

Abstract
The preparation of 2′,4′,6′-[3H3]-(R,R)-4-methoxyfenoterol, a tritium-labeled derivative of (R,R)-4-methoxyfenoterol was demonstrated on a 15 mCi scale providing material with a specific activity of 57 Ci/mmol.
Introduction
(R,R)-4-Methoxyfenoterol (1) is a potent β2-adrenoreceptor agonist identified as a potential drug for the treatment of congestive heart failure. The drug candidate is one of four stereoisomers of methoxyfenoterol, and is one of nine synthetic analogs of fenoterol synthesized in our laboratory [1]. The compounds have been prepared in support of a broad study into the pharmacological effects of fenoterol analogs and the role of stereochemistry on receptor activity [2].
To assist in elucidating the pharmacology of the drug it was necessary to synthesize an isotopically modified derivative. The use of tritium provides the sensitivity required for the detection of ligand displacement at the β2-adrenergic receptor in the high affinity state. Tritium also provides the advantage of minimally disrupting the molecule, as well as the benefit of allowing multiple synthetic routes to accessing a tritium labeled species.
Our first strategy was to tritium-label at the 4-methoxy position. This route seemed desirable, as it would give three tritium atoms associated with the molecule, satisfying the required specific activity of 30–100 Ci/mmol (each tritium atom gives a theoretical specific activity of 28.6 Ci/mmol). Because prior attempts at demethylating 4-methoxyfenoterol using BBr3, sodium thiomethoxide or aqueous HBr have been problematic in our lab, the [3H]-methyl group would have to be introduced earlier in the synthesis.
A second consideration for the radiolabeling of 1 was that the method could be applied to existing and new analogs. Devising a method to label 1 at the 4-methoxy position would give us little benefit in the labeling of next generation compounds. One common component in each of the nine synthetic fenoterol analogs is the 3′,5′-dihydroxy ring. This side of the molecule is not only conserved in each currently synthesized analog, but also likely to be present in next generation analogs, as modifications thus far have occurred only at the aminoalkyl portion of the molecule.
The most obvious approach was exchanging tritium for hydrogen at the 2′,4′,6′-positions of the dihydroxy ring using established halogenation/catalytic tritiation radiochemistry. This report describes the convenient preparation of [3H3]-(R,R)-4-methoxyfenoterol HCl (3) using this approach.
Results and Discussion
To test the halogenation/catalytic hydrogenation approach, a sample of the available drug product, (R,R)-4-methoxyfenoterol semi-fumarate (1·fum) was bromine was brominated with 3 equivalents of N-bromosuccinamide (NBS) in DMF resulting in a mixture of the 4′-bromo and 2′,4, ′6-tribromo species, which are separated on a column of silica gel [3]. We estimated that performing the bromination on the base form of 1 would provide a better conversion to the tribromo product. Also, a concern that treating the fumarate salt with tritium gas would result in tritiated diacid prompted us to synthesize the base form of 1 (Scheme I) using our reported method [1]. In brief, (R)-(−)-3′,5′-dibenzyloxyphenylbromohydrin (4) was converted to the epoxide (5) in the presence of K2CO3 in THF-methanol, extracted and then heated neat at 120 °C with (R)-(−)-1-(4′-methoxyphenyl)-2-benzylaminopropane (6) until completion. Attempts to brominate this benzylated product (7) using NBS or pyridinium tribromide were unsuccessful, so the residue was then debenzylated with hydrogen gas in ethanol and then purified on silica gel, yielding the base form of 1 in 75% yield, ee = 93.1%.
Scheme I.
