

Abstract
The class B, type I scavenger receptor, SR-BI, binds high density lipoprotein (HDL) and mediates the selective uptake of HDL cholesteryl ester (CE) by cultured transfected cells. The high levels of SR-BI expression in steroidogenic cells in vivo and its regulation by tropic hormones provides support for the hypothesis that SR-BI is a physiologically relevant HDL receptor that supplies substrate cholesterol for steroid hormone synthesis. This hypothesis was tested by determining the ability of antibody directed against murine (m) SR-BI to inhibit the selective uptake of HDL CE in Y1-BS1 adrenocortical cells. Anti-mSR-BI IgG inhibited HDL CE-selective uptake by 70% and cell association of HDL particles by 50% in a dose-dependent manner. The secretion of [3H]steroids derived from HDL containing [3H]CE was inhibited by 78% by anti-mSR-BI IgG. These results establish mSR-BI as the major route for the selective uptake of HDL CE and the delivery of HDL cholesterol to the steroidogenic pathway in cultured mouse adrenal cells.
The cellular uptake of lipoprotein lipids is an essential process in the regulation of plasma cholesterol levels and the delivery of cholesterol to a variety of cell types. In humans, the primary pathway for cellular cholesterol uptake involves the low density lipoprotein (LDL) receptor (1, 2) and other members of the LDL receptor family (3). These receptors function via endocytic uptake and lysosomal degradation of lipoprotein particles to release cholesterol and other lipids to the cell (1, 2). An alternate process, occurring primarily with high density lipoprotein (HDL), is the selective uptake pathway in which HDL cholesteryl ester (CE) is taken into the cell without the uptake and lysosomal degradation of the HDL particle (4–9). The selective uptake pathway is active in a variety of human and other mammalian cell types (4, 10–15), but is particularly active in steroidogenic tissues of rats and mice (5, 8, 9). Adrenal and ovarian cells derive the majority of their precursor cholesterol for steroid synthesis and for CE storage from this route of HDL processing (7, 16–21). Although the biochemical mechanism of selective uptake is unclear, the recent discovery that both the murine and human scavenger receptor class B, type I (mSR-BI and hSR-BI) can mediate the selective uptake of HDL CE in transfected cells suggests that SR-BI may be responsible for this activity in steroidogenic cells (22–24). SR-BI is expressed in vivo in those tissues and cell types that exhibit high rates of HDL CE-selective uptake, namely the liver and steroidogenic cells (22, 24–27). In addition, in vivo studies in mice and rats show that SR-BI regulation by tropic hormones in the ovary, adrenal gland, and testicular Leydig cells is consistent with this receptor playing a key role in the delivery of HDL CE (26, 27). In this study, the function of SR-BI in steroidogenic cells was tested directly with antibody raised against a portion of the extracellular domain of the protein. The results establish that SR-BI serves as the major route for the selective uptake of HDL CE and for the delivery of HDL cholesterol to the steroidogenic pathway in cultured adrenal cells.
MATERIALS AND METHODS
Preparation of Antibodies to mSR-BI.
Rabbit polyclonal antibodies were raised to a glutathione-S transferase (GST) fusion protein containing mSR-BI amino acid residues 174–356. This corresponds to approximately 45% of the putative extracellular domain (amino acid residues 33–439) of the receptor. For this purpose, oligonucleotides (sense XmaI primer, 5′-GATGGCCCGGGCCGCACAGTTGGTGAGATCC-3′ and antisense XhoI primer, 5-′GGATAGCCCTCGAGTTCTGACAACACAGGGTCGGC-3′) were used to PCR amplify bases 520–1,068 from the ORF of mSR-BI under the following conditions: 2.5 mM MgCl2, 0.01% gelatin, 62.5 μM dNTPs, 0.5 μM sense XmaI primer, 0.5 μM antisense XhoI primer, 20 ng pcDNA3-mSR-BI, 1× PCR reaction buffer, and 1 unit Taq DNA polymerase (Boehringer Mannheim). PCR reactions were carried out with a 1 cycle denaturation program (95°C for 5 min), a 35 cycle amplification program (95°C for 45 sec, 58°C for 45 sec, and 72°C for 60 sec), and a 1 cycle extension program (72°C for 7 min). The PCR product and pGEX-4T-1 (Pharmacia) were cut with XhoI and XmaI (New England Biolabs), gel purified, and ligated overnight. Ligation products were transformed into Max efficiency DH5α competent cells (GIBCO/BRL) and selected on Luria broth plates containing 100 μg/ml ampicillin. The desired plasmid, pGEX-4T-1-mSR-BI EC, was identified by restriction enzyme mapping, and the entire mSR-BI coding region and cloning junctions were sequenced.
