

Abstract
Maple syrup urine disease (MSUD; OMIM 248600) is an inborn error of metabolism of the branched chain α-ketoacid dehydrogenase (BCKDH) complex that is treated primarily by dietary manipulation of branched-chain amino acids (BCAA). Dietary restriction is lifelong and compliance is difficult. Liver transplantation significantly improves outcomes; however, alternative therapies are needed. To test novel therapies such as hepatocyte transplantation (HTx), we previously created a murine model of intermediate MSUD (iMSUD), which closely mimics human iMSUD. LacZ-positive murine donor hepatocytes were harvested and directly injected (105 cells/50 µl) into liver of iMSUD mice (two injections at 1–10 days of age). Donor hepatocytes engrafted into iMSUD recipient liver, increased liver BCKDH activity, improved blood total BCAA/alanine ratio, increased body weight at weaning, and extended the lifespan of HTx-treated iMSUD mice compared to phosphate-buffered saline (PBS)–treated and untreated iMSUD mice. Based on these data demonstrating partial metabolic correction of iMSUD in a murine model, coupled to the fact that multiple transplants are possible to enhance these results, we suggest that HTx represents a promising therapeutic intervention for MSUD that warrants further investigation.
Introduction
Maple syrup urine disease (MSUD; OMIM 248600) is an inborn error of metabolism with limited options for effective treatment. The disease occurs through dysfunction of the branched chain α-ketoacid dehydrogenase (BCKDH) mitochondrial enzyme complex resulting in an inability to correctly process branched-chain α-ketoacids.1,2,3,4,5 Branched-chain α-ketoacids (ketoisocaproate, ketomethylvalerate, and ketoisovalerate) are derived from the branched-chain amino acids (BCAAs) leucine, isoleucine, and valine, respectively.1,2,3 Toxic accretion of BCAAs and their related metabolites in the cerebrospinal fluid, brain, blood, and other tissues, results in numerous and serious complications (e.g., ketoacidosis, neurological dysfunction, seizures, and infant death) without immediate lifelong treatment.3,6 However, it is known that 100% activity of whole-body BCKDH is not needed to correct the disease phenotype; 30–40% or greater residual whole-body BCKDH activity is rarely associated with symptoms.3 Importantly, orthotopic liver transplantation in classic MSUD patients results in a full or partial metabolic correction of the disease phenotype.7,8,9,10 These clinical observations suggest that restoring enzyme activity to a small percentage of the total liver mass in a patient with MSUD might ameliorate the clinical and biochemical manifestations, possibly converting a severe case into a more manageable variant.
Recent clinical interventions have used whole liver transplantation to correct MSUD in children.7,8,9,10 However, hepatocyte transplantation (HTx) offers numerous benefits over orthotopic liver transplantation. HTx is a less invasive procedure with less associated morbidity, has fewer and less serious complications, and a reduced recovery time.11 HTx is estimated to cost ~10% of orthotopic liver transplantation11 and transplantation of up to 5% of patient liver cell number has been accomplished with few adverse effects.12 Importantly, repeated HTx treatments are possible and cryopreservation of donor hepatocytes could be employed so that cells are readily available upon demand. In addition, one donor liver could be used to transplant multiple HTx recipients, which may help to relieve shortages of donor organs.
Proof-of-concept studies for HTx have been accomplished in both humans and laboratory animals. For example, HTx has had initial successes in the treatment of inherited and metabolic liver disorders such as Crigler–Najjar syndrome,13,14,15,16 glycogen storage disease, type 1a and 1b,17,18 Wilson's disease,19 Factor VII,12 a urea cycle disorder,20,21 and hyperoxaluria type I.22 HTx has been successful at improving metabolic defects, such as albumin secretion23,24 and tyrosinemia.25 It has also been investigated and implemented as a “bridge” therapy to support liver function in times of liver injury or acute failure as the result of a variety of liver diseases.26,27,28,29 We hypothesized that HTx could be used successfully to increase liver BCKDH activity and alleviate the disease phenotype in a murine model of iMSUD, which expresses C-terminal myc tagged human BCKDH E2 on a mouse BCKDH E2 knockout background.30 This report summarizes our successful intervention with HTx in iMSUD mice.