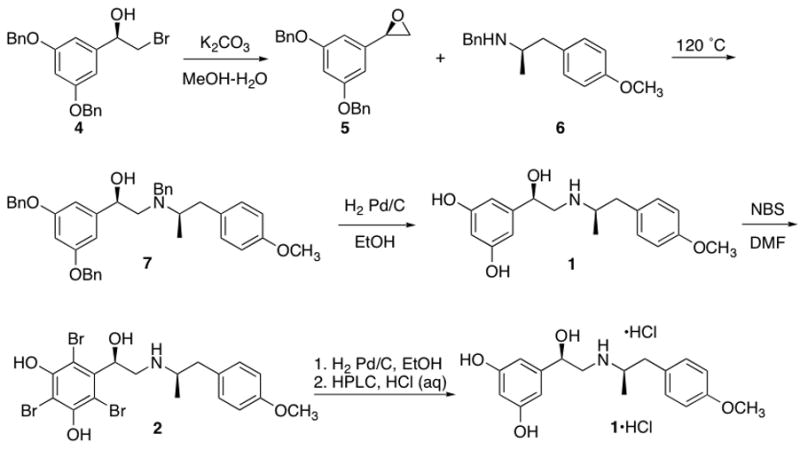
Bromination commenced by treating 1 with 3 eq of NBS in DMF at −30 to −10 °C until completion (~1 h). Silica gel chromatography (methanol-chloroform) gave pure (R,R)-2′,4′,6′-tribromo-4-methoxyfenoterol (2) in 76% yield. Conditions for the catalytic reduction were carried out in-house using hydrogen gas in place of tritium gas. Ethanol was used as the reaction solvent at 1 ATM of hydrogen gas in the presence of ~20% wt of palladium on carbon, which provided a complete conversion back to 1 after 16 h. Purification on a reverse-phase HPLC column using aqueous HCl and methanol gave a material with a final purity of 96.8% and also provided a convenient method to convert the base to the HCl salt (1·HCl).
We had concerns about using a protic solvent such as ethanol because of the possibility of tritium exchanging with the solvent at a higher rate than with bromine and resulting in a species with low specific activity. We investigated the feasibility of using DMF and 1,4-dioxane in place of ethanol, both resulted in no reaction. Fenoterol analogs have shown to be unstable to base, so the use of a basic proton scavenger in the solvent selection experiments was not investigated. We estimated that tritiation of 2 in high concentration in ethanol would commence at a rate sufficient to provide a suitable specific activity.
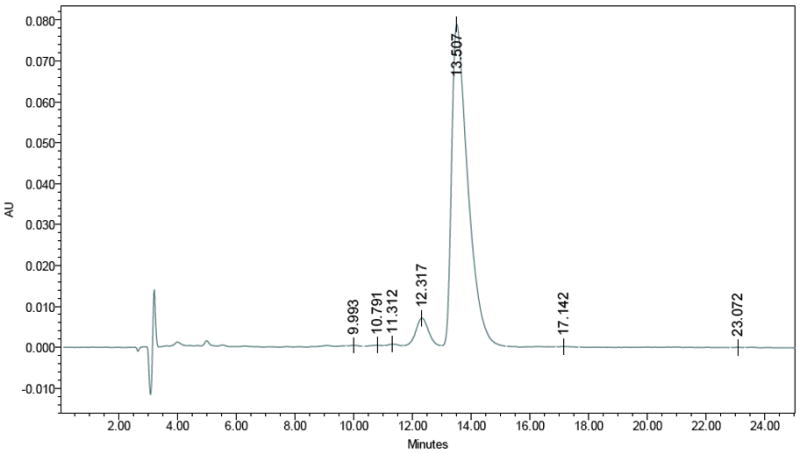
To address concerns about stereochemistry and racemization, a chiral HPLC method was developed to quantify enantiomeric purity (Figures 1 and 2). The enantiomeric excess of the starting (R,R)-4-methoxyfenoterol base was measured by chiral HPLC and compared to the debromination product. The result is an unchanged stereochemical designation and enantiomeric purity through the process.
Figure 1.
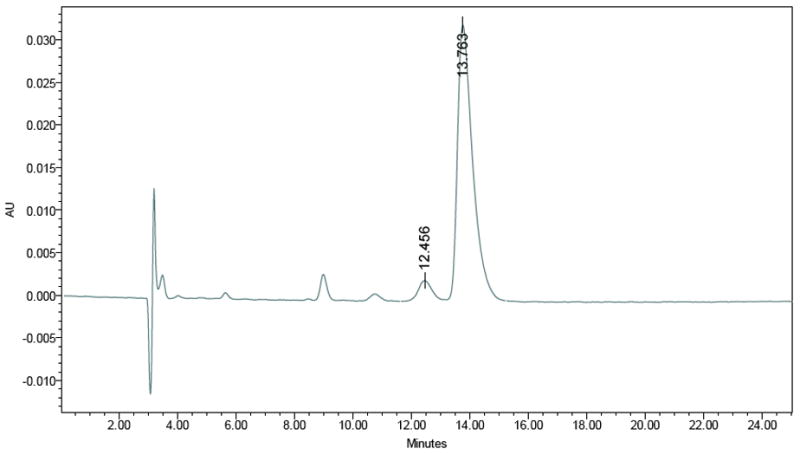
Figure 2.
After we defined the hot synthesis method, 3 was prepared. The hot run was carried out at high concentration (0.1mL/mg) using a large excess (10 Ci) of tritium gas to achieve a specific activity of 57 Ci/mmol (Scheme II). As with the cold run, purification was carried out on a reverse-phase HPLC column using aqueous HCl, providing 15 mCi of the HCl salt with a final radiochemical purity of 99.7%. Compound 3 was analyzed by reverse-phase HPLC, MS and 3H NMR (Figures 3–5).
Scheme II.
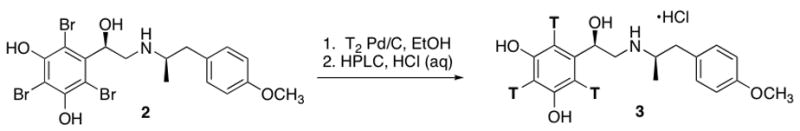
Figure 3.
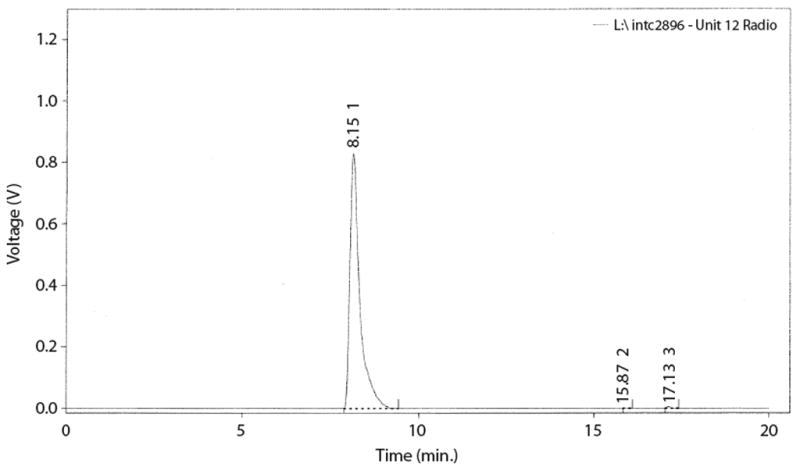
Figure 5.
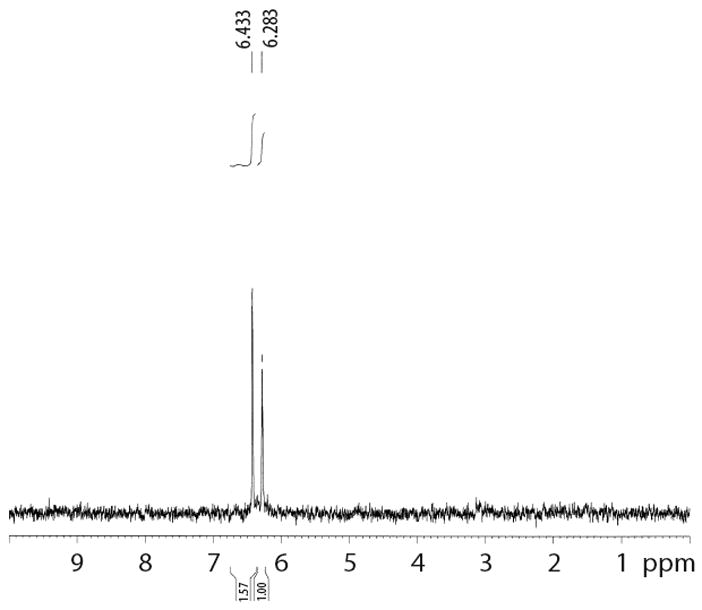
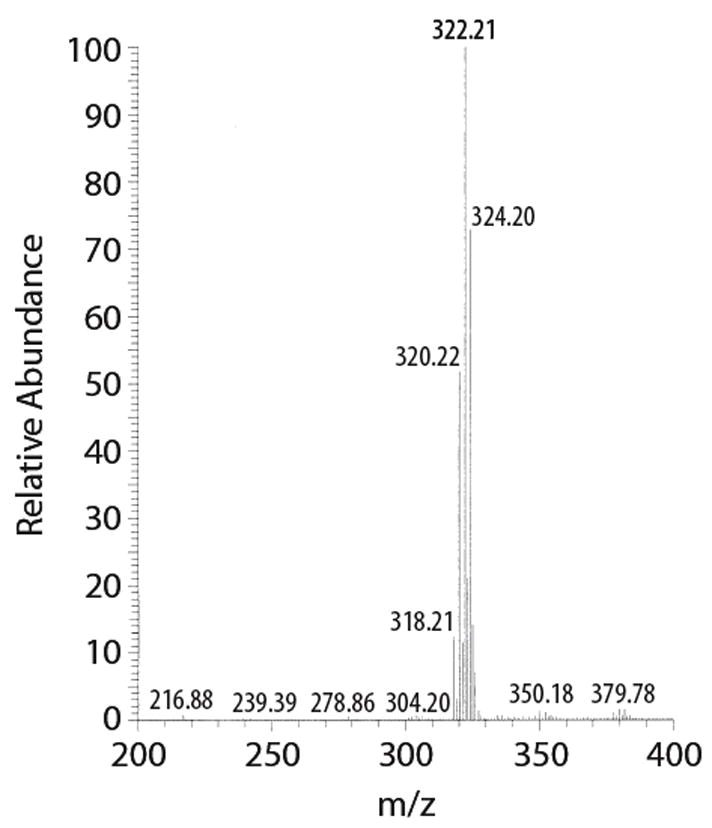
Both the mass spectrum and the tritium NMR of 3 are consistent with a specific activity of 57 Ci/mmol. The mass spectrum (Figure 4) shows peaks at 318 for T0, 320 for T1, 322 for T2 and 324 for T3, the ratio being 12/53/100/73. In the tritium NMR (Figure 5) the integrals of the peaks at 6.28 ppm (s, 1T) and 6.43 ppm (s, 2T) have a ratio of 1.57/1 instead of 2/1 expected for 100% enrichment.
Figure 4.
Radioligand 3 stored at 1 mCi/mL in ethanol at −20 °C was found to be unstable to the effects of radiolysis. At this concentration the sample degraded to 89.7% radiopurity in 12 days and then 82.8% after 62 days. A 1 mCi sample was repurified by HPLC giving 0.32 mCi of 97.5% radiopure 3. Samples stored at −20 °C at a concentration of 64 μCi/mL in acetonitrile/0.001M HCl (pH 3) (1/4) remained stable after 50 days.
Experimental
General Methods
For the analysis of unlabeled compounds: 1H NMR spectra were recorded on a Varian Mercury VMX 300-MHz spectrophotometer. Low resolution mass spectra were obtained on a Finnigan LCQDuo LC MS/MS atmospheric pressure chemical ionization (API) quadrupole ion trap MS system equipped with an electrospray (ESI) probe. Reverse-phase and chiral HPLC data were obtained using a Waters 2690 Separations Module with PDA detection. For the analysis of 3: 3H NMR spectra were run on a Bruker 300 MHz spectrophotometer. LC/MS data was obtained using a Thermo Finnigan LCQ Deca XP.
(R,R)-2′,4′,6′-tribromo-4-methoxyfenoterol (2)
A 123 mg (0.390 mmol) sample of (R,R)-methoxyfenoterol base (1) is dissolved in 12 mL of dry DMF and cooled to −30 °C. Added in one portion under argon is 207 mg (1.16 mmol, 3 eq) of freshly recrystallized N-bromosuccinamide. The bath temperature is brought to −10 °C and the solution is stirred for 1 h. The solvents are removed under high vacuum to give 369 mg. Silica gel chromatography eluting with 20–50% methanol-chloroform gives 163 mg (76%) of 2.
RP-HPLC: Column: ACE-5 C18, 100 × 4.6 mm; Isocratic: (8/2) 0.1% TFA-H2O/ACN; Flow: 1.0 mL/min; Detection: 275 nm; Retention time: Rt = 8.796 min, 95.0% pure.
MS: m/z (rel. intensity): 555 (100, M+H).
1H NMR: (300 MHz, CD3OD) δ1.24 (d, 3H, J = 6.3 Hz), 2.71 (dd, 1H, J = 9.0, 13.2 Hz), 3.06 (dd, 1H, J = 5.4, 13.5 Hz), 3.32–3.47 (m, 2H), 3.77 (s, 3H), 4.03 (dd, 1H, J = 10.8, 12.3 Hz), 5.19 (dd, 1H, J = 3.3, 11.1 Hz), 6.89 (d, 2H, J = 8.7 Hz) 7.18 (d, 2H, J = 9.0 Hz) ppm.
(R,R)-4-Methoxyfenoterol HCl (1·HCl)
A 7.7 mg (0.014 mmol) sample of 2 is dissolved in 0.8 mL of absolute ethanol and treated with 3.0 mg of 10% palladium on carbon (Strem Co, ~50% paste in water). The vial is fitted with a balloon filled with hydrogen gas and then its contents stirred at RT for 16 h. The mixture is diluted with 2 mL of MeOH and filtered through a 0.45 micron syringe filter and the solvent removed to give 4.2 mg of 1. Purification: Column: ACE-5 C18, 100 × 4.6 mm; Isocratic 8/2 0.001M HCl/ACN; 1.0 mL/min; Detection: 275 nm. Pure fractions from multiple injections are combined and lyophilized to afford the HCl salt (1·HCl) as a white solid.
RP-HPLC: Column: ACE-5 C18, 100 × 4.6 mm; Isocratic: (8/2) 0.1% TFA-H2O/ACN; Flow: 1.0 mL/min; Detection: 275 nm; Retention time: Rt = 6.191 min, 96.83 % pure (Rt of 1 standard = 6.791).
Chiral-HPLC: Column: Astec Chirobiotic-V, 250 × 4.6 mm; Isocratic: (6/4) 20 mM NH4NO3/methanol; Flow: 1.0 mL/min; Detection: 275 nm; Retention time: Rt = 13.507, ee = 93.1 (Rt of 1 standard = 13.763, ee = 93.4%).
MS: m/z (rel. intensity): 318 (100, M+H).
1H NMR: (300 MHz, CD3OD) δ1.22 (d, 1H, J = 6.6 Hz), 2.70 (dd, 1H, J = 9.9, 13.5 Hz), 3.07–3.12 (m, 1H), 3,13–3.18 (m, 2H), 3.44–3.56 (m, 2H), 3.77 (s, 3H), 4.74–4.85 (m, 1H), 6.22 (t, 1H, J = 2.1 Hz), 6.67 (d, 2H, J = 2.4 Hz), 6.90 (d, 2H, J = 8.7 Hz), 7.16 (d, 2H, J = 8.4 Hz) ppm.
2′,4′,6′-[3H3]-(R,R)-4-Methoxyfenoterol (3)
A solution of 2 (2.0 mg, 3.6 μmol) in absolute ethanol (0.200 mL) was treated with 10% palladium on carbon (1 mg). Carrier free tritium gas (10 Ci) was introduced at 1 atm stirring at RT for 18 h. The mixture was filtered and then back-exchanged with methanol several times giving 50 mCi of crude product. This was purified by gradient HPLC to obtain 15 mCi of 99.7% radiopure 3 (7.3%).
RP-HPLC (Purification and analysis): Column: Supelco Discovery C18 (5 micron), 250 × 4.6 mm; Solvent A: ACN, Solvent B: 0.001M HCl (pH 3); Program: 10–100% B over 15 minutes; Flow: 1.0 mL/min; Detection: 275 nm. Retention time: Rt = 8.15 min for the both the tritium labeled product and the standard. MS: m/z (rel. intensity): 322 (100, M+H, T2), 324 (73, M+H, T3). 3H NMR: (300 MHz, CD3OD) δ6.28 (s, 1T), 6.43 (s, 2T) ppm.
Acknowledgments
This work was supported by the National Institutes on Aging (NIA) under contract number N01AG-3-1009 and by funds from the Intramural Research Program of the National Institutes on Aging.
References
- 1.Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, Woo A, Zhu W, Xiao RP, Abernethy DR, Wainer IW. J Med Chem. 2007:12. doi: 10.1021/jm070030d. [DOI] [PubMed] [Google Scholar]
- 2.Woo AYH, Wang TB, Zeng X, Zhu W, Abernethy DR, Wainer IW, Xiao RP. Mol Pharmacol. 2009:75. doi: 10.1124/mol.108.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitchell RH, Lai YH, Williams RV. J Org Chem. 1979:44. [Google Scholar]