005;6(8):823–834. doi: 10.2217/14622416.6.8.823. [DOI] [PubMed] [Google Scholar]
- 97.Bishop CM. Pattern Recognition and machine learning. Springer; 2006. [Google Scholar]
- 98.Hastie T, Tibshirani R, Friedman J. The Elements of statistical learning: data mining, inference, and prediction. Springer-Verlag; 2001. [Google Scholar]
- 99.Good P. Permutation Tests: a practical guide to resampling methods for testing hypotheses. Springer-Verlag; 2000. [Google Scholar]
- 100.Pattin KA, White BC, Barney N, et al. A computationally efficient hypothesis testing method for epistasis analysis using multifactor dimensionality reduction. Genet. Epidemiol. 2008;33(1):87–94. doi: 10.1002/gepi.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thornton-Wells TA, Moore JH, Haines JL. Genetics, statistics and human disease: analytical retooling for complexity. Trends Genet. 2004;20(12):640–647. doi: 10.1016/j.tig.2004.09.007. •• Overview of novel methodology for analysis of complex traits, mostly focusing on case–control designs.
- 102.Hung SL, Adeli H. A parallel genetic/neural network learning algorithm for MIMD shared memory machines. IEEE Trans. Neural Netw. 1994;5(6):900–909. doi: 10.1109/72.329686. [DOI] [PubMed] [Google Scholar]
- 103.Lee SW. Off-line recognition of totally unconstrained handwritten numerals using multilayer cluster neural network. IEEE Trans. Pattern Anal. Mach. Intell. 1996;18:648–652. [Google Scholar]
- 104.Likartsis A, Vlachavas I, Tsoukalas LH. A new hybrid neural-genetic methodology for improving learning. Proceedings of the Ninth IEEE International Conference on Tools with Artificial Intelligence; 1997. pp. 32–36. [Google Scholar]
- 105.Yang JM, Kao CY, Horng JT. Evolving neural induction regular language using combined evolutionary algorithms: ISAI/IFIS 1996; Mexico-USA Proceedings of Collaboration in Intelligent Systems Technologies; 1996. pp. 162–169. [Google Scholar]
- 106.Zhang P, Sankai Y, Ohta M. Hybrid adaptive learning control of nonlinear system. Proceedings of the 1995 American Control Conference; 1995. pp. 2744–2748. [Google Scholar]
- 107.Belew RK, McInerney J, Schraudolph NN. Evolving networks: using the genetic algorithm with connectionist learning. Computer Science & Engineering Department Technical report. 1990 [Google Scholar]
- 108.Chen YM, O’Connell RM. Active power line conditioner with a neural network control. IEEE Transactions on Industry Applications. 1997;33:1131–1136. [Google Scholar]
- 109.Topchy A, Lebedko OA. Evolving networks: using the genetic algorithm with connectionist learning. Nucl. Instrum. Methods Phys. Res. 1997;389:240–241. [Google Scholar]
- 110.Cantu-Paz E, Kamath C. Evolving neural networks to identify bent-double galaxies in the FIRST survey. Neural Netw. 2008;16:507–517. doi: 10.1016/S0893-6080(03)00020-0. [DOI] [PubMed] [Google Scholar]
- 111.Skinner AJ, Broughton JQ. Neural networks in computational materials science: training algorithms. Modelling and Simulation in Materials Science and Engineering. 1995;3(3):371–390. [Google Scholar]
- 112.Yan W, Zhu Z, Hu R. A hybrid genetic/BP algorithm and its application for radar target classification. Proceedings of the IEEE 1997 National Aerospace and Electronics Conference; 1997. pp. 981–984. [Google Scholar]
- 113.Motsinger AA, Lee SL, Mellick G, Ritchie MD. GPNN: power studies and applications of a neural network method for detecting gene–gene interactions in studies of human disease. BMC Bioinformatics. 2006;7:39. doi: 10.1186/1471-2105-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Motsinger AA, Dudek SM, Hahn LW, Ritchie MD. Grammatical evolution for the optimization of neural networks for genetic association studies. Bioinformatics. 2008;32(4):325–340. [Google Scholar]
- 115.Motsinger AA, Reif DM, Dudek SM, Ritchie MD. Understanding the evolutionary process of grammatical evolution neural networks for feature selection in genetic epidemiology; IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology; 2006. pp. 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Motsinger AA, Reif DM, Fanelli TJ, Davis AC, Ritchie MD. Linkage disequilibrium in genetic association studies improves the performance of grammatical evolution neural networks; IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology; 2007. pp. 1–8. [PMC free article] [PubMed] [Google Scholar]