Results
Donor hepatocytes engrafted recipient liver at a low level
LacZ-positive donor hepatocytes [1 × 105 cells in 50 µl phosphate-buffered saline (PBS)] expressing wild-type BCKDH were transplanted by transdermal injection directly into the liver parenchyma of 10 iMSUD neonatal mouse pups. Nine iMSUD mouse pups were instead injected with PBS to serve as a treatment control. A first injection was administered between 1 and 7 days of age and a second injection administered ~3 and 7 days later. All injections occurred within the first 10 postnatal days. Preliminary data revealed that the LacZ gene was silenced in most hepatocytes isolated from older donors (data not shown). Therefore, other methods were used to determine cell engraftment at the time of killing.
To verify donor cell engraftment following HTx into iMSUD livers, PCR was performed with donor cell–specific LacZ primers (~500-bp product) on recipient liver DNA collected on the date of killing. Control primers (374 bp product) served to verify the quality of the DNA; all DNA samples analyzed displayed a band from control primers (Figure 1a). LacZ-positive donor liver DNA samples (D) produced a band at ~500 bp, while LacZ-negative samples [control (C), untreated iMSUD (U), and iMSUD–PBS (P)] did not exhibit a LacZ-specific product (Figure 1a). All iMSUD–HTx DNA samples but one displayed the ~500-bp LacZ product (Figure 1a) derived from donor hepatocyte DNA.
Figure 1.

Verification of liver engraftment by PCR and qRT-PCR at killing. (a) PCR using liver DNA was performed with donor cell–specific LacZ primers (~500-bp product) to verify engraftment at the time of killing. Control primers (374 bp product) served to verify the quality of the DNA. Samples were designated as follows: hepatocyte donors (D), wild-type control (C), untreated iMSUD (U), iMSUD–PBS (P), iMSUD–HTx (HTx), and a blank, no DNA control (B). Band size was determined with a 100-bp Ladder (Invitrogen). (b) The E2 mouse gene sequence (top, bp 444–617), deleted to create the classic MSUD model,30 was aligned to the corresponding E2 human sequence (bottom, bp 415–588; 84% identity to mouse). Vertical lines connecting human and mouse base pairs indicate a match while the absence of a line indicates a mismatch. Primers chosen for SYBRgreen real-time qRT-PCR analysis are bolded and highlighted in grey; primers spanned mouse E2 exons 4/5. The bolded ATT codon designates the start of exon 5. (c) Mouse RNA samples were analyzed at the time of killing using qRT-PCR SYBRgreen E2-specific primers (b) and normalized to mouse β-actin. Quantified E2/β-actin RNA from iMSUD–HTx, untreated iMSUD (contained only transgenic human E2), and human hepatocyte samples were normalized to wild-type controls. N.D., RNA levels were not detectable confirming primer specificity to mouse E2. Data are shown as the mean ± SEM [*compared to iMSUD and human (P < 0.01), and control (P < 0.0001)]. The number displayed below each bar indicates the n value. HTx, hepatocyte transplantation; iMSUD, intermediate maple syrup urine disease; PBS, phosphate-buffered saline; qRT-PCR, real-time quantitative reverse transcription PCR.
Real-time quantitative reverse transcription PCR (qRT-PCR) was also employed to verify the presence of donor cells in the liver of HTx mice at killing and to estimate the extent of engraftment. Because iMSUD mice express only transgene-derived human E2 (the endogenous mouse E2 gene was inactivated by gene targeting30), we screened for the presence of donor hepatocytes (which have an intact mouse E2 gene) using mouse-specific E2 primers (Figure 1b) and Real-time qRT-PCR. Human liver RNA samples (n = 6), as well as liver RNA samples from untreated iMSUD (n = 5; Figure 1c) and iMSUD–PBS treated mouse samples (data not shown) all had undetectable levels of mouse E2 confirming primer specificity to mouse RNA. In contrast, samples from iMSUD–HTx treated mice (n = 7) contained a small, but readily detectable amount of mouse E2 RNA that was estimated to be 2.83 ± 1.11% (mean ± SEM) of wild-type control mouse levels (n = 6; Figure 1c).
HTx increased liver BCKDH activity
BCKDH enzyme activity analyzed at killing in liver from PBS treated iMSUD mice was 75.80 ± 6.04 pmol α-ketoisocaproic acid oxidized/min/mg protein, or 6.23% of control activity (Figure 2; n = 8, P < 0.005). This value was not different from untreated iMSUD mice (data not shown) and agrees with data reported previously.30 HTx treatment of iMSUD mice increased liver BCKDH activity to 174.75 ± 26.71 pmol α-ketoisocaproic acid oxidized/min/mg protein, or 14.36% of control activity (n = 7, Figure 2); this level of activity was greater than untreated iMSUD liver (P < 0.005), but was not normalized to wild-type control levels (n = 5, P < 0.005).