For purification of the fusion protein, pGEX-4T-1-mSR-BI EC was transformed into TG-1 cells, and GST-mSR-BI EC fusion protein was isolated by a modified version of the protocol of Smith and Johnson (28, 29). Following induction with isopropylthiogalactoside, cells were lysed by sonication in 10 mM Tris⋅HCl (pH 7.4), 100 mM NaCl, 1 mM MgCl2, 5 mM DTT, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 0.2 mM phenylmethylsulfonyl fluoride. The lysate was centrifuged for 10 min at 10,000 × g, and the pellet containing the fusion protein was washed twice by resuspension in 0.2 M Tris⋅HCl (pH 8), 0.5 M NaCl, 5 mM DTT (TN buffer), followed by centrifugation as above. The pellet was extracted with 8 M urea/5 mM DTT for 1–3 hr at 4°C, dialyzed against TN buffer, cleared by centrifugation, and incubated with glutathione agarose (Sigma) for 1–2 hr at 4°C. The glutathione agarose was washed with TN buffer, and the fusion protein was eluted in TN buffer containing 20 mM glutathione. Two male New Zealand White rabbits (Rb355 and Rb356) were immunized with 300 μg of fusion protein in Freund’s complete adjuvant and boosted with 150 μg of fusion protein in incomplete Freund’s adjuvant at weeks 2, 3, and 7. Thereafter, rabbits were boosted three times at monthly intervals with an SDS/10% polyacrylamide gel slice containing 250 μg of the SR-BI fragment that had been cleaved from the fusion protein by thrombin digestion. Ten days after the last boost, rabbits were exsanguinated, and IgG was prepared by chromatography on protein A-agarose (Bio-Rad) (30). Control or nonimmune IgG was prepared from two rabbits that had not been immunized. Prior to incubation with cells, IgG was dialyzed against 25 mM ammonium bicarbonate (pH 7.4), lyophilized, reconstituted in F10 serum-free medium, and cleared by centrifugation. Protein concentration was determined according to Lowry et al. (31).
Characterization of Rb355 and Rb356 mSR-BI EC IgG by Western Blotting.
Postnuclear supernatant was isolated from ldlA[mSR-BI] and Y1-BS1 cells as described (22, 27), except that lysis buffer also contained 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 0.2 mM phenylmethylsulfonyl fluoride. Proteins (20 μg) were resolved on an SDS/8% PAGE gel, transferred to a nitrocellulose membrane, and probed with IgG as described (27). Antibody binding was visualized by chemiluminescence detection (Amersham) using Reflection autoradiography film (NEN/Dupont).
Preparation of [125I]Dilactitol Tyramine-[3H]Cholesteryl Oleolyl Ether hHDL3 ([125I,3H]hHDL3), [3H]Cholesteryl Oleate hHDL3 ([3H] hHDL3), and [3H]Cholesteryl Oleate Recombinant (r) HDL ([3H] rHDL).
Human (h) HDL3 (1.125 g/ml < ρ < 1.210 g/ml) labeled with [125I]dilactitol tyramine and [3H]cholesteryl oleolyl ether was prepared as described (18). The specific activity of the [125I,3H]hHDL3 ranged from 46–70 dpm/ng protein for 125I and from 6–28 dpm/ng protein for 3H. The specific activity of [3H]hHDL3, prepared as described (17, 18), ranged from 3–5 dpm/ng protein. The specific activity of [3H]rHDL, prepared as described (21) was 60 dpm/ng protein.
Determination of HDL Cell Association, Selective CE Uptake, and Apolipoprotein Degradation.
Y1-BS1 murine adrenocortical cells (32) were maintained and experiments were performed in a 37°C humidified 95% air/5% CO2 incubator as described (27). For all experiments, 6-well plates (Costar), which had been treated with 100 μg/ml poly d-lysine (Becton Dickinson), were seeded with Y1-BS1 cells at a density of 1.5 × 106 cells per well. After 48 hr, medium was removed and replaced with 1.5 ml Ham’s F-10 complete medium plus or minus 100 nM Cortrosyn (Organon), a synthetic 1–24adrenocorticotropic hormone (ACTH) analogue. After 24 hr, medium was replaced with serum-free Ham’s F-10 medium lacking ACTH and containing no additions or 6 mg/ml IgG consisting of varying proportions of nonimmune IgG and anti-mSR-BI IgG. After a 1-hr preincubation, [125I,3H]hHDL3 was added at 10 μg protein/ml (except where indicated), and the incubation was continued for 2 or 4 hr. Cells were washed three times with 0.1% BSA in PBS (pH 7.4), one time with PBS (pH 7.4), lysed with 1.25 ml 0.1 N NaOH, and passed five times through a 28.5-gauge needle. The lysate was then processed to determine trichloroacetic acid soluble and insoluble 125I radioactivity and organic solvent-extractable 3H radioactivity as described (17, 18). Trichloroacetic acid insoluble 125I radioactivity represents cell-associated HDL apolipoprotein, which is the sum of cell surface bound apolipoprotein and endocytosed apolipoprotein that is not yet degraded. Trichloroacetic acid soluble 125I radioactivity represents endocytosed and degraded apolipoprotein that is trapped in lysosomes due to the dilactitol tyramine label (18, 33). The sum of the 125I-degraded and 125I cell-associated undegraded apolipoprotein expressed as CE equivalents was subtracted from the CE measured as extractable 3H radioactivity to give the selective uptake of HDL CE (17, 18). Values for these parameters are expressed as nanograms of HDL cholesterol/mg cell protein. The HDL concentration dependence for each of these parameters was modeled by a simple binding isotherm composed of a high-affinity saturable process and a low-affinity nonsaturable process:
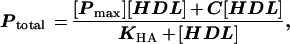 |
where Ptotal is the measured parameter, [Pmax] is the high-affinity parameter at saturating levels of HDL, KHA is the apparent high-affinity Km, and C is the slope of the low-affinity nonsaturable process. For each parameter, Ptotal was resolved into high- and low-affinity components by determining C and subtracting C [HDL] from Ptotal to generate the curve for the high-affinity HDL concentration dependence.