- 117.Motsinger-Reif AA, Dudek SM, Hahn LW, Ritchie MD. Comparison of approaches for machine-learning optimization of neural networks for detecting gene–gene interactions in genetic epidemiology. Gen. Epidemiol. 2008;32(4):325–340. doi: 10.1002/gepi.20307. [DOI] [PubMed] [Google Scholar]
- 118.Motsinger-Reif AA, Fanelli TJ, Davis AC, Ritchie MD. Power of grammatical evolution neural networks to detect gene–gene interactions in the presence of error. BMC. Res. Notes. 2008;1:65. doi: 10.1186/1756-0500-1-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ritchie MD, White BC, Parker JS, Hahn LW, Moore JH. Optimization of neural network architecture using genetic programming improves detection and modeling of gene–gene interactions in studies of human diseases. BMC Bioinformatics. 2003;4(1):28. doi: 10.1186/1471-2105-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ritchie MD, Coffey CSMJH. Genetic programming neural networks: a bioinformatics tool for human genetics. Lect. Notes Comput. Sci. 2004;3102:438–448. doi: 10.1016/j.asoc.2006.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang DS, Liu KH, Xu CG. A genetic programming based approach to the classification of multiclass microarray datasets. Bioinformatics Btn. 2008;644 doi: 10.1093/bioinformatics/btn644. [DOI] [PubMed] [Google Scholar]
- 122.Mukhopadhyay A, Maulik U, Bandyopadhyay S. Refining genetic algorithm based fuzzy clustering through supervised learning for unsupervised cancer classification. Lect. Notes Comput. Sci. 2009;5483:191–202. [Google Scholar]
- 123.Tavares J, Mesmoudi S, Talbi E. On the efficiency of local search methods for the molecular docking problem. Lect. Notes Comput. Sci. 2009;5483:104–115. [Google Scholar]
- 124.Vullo A, Passerini A, Frasconi P, Costa F, Pollastri G. On the convergence of protein structure and dynamics. statistical learning studies of pseudo folding pathways. Lect. Notes Comput. Sci. 2008;4973:200–211. [Google Scholar]
- 125. Poli R, Langdon WB, McPhee NF. A field guide to genetic programming. Lulu enterprises; 2008. •• Introduction to evolutionary computing using genetic programming. Available free in PDF format at the URL referenced in[205].
- 126.Witten IH, Frank E. Data mining: practical machine learning tools and techniques. CA, USA: Morgan Kaufmann; 2005. [Google Scholar]
- 127.Bush WS, Dudek SM, Ritchie MD. Biofilter: A knowledge-integration system for the multi-locus analysis of genomewide association studies. Pac. Symp. Biocomput. 2009;14:368–379. [PMC free article] [PubMed] [Google Scholar]
- 128.Moore JH, White BC. Genomewide genetic analysis using genetic programming: the critical need for expert knowledge. Genetic Programming Theory and Practice. 2007;4:11–28. [Google Scholar]
- 129.White BC, Gilbert JC, Reif DM, Moore JH. A statistical comparison of grammatical evolution strategies in the domain of human genetics; Proceedings of the IEEE Congress on Evolutionary Computing; 2005. pp. 676–682. [Google Scholar]
- 130.Greene CS, White BC, Moore JH. Sensible initialization using expert knowledge for genomewide analysis of epistasis using genetic programming; Proceedings of the IEEE Congress on Evolutionary Computing; 2009. pp. 676–682. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology: the gene ontology consortium. Nat. Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chanock SJ, Manolio T, Boehnke M, et al. Replicating genotype-phenotype associations. Nature. 2007;447(7145):655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 134.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N. Engl. J. Med. 2008;359(8):789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 135.Roden DM, Pulley JM, Basford MA, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008;84(3):362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McCarty CA, Chapman-Stone D, Derfus T, Giampietro PF, Fost N. Community consultation and communication for a population-based DNA biobank: the Marshfield clinic personalized medicine research project. Am. J. Med. Genet. 2008;146A(23):3026–3033. doi: 10.1002/ajmg.a.32559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 201.Structure. 2009 http://pritch.bsd.uchicago.edu/structure.html.
- 202.Eigensoft. http://genepath.med.harvard.edu/~reich/EIGENSTRAT.htm.
- 203.Plink. http://pngu.mgh.harvard.edu/~purcell/plink.
- 204.A language and environment for statistical computing: foundation for StatIstical computing. www.R-project.org.
- 205.A field guide to genetic programming. www.gp-field-guide.org.uk.
- 206.Weka. www.cs.waikato.ac.nz/ml/weka.