Figure 2.

HTx increased iMSUD liver BCKDH activity. BCKDH enzyme activity was assessed on the day of killing, and shown as the mean ± SEM (*P < 0.005 compared to control; **P < 0.005 compared to control and iMSUD–PBS). The number displayed below each bar indicates the n value. BCKDH, branched chain α-ketoacid dehydrogenase; HTx, hepatocyte transplantation; iMSUD, intermediate maple syrup urine disease; KIC, α-ketoisocaproic acid; PBS, phosphate-buffered saline.
Neonatal HTx improved blood BCAA profiles in iMSUD mice at weaning and beyond, though profiles are elevated at the time of killing.
For rapid evaluation of blood amino acids at weaning, we employed tandem mass spectrometry analysis of mouse blood spots on filter paper comparable to methodology employed for human newborn screening.31 Blood spots were collected from all mice at weaning (i.e., ~21 days of age) and in a subset HTx-treated animals at additional time points between 21 and 30 days of age until killing. Analysis of blood BCAA/alanine (ala) ratios revealed an effect of treatment [analysis of variance (ANOVA): F(3,20) = 242, P < 0.0001]. At these time points, this type of analysis revealed a substantial decrease in BCAA/ala of iMSUD–HTx mice (3.91 ± 0.40, n = 9) compared to untreated iMSUD mice [15.75 ± 0.48, n = 5; P < 0.0001 (Figure 3a)]. However, iMSUD–HTx blood BCAA/ala levels were not completely normalized to control levels; iMSUD–HTx BCAA/ala levels were approximately fourfold above controls (P < 0.0001). The blood BCAA/ala ratio of controls, either HTx-treated (n = 5) or untreated (n = 5), was an average of 1.02 ± 0.13 (Figure 3a), which agrees with data reported previously.30 PBS-treated iMSUD BCAA/ala values were also severely elevated (data not shown) and were not different from untreated iMSUD values. Figure 3b displays individual repeated measurement results for each animal [weaning (circles), between 21 and 30 days of age (squares)].
Figure 3.

HTx improved blood BCAA/alanine ratios in iMSUD mice before killing. (a) Blood was collected from live mice at weaning and at several additional time points between 21 and 30 days of age and analyzed by tandem mass spectrometry. Results are represented as the mean ± SEM (*P < 0.0001 compared to iMSUD–HTx and control; **P < 0.0001 compared to iMSUD). The number displayed below each bar indicates the n value. (b) Data from a was broken down to show individual repeated measurement results for each animal collected at weaning (circles) and between 21 and 30 days of age (squares). The horizontal line indicates mean value. BCAA, branched-chain amino acids; HTx, hepatocyte transplantation; iMSUD, intermediate maple syrup urine disease.
At killing, comprehensive serum amino acids were assessed with standard ion-exchange high-performance liquid chromatography and ninhydrin postcolumn detection.32 As depicted in Figure 4a–c, untreated iMSUD animals (18–24 days of age) demonstrated significant increases of valine (923.5 ± 194.7 µmol/l, n = 14; P < 0.01), isoleucine (593.4 ± 66.49 µmol/l, n = 14; P < 0.001), leucine (992.8 ± 234.2 µmol/l, n = 14; P < 0.01), alloisoleucine (76.76 ± 16.13 µmol/l, n = 14; P < 0.001), and glycine (819.5 ± 67.51 µmol/l, n = 15; P < 0.001) in serum compared to controls [n = 14: (val) 253.9 ± 23.07 µmol/l, (ile) 113.3 ± 19.67 µmol/l, (leu) 207.6 ± 27.11 µmol/l, (alloile) undetectable levels, and (gly) 388.4 ± 15.44 µmol/l (n = 13)]. Isoleucine values for iMSUD–HTx animals (35–37 days of age, n = 4) were also significantly elevated compared to controls [484.4 ± 149.9 µmol/l; P < 0.01 (Figure 4a)]. However, HTx-treated animals revealed no significant change in valine (951.4 ± 280 µmol/l), leucine (484.4 ± 149.9 µmol/l), alloisoleucine (32.75 ± 14.85 µmol/l), or glycine (589.4 ± 64.44 µmol/l) compared to both controls and untreated iMSUD (Figure 4a–c). Alanine remained unchanged for all groups (Figure 4d; control: 35 days of age, n = 13, 555.8 ± 25.5 µmol/l; iMSUD: 18–24 days of age, n = 12, 460.2 ± 26.45 µmol/l; iMSUD–HTx: 35–37 days of age, n = 4, 469.2 ± 147.3 µmol/l).