Determination of [3H] Steroid Production.
Y1-BS1 cells were preincubated as above with or without 6 mg/ml IgG for 1 hr prior to addition of [3H]hHDL3 at 25 μg protein/ml or [3H]rHDL at 5 μg protein/ml. The incubation was continued for 24 hr in the presence of the indicated IgG and 100 nM 1–24ACTH. Medium was removed, a [14C]progesterone recovery standard (New England Nuclear) was added, and the sample was extracted with CH2Cl2 as described (34). Steroids were separated on a Brownlee reverse-phase C18 column (OD-300, Aquapore ODS, 25 cm × 4.6 mm) in a mobile phase of methanol:acetonitrile:water (11:45:44), and the peaks corresponding to 11β,20α-dihydroxy-4-pregnene-3-one, 11β-hydroxyprogesterone, and progesterone were collected and counted by liquid scintillation spectrometry. Measured values for [3H] steroids were corrected for recovery losses and normalized for cell protein. Values for samples incubated with IgG were expressed as a percentage of the control samples with no IgG. Control values for [3H] steroid secretion with several preparations of [3H]hHDL3 ranged from 19,000–34,000 dpm/mg cell protein. Control values for samples incubated with [3H]rHDL were 173,000 dpm/mg cell protein.
Determination of DiI-HDL Uptake into ldlA[mSR-BI] Cells.
ldlA[mSR-BI] cells or ldlA cells (22) were plated at a density of 1.5 × 104 per well in 24-well plates in 1 ml Ham’s F-12 complete media (5% heat-inactivated fetal bovine serum/2 mM l-glutamine/50 units/ml penicillin/50 μg/ml streptomyocin, either with or without 0.5 mg/ml G418, respectively). After an overnight incubation at 37°C in a humidified 95% air/5% CO2 incubator, the medium was replaced with 1 ml of DMEM/F-12 serum-free medium (2 mM l-glutamine/50 units/ml penicillin/50 μg/ml streptomyocin). After 24 hr, the medium was removed, and the cells were washed with 0.5 ml DMEM/F-12 serum-free medium. Each well-received 0.2 ml of DMEM/F-12 serum-free media supplemented with or without 6 mg/ml IgG. After a 2-hr incubation at 37°C, DiI-labeled HDL (22) was added to 10 μg protein/ml with or without unlabeled HDL at 400 μg protein/ml, and the incubation was continued for 2 hr at 37°C. After washing two times with PBS, cells were removed by trypsin treatment for 3 min followed by quenching with new-born-calf lipoprotein-deficient serum. Fluorescence intensities were measured on a Becton Dickinson FACStar Plus flow cytometer. DiI was excited with 100 mW of 514 nm light from a Coherent Innova 90–5 argon ion laser. Emitted light was collected using a 575 DF/26 filter. Note that for this experiment (using ldlA[mSR-BI] cells) the ratios of the amounts of antibody per cell were much higher than for those for experiments carried out with Y1-BS1 cells (described above). These differences appear to account for differences in apparent KI values measured for antibody inhibition for the two cell types (see below).
RESULTS AND DISCUSSION
Characterization of Anti-mSR-BI Antibody.
Previous studies have shown that SR-BI is regulated by ACTH in the murine adrenal gland in vivo (27) and in murine Y1 (35) and Y1-BS1 (27) adrenocortical cell lines, suggesting that SR-BI may be responsible, at least in part, for HDL CE-selective uptake and the provision of HDL cholesterol to the steroidogenic pathway. To test this hypothesis, a polyclonal antibody was raised to a proposed extracellular domain of mSR-BI (36) with the aim of interfering with HDL binding or otherwise disrupting SR-BI function. The Western blots in Fig. 1 show that 355 and 356 anti-mSR-BI IgG recognized an ≈82-kDa band in the postnuclear supernatant of ldlA[mSR-BI] cells, a stably transfected Chinese hamster ovary cell line expressing mSR-BI (Fig. 1 C and D, lanes 1) (22). The mobility of this band was identical to that of mSR-BI detected by a previously characterized anti-peptide antibody directed against the C terminus of the protein (22) (data not shown). In addition to the 82-kDa mSR-BI band, both 355 and 356 anti-mSR-BI IgG detected low levels of background bands, which were present at comparable levels in ldlA[mSR-BI] and Y1-BS1 cells (C and D) and in untransfected ldlA cells (data not shown). The background bands were not related to mSR-BI expression in transfected cells and were not altered by ACTH treatment in Y1-BS1 cells, indicating no functional relationship to the selective uptake process. Neither preimmune IgG (data not shown) nor nonimmune IgG from two other rabbits (Fig. 1 A and B, lanes 1) detected the SR-BI band. In addition, 356 anti-mSR-BI did not detect the SR-BI band in nontransfected ldlA cells (data not shown). Both 355 and 356 anti-mSR-BI IgG recognized SR-BI in Y1-BS1 adrenocortical cells (C and D, lanes 2) and readily detected the previously described (27) induction of SR-BI expression when these cells were treated with ACTH (C and D, lanes 2 vs. 3). Neither preimmue IgG (data not shown) nor nonimmune IgG recognized SR-BI in Y1-BS1 cells (A and B, lanes 2 and 3). From these results, it was concluded that 355 and 356 anti-mSR-BI IgG specifically recognized mSR-BI with 356 anti-SR-BI having a significantly higher titer. To evaluate possible immunoreactivity with the related class B scavenger receptor CD36 (25), 356 anti-mSR-BI was tested by Western blot analysis with extracts from cells overexpressing rat CD36 and with the entire extracellular domain of CD36 expressed by means of a baculovirus vector. No immunoreactivity with CD36 was detected (data not shown).
Figure 1.
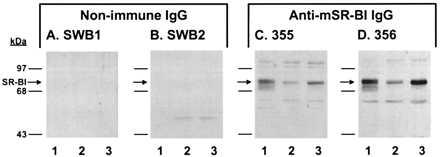
Western blot analysis of anti-mSR-BI antibodies. Postnuclear supernatant (20 μg protein) from ldlA[mSR-BI] cells (lane 1), and Y1-BS1 cells treated without (lane 2) or with (lane 3) 1–24ACTH were separated by SDS/8% PAGE and transferred to nitrocellulose membranes. The membranes were incubated overnight at 4°C in the presence of either SWB1 nonimmune IgG (A), SWB2 nonimmune IgG (B), 355 anti-mSR-BI IgG (C), or 356 anti-mSR-BI IgG (D) at 4 μg/ml. IgG binding was visualized by enhanced chemiluminescence.
Anti-mSR-BI IgG Inhibits DiI-HDL Uptake by ldlA[mSR-BI] Cells.
We have previously shown that mSR-BI mediates selective uptake of the fluorescent lipid, DiI, from DiI-HDL (22). To test the ability of the 356 antibody to disrupt SR-BI function, ldlA[mSR-BI] cells, which had been preincubated with increasing concentrations of 356 anti-SR-BI IgG, were exposed to DiI-HDL (10 μg protein/ml), and the accumulation of DiI was measured by flow cytometry. The total IgG concentration in the incubation medium was held constant at 6 mg/ml and the proportion of 356 anti-SR-BI IgG and nonimmune IgG was varied. As shown in Fig. 2A, 356 anti-mSR-BI IgG inhibited the uptake of DiI-HDL in a dose-dependent manner, reaching 85% inhibition at the highest concentration tested. Because a similar inhibition was produced with excess unlabeled HDL (400 μg protein/ml) (Fig. 2B), the 356 antibody appears to have blocked most of the high-affinity interactions between HDL and the ldlA[mSR-BI] cells. In addition, comparison of DiI-HDL uptake by ldlA[mSR-BI] cells incubated with or without 6 mg/ml nonimmune IgG (Fig. 2B) showed that IgG alone had no effect on DiI uptake. These results indicate that 356 anti-mSR-BI IgG effectively disrupts mSR-BI-mediated selective lipid uptake.
Figure 2.
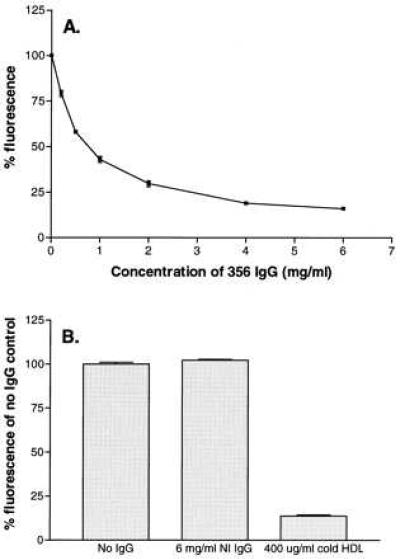
Effects of 356 anti-mSR-BI IgG on DiI uptake from DiI HDL by ldlA[mSR-BI] cells. (A) ldlA[mSR-BI] cells were incubated for 2 hr with DiI-HDL (10 μg protein/ml) in medium containing the indicated concentration of 356 anti-mSR-BI IgG and complementary amounts of nonimmune IgG to give a final IgG concentration of 6 mg/ml. Cells were then washed and processed for fluorescence determination by flow cytometry as described. Samples containing 6 mg/ml nonimmune IgG were taken as the 100% control value (arbitrary scale). (B) The uptake of DiI-HDL in the presence of no IgG (100% value) is shown in comparison with cells incubated with 6 mg/ml nonimmune IgG or with excess unlabeled HDL. Error bars represent the range of duplicate determinations.
Anti-mSR-BI IgG Inhibits HDL-Selective CE Uptake and Cell Association of HDL in Y1-BS1 Cells.
To examine the effects of the anti-mSR-BI antibody on the selective uptake of HDL CE, ACTH-treated Y1-BS1 cells were incubated with [125I,3H]hHDL3, which contains [3H]cholesteryl oleolyl ether as a tracer for CE and [125I]dilactitol tyramine-labeled apoproteins as a tracer for cell-associated apolipoprotein, as well as apolipoprotein degraded in lysosomes (18, 33). Fig. 3 shows the dependence on HDL concentration for selective CE uptake (A) and for cell association of HDL (B), with the data for each parameter expressed on the basis of HDL cholesterol. In each case, the experimentally measured values (circles) show an HDL concentration dependence indicative of both high- and low-affinity components. The high-affinity component was resolved as described, and is illustrated in Fig. 3 (triangles). Based on the assumption that SR-BI would reflect the high-affinity component, antibody inhibition experiments were carried out at 10 μg protein/ml HDL, a concentration at which 90% of the selective uptake is due to the high-affinity component. Note that as previously reported (4, 37), HDL cholesterol taken up through the selective uptake pathway exceeded by a factor of 40 the HDL cholesterol accounted for by cell association of HDL apolipoprotein (compare A and B). The amount of HDL cholesterol accounted for by degraded apolipoprotein was even less (≈1% of the selective CE uptake) (data not shown), illustrating that there was very little HDL apolipoprotein degradation.
Figure 3.
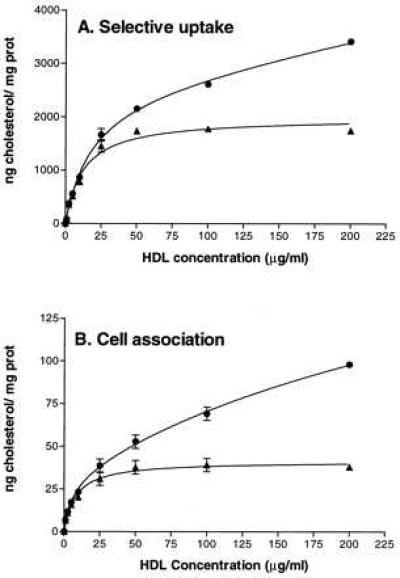
Selective CE uptake and cell association of [125I,3H]hHDL3 by Y1-BS1 cells. Y1-BS1 cells were incubated with the indicated concentrations of [125I,3H]hHDL3 for 4 hr, after which the cells were processed to determine selective CE uptake (A) and cell association of HDL apolipoprotein (B). The high-affinity (▴) component for each of these parameters was resolved from the total measured value (•) as described. Error bars represent the range of duplicate determinations.
Fig. 4A shows that 356 anti-mSR-BI IgG caused a dose-dependent decrease in HDL-selective CE uptake, which reached 70% inhibition of the total uptake (high plus low affinity) at the highest IgG concentration tested. In similar experiments with the lower titer anti-mSR-BI antibody, the maximum dose-dependent inhibition at 6 mg/ml 355 anti-mSR-BI IgG was 31% (data not shown). As shown in Fig. 4C, the addition of 6 mg/ml nonimmune IgG alone had no effect on HDL-selective CE uptake (open bars). In addition, when the Y1-BS1 cells were exposed to both [125I,3H]hHDL3 (10 μg protein/ml) and a 50-fold excess of unlabeled HDL3, total selective uptake was reduced to 7% of control (Fig. 4C). This result indicates that approximately 90% of the selective uptake at 10 μg/ml HDL corresponded to the high-affinity component as predicted by the analysis in Fig. 3. Thus, 356 anti-mSR-BI at a concentration of 6 mg/ml inhibited 75% of the high-affinity selective CE uptake. These data indicate that SR-BI is responsible for most of the high-affinity HDL-selective CE uptake in cultured adrenocortical cells.
Figure 4.
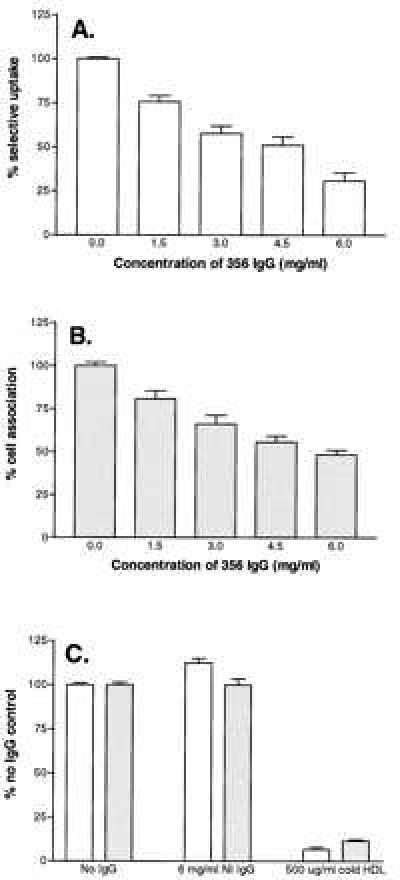
Effects of 356 anti-mSR-BI IgG on HDL-selective CE uptake and HDL cell association. Y1-BS1 cells were incubated for 2 hr with [125I,3H]hHDL3 (10 μg protein/ml) in medium containing the indicated concentration of 356 anti-mSR-BI IgG and complementary amounts of nonimmune IgG to give a final IgG concentration of 6 mg/ml. Cells were processed to determine HDL-selective CE uptake (A) and cell association of HDL apolipoprotein (B). The 100% of control value in each case refers to samples incubated with 6 mg/ml nonimmune IgG. (A and B) Results for 0.0 mg/ml and 6.0 mg/ml anti-mSR-BI IgG are the means of 22 samples (±SEM) from seven experiments. The results for the intermediate anti-mSR-BI IgG concentrations are the means of eight samples (±SEM) from four experiments. (C) HDL-selective CE uptake (open bars) and cell-associated HDL apolipoprotein (stippled bars) in the presence of no IgG (100% of control value) in comparison with cells incubated with 6 mg/ml nonimmune IgG or with excess unlabeled HDL (500 μg protein/ml). For the no IgG samples, the results are the means of 20 samples (±SEM) from seven experiments. The results for the 6 mg/ml NI (nonimmune) IgG are the means of 22 samples (±SEM) from seven experiments, and the 500 μg/ml cold HDL results are the means of 10 samples (±SEM) from 4 experiments.
Fig. 4B shows that 356 anti-mSR-BI IgG caused a dose-dependent decrease in cell association of HDL, which reached 50% inhibition at the highest IgG concentration tested. Nonimmune IgG alone had no effect on cell association of HDL, and excess unlabeled HDL reduced cell association of HDL by 85% (Fig. 4C). Thus, approximately 57% of the high-affinity cell association of HDL was blocked by 356 anti-SR-BI IgG. Because most of the cell association of HDL is believed to reflect cell surface bound lipoprotein particles, this result suggests that 356 anti-mSR-BI inhibits HDL-selective CE uptake primarily by interfering with HDL binding to SR-BI. Interestingly, at all antibody or HDL concentrations examined (data not shown), the inhibition of binding was consistently less than the inhibition of selective uptake. This result may indicate either that there are multiple sites on SR-BI for HDL binding or that HDL may bind with high affinity to cell surface sites other than SR-BI.
Anti-mSR-BI IgG Inhibits the Delivery of HDL CE to the Steroidogenic Pathway.
Having established that the anti-mSR-BI IgG blocks HDL binding to SR-BI and SR-BI-mediated selective lipid uptake, we used this blocking antibody to determine whether SR-BI is directly involved in providing substrate cholesterol to the steroidogenic pathway. In the presence and absence of the antibody, Y1-BS1 cells were exposed to [3H]hHDL3 particles containing [3H]cholesteryl oleate, and the types and amounts of the secreted radiolabeled steroids were determined using HPLC. The HPLC absorbance profile in Fig. 5A shows that, as previously reported (34, 38), there is one major steroid product of Y1 cells, 11β,20α-dihydroxy-4-pregnene-3-one (elution at 5 ml), as well as minor amounts of others, including 11β-hydroxyprogesterone (elution at 6 ml). The profile of radiolabeled steroids produced by cells incubated with [3H]hHDL3 (Fig. 5B) was coincident with the absorbance profile. Both the mass of secreted steroids (A) and the radioactive steroids (B) were eliminated when the cells were incubated with aminoglutethimide, an inhibitor of steroid production that inhibits the p450 side-chain cleavage enzyme (39) (Fig. 5). Using this assay, radiolabeled steroids secreted by Y1-BS1 cells in response to 25 μg protein/ml [3H]hHDL3 were quantified in cells incubated with no antibody or with 6 mg/ml of either nonimmune IgG or 356 anti-SR-BI IgG. As shown in Table 1, anti-mSR-BI IgG inhibited [3H]steroid production by 67% versus control or nonimmune IgG (P < 0.0001), whereas nonimmune IgG had no significant effect versus control (P > 0.2). To test antibody inhibition of steroid secretion at a lower HDL concentration, high specific activity recombinant [3H]rHDL was used. Table 1 shows that 356 anti-mSR-BI IgG inhibited [3H] steroid production by 78% versus control or nonimmune IgG (P < 0.0001) with [3H]rHDL at 5 μg protein/ml, whereas nonimmune IgG had no effect versus control (P > 0.15). These data indicate that SR-BI is responsible for the delivery of most of the HDL CE to the steroidogenic pathway in Y1-BS1 adrenocortical cells.
Figure 5.
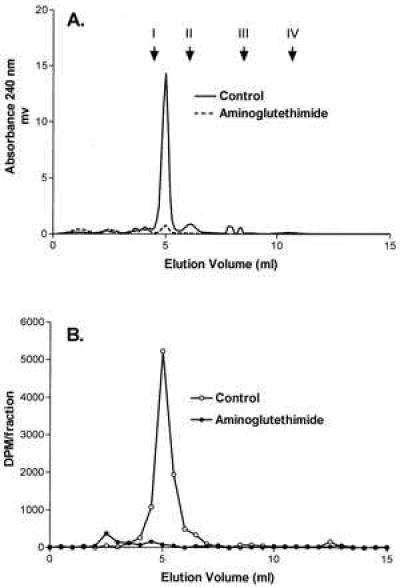
Secretion of [3H] steroid by 1–24ACTH-stimulated Y1-BS1 cells incubated with [3H]hHDL3. Y1-BS1 cells were incubated for 24 hr with 25 μg protein/ml [3H]hHDL3 in the presence or absence of 1 mM aminogluthethimide. Steroids were extracted from the medium with CH2Cl2 and separated by HPLC as described. (A and B) Absorbance profile at 240 nm and the radioactivity profile, respectively. Arrows in A indicate the elution position of standards: corticosterone (I), 11β-hydroxyprogesterone (II), 20α-hydroxyprogesterone (III), and progesterone (IV).
Table 1.
Anti-SR-B1 IgG inhibits the production of [3H]steroid derived from [3H]HDL
| HDL | [3H]steroid, % control ± SD
|
||
|---|---|---|---|
| Control | Nonimmune IgG | 356 anti-SR-BI IgG | |
| [3H]hHDL3 | 100 ± 11.3 | 90.2 ± 11 | 33.2 ± 7.1* |
| 25 μg/ml | (n = 7) | (n = 4) | (n = 4) |
| [3H]rHDL | 100 ± 3.8 | 92.9 ± 6.6 | 21.7 ± 0.8* |
| 5 μg/ml | (n = 3) | (n = 3) | (n = 3) |
Differs from the control or nonimmune IgG, P < 0.0001.
The selective uptake of HDL CE occurs in a variety of human and other mammalian cell types and appears to be an important pathway for the movement of plasma HDL CE into the liver, as well as steroidogenic cells. In rodents that lack plasma CE transfer protein, the selective uptake pathway is the predominant means by which plasma HDL CE is delivered to either the liver or steroidogenic cells (5, 7, 8). In mice lacking apoA-I, CE accumulation in steroidogenic cells is dramatically reduced, and ACTH-stimulated corticosteroid production is blunted, illustrating the dependence of adrenocortical cells on the HDL-selective CE uptake pathway (16). HDL tracer studies and kinetic modeling analysis suggest that the selective uptake of HDL CE also occurs in animals expressing high levels of CE transfer protein, although in that situation much of the HDL CE is thought to be transferred to LDL prior to hepatic clearance (10).
Despite the widespread nature of the HDL CE-selective uptake pathway, little is known about the biochemical mechanism by which CE is transferred into the cell without the uptake and degradation of the HDL particle. Recent studies showing that mSR-BI mediates the selective uptake of HDL CE in transfected Chinese hamster ovary cells provide an important link between a defined-cell surface receptor and the selective uptake pathway (22). Overexpression of mSR-BI in mouse liver in vivo enhances the clearance of HDL cholesterol from plasma (40), whereas reduced expression of mSR-BI due to targeted mutagenesis in mice (≈50% or 0% hepatic expression in heterozygous or homozygous mutants, respectively) results in increased HDL cholesterol in plasma (41), suggesting that SR-BI is important for hepatic HDL metabolism (40). The results of this study provide strong evidence that mSR-BI is an HDL receptor that provides substrate cholesterol for steroid hormone synthesis in adrenocortical cells. This conclusion is supported by the observation that SR-BI-deficient knockout mice have substantially reduced adrenal cholesterol stores (41) compared with normal controls. The high level of expression of SR-BI in the ovary and testis in mice and rats suggests that SR-BI may have this role in these tissues as well, although this remains to be tested. Interestingly, hSR-BI (CLA-1) (23) also has been shown to mediate HDL-selective CE uptake in transfected cells and to be expressed at high levels in the adrenal gland of humans (24). This result raises the possibility that hSR-BI-mediated uptake of HDL CE may be a significant source of precursor cholesterol for steroid production in humans as it is in mice. However, additional studies will be required to evaluate the roles of SR-BI and the HDL CE-selective uptake pathway in steroidogenic cells of humans and other species that transport most of their plasma cholesterol in LDL particles.
Acknowledgments
We are grateful to Glenn Paradis and Mike Jennings of the Massachusetts Institute of Technology Flow Cytometry Core Facility for performing the flow cytometry. We thank Nada Abumrad for supplying CD36-expressing cells and baculovirus-derived CD36 and Margery Connelly for performing Western blots. This work was supported by Grants HL 32868, HL58012, HL 41484, and HL 52212 from the National Institutes of Health, National Heart, Lung, and Blood Institute, and by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs. B.T. is a Medical Research Council of Canada Postdoctoral Fellow.
ABBREVIATIONS
- SR-BI
scavenger receptor, class B, type I
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- CE cholesteryl ester
ACTH, adrenocorticotropic hormone
References
- 1.Brown M S, Goldstein J L. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein J L, Brown M S, Anderson R G, Russell D W, Schneider W J. Annu Rev Cell Biol. 1985;1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 3.Krieger M, Herz J. Annu Rev Biochem. 1994;63:601–637. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 4.Pittman R C, Knecht T P, Rosenbaum M S, Taylor C A., Jr J Biol Chem. 1987;262:2443–2450. [PubMed] [Google Scholar]
- 5.Glass C, Pittman R C, Civen M, Steinberg D. J Biol Chem. 1985;260:744–750. [PubMed] [Google Scholar]
- 6.Reaven E, Chen Y-D I, Spicher M, Azhar S. J Clin Invest. 1984;74:1384–1397. doi: 10.1172/JCI111549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gwynne J T, Hess B. J Biol Chem. 1980;255:10875–10883. [PubMed] [Google Scholar]
- 8.Glass C, Pittman R C, Weinstein D B, Steinberg D. Proc Natl Acad Sci USA. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein Y, Dabach Y, Hollander G, Halperin G, Stein O. Biochim Biophys Acta. 1983;752:98–105. doi: 10.1016/0005-2760(83)90237-0. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg D I, Beltz W F, Pittman R C. J Clin Invest. 1991;87:331–346. doi: 10.1172/JCI114991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pieters M N, Schouten D, Bakkeren H F, Esbach B, Brouwer A, Knook D L, Van Berkel T J C. Biochem J. 1991;280:359–365. doi: 10.1042/bj2800359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rinninger F, Greten H. Biochim Biophys Acta. 1990;1043:318–326. doi: 10.1016/0005-2760(90)90033-t. [DOI] [PubMed] [Google Scholar]
- 13.Rinninger F, Pittman R C. J Lipid Res. 1988;29:1179–1194. [PubMed] [Google Scholar]
- 14.Pittman R C, Glass C K, Atkinson D, Small D M. J Biol Chem. 1987;262:2435–2442. [PubMed] [Google Scholar]
- 15.Rinninger F, Pittman R C. J Lipid Res. 1987;28:1313–1325. [PubMed] [Google Scholar]
- 16.Plump A S, Erickson S K, Weng W, Partin J S, Breslow J L, Williams D L. J Clin Invest. 1996;97:2660–2671. doi: 10.1172/JCI118716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azhar S, Tsai L, Reaven E. Biochim Biophys Acta. 1990;1047:148–160. doi: 10.1016/0005-2760(90)90041-u. [DOI] [PubMed] [Google Scholar]
- 18.Azhar S, Stewart D, Reaven E. J Lipid Res. 1989;30:1799–1810. [PubMed] [Google Scholar]
- 19.Azhar S, Cooper A, Tsai L, Maffe W, Reaven E. J Lipid Res. 1988;29:869–882. [PubMed] [Google Scholar]
- 20.Nestler J E, Bamberger M, Rothblat G H, Strauss J F I. Endocrinology. 1985;117:502–510. doi: 10.1210/endo-117-2-502. [DOI] [PubMed] [Google Scholar]
- 21.Reaven E, Tsai L, Azhar S. J Lipid Res. 1995;36:1602–1617. [PubMed] [Google Scholar]
- 22.Acton S, Rigotti A, Landschulz K T, Xu S, Hobbs H H, Krieger M. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 23.Calvo D, Vega M A. J Biol Chem. 1993;268:18929–18935. [PubMed] [Google Scholar]
- 24.Murao K, Terpstra V, Green S R, Kondratenko N, Steinberg D, Quehenberger O. J Biol Chem. 1997;272:17551–17557. doi: 10.1074/jbc.272.28.17551. [DOI] [PubMed] [Google Scholar]
- 25.Acton S, Scherer P E, Lodish H F, Krieger M. J Biol Chem. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 26.Landschulz K T, Pathak R K, Rigotti A, Krieger M, Hobbs H H. J Clin Invest. 1996;98:984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rigotti A, Edelman E R, Seifert P, Iqbal S N, DeMattos R B, Temel R E, Krieger M, Williams D L. J Biol Chem. 1996;271:33545–33549. doi: 10.1074/jbc.271.52.33545. [DOI] [PubMed] [Google Scholar]
- 28.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 29.Koff A, Giordano A, Desai D, Yamashita K, Harper J W, Elledge S, Nishimoto T, Morgan D O, Franza B R, Roberts J M. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- 30.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 31.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 32.Schimmer B P. Functionally Differentiated Cell Lines. New York: Liss; 1981. pp. 61–92. [Google Scholar]
- 33.Glass C K, Pittman R C, Keller G A, Steinberg D. J Biol Chem. 1983;258:7161–7167. [PubMed] [Google Scholar]
- 34.Cheng B, Kowal J. J Chromatogr. 1988;432:302–307. doi: 10.1016/s0378-4347(00)80657-x. [DOI] [PubMed] [Google Scholar]
- 35.Wang N, Weng W, Breslow J L, Tall A R. J Biol Chem. 1996;271:21001–21004. doi: 10.1074/jbc.271.35.21001. [DOI] [PubMed] [Google Scholar]
- 36.Rigotti A, Trigatti B L, Babbit J, Penman M, Xu S, Krieger M. Curr Opin Lipidol. 1997;8:181–188. doi: 10.1097/00041433-199706000-00009. [DOI] [PubMed] [Google Scholar]
- 37.Knecht T P, Pittman R C. Biochim Biophys Acta. 1989;1002:365–375. doi: 10.1016/0005-2760(89)90351-2. [DOI] [PubMed] [Google Scholar]
- 38.Kowal J, Fieldler R. Arch Biochem Biophys. 1968;128:406–421. doi: 10.1016/0003-9861(68)90047-7. [DOI] [PubMed] [Google Scholar]
- 39.Kowal J. Endocrinology. 1969;85:270–279. doi: 10.1210/endo-85-2-270. [DOI] [PubMed] [Google Scholar]
- 40.Kozarsky K F, Donahee M H, Rigotti A, Iqbal S N, Edelman E R, Krieger M. Nature (London) 1997;387:414–417. doi: 10.1038/387414a0. [DOI] [PubMed] [Google Scholar]
- 41.Rigotti, A., Trigatti, B. L., Penman, M., Rayburn, H., Herz, J. & Krieger, M. (1997) Proc. Natl. Acad. Sci. USA 94, in press. [DOI] [PMC free article] [PubMed]