Figure 4.

Serum amino acids and alloisoleucine in iMSUD–HTx mice suggest catabolism at killing. Blood was collected by cardiac puncture at killing and the serum analyzed by ion-exchange HPLC for (a) BCAAs (valine, isoleucine, and leucine), (b) alloisoleucine, (c) glycine, and (d) alanine. Results are represented as the mean ± SEM (*P < 0.01 compared to controls; **P < 0.001 compared to controls). The number displayed below each bar indicates the n value. BCAA, branched-chain amino acids; HPLC, high-performance liquid chromatography; HTx, hepatocyte transplantation; iMSUD, intermediate maple syrup urine disease.
HTx increased body weight at weaning and survival of iMSUD mice
Following HTx, body weights were monitored at weaning. ANOVA revealed an effect of treatment [F(2,28) = 59, P < 0.0001]. Figure 5a shows that body weight of iMSUD–HTx mice was increased compared to iMSUD–PBS (10.21 ± 0.19 g, n = 7 vs. 7.22 ± 0.45 g, n = 9, respectively; P < 0.0001) but was less than controls (11.79 ± 0.23 g, n = 15; P < 0.001).
Figure 5.

HTx improved body weight and lengthened survival in iMSUD mice. (a) Body weight of control (n = 15), iMSUD–PBS (n = 9), and iMSUD–HTx (n = 6) mice was assessed at weaning. The horizontal line indicates mean value (*P < 0.0001 compared to iMSUD–HTx and control; **P < 0.001 compared to control). (b) Survival was compared between iMSUD–PBS (PBS), iMSUD (untreated), and iMSUD–HTx (HTx) mice (*P < 0.0001 compared to untreated and PBS). N values are indicated below their respective data label in the graph. HTx, hepatocyte transplantation; iMSUD, intermediate maple syrup urine disease; PBS, phosphate-buffered saline.
All PBS-treated and untreated iMSUD animals died or were found moribund and humanely killed on or before day 24. In stark contrast, survival of HTx animals [70% survival at 37 days (n = 7/10)] was significantly improved over PBS-treated animals (P < 0.0001) and untreated animals (P < 0.0001, Figure 5b). The three iMSUD–HTx animals which perished before 37 days were found dead in their cage without prodrome. Long-term survival of HTx iMSUD mice was not determined as surviving mice were killed by 37 days of age for tissue collection.
Discussion
The objective of this study was to explore the feasibility of HTx for the management of MSUD. We hypothesized that liver-directed HTx therapy would increase BCKDH E2 expression, restore liver BCKDH activity, and improve the disease phenotype in the iMSUD mouse model. Neonatal HTx was determined to be the most beneficial time point for treatment for this model. Although indistinguishable from unaffected littermates at birth,30 by weaning age iMSUD mice had a significantly lower body weight33 and significantly elevated BCAA/ala in serum and brain compared to controls that was accompanied by neurological dysfunction.30,34 The results of HTx in the iMSUD mouse model show that the use of cellular therapy has promise to assist in the management of MSUD. Several lines of evidence support this conclusion.
First, whether the donor hepatocytes will successfully engraft into iMSUD recipient liver was investigated. Before HTx, we determined that >80% of donor hepatocytes isolated from 1.5 week-old mice expressed LacZ (data not shown). However, we discovered that the LacZ gene was silenced in most hepatocytes isolated from older donors (data not shown). Therefore, engraftment was verified by two separate experimental approaches: qRT-PCR and PCR. Results show that donor transplanted hepatocytes engrafted into the recipient liver (Figure 1a–c). Furthermore, qRT-PCR revealed mouse E2 RNA expression in iMSUD–HTx livers to be 2.83% of normal, which suggested a small but significant repopulation by HTx. These data provided the foundation to carry on further studies.
Second, HTx increased the activity of BCKDH in the recipient liver (Figure 2). Although the increase was nowhere close to normal enzyme activity, considering the small number of hepatocytes administered and engrafted, it was substantial and significant (14.36% of control in HTx iMSUD liver versus 6.23% of control in PBS-treated iMSUD liver).
Third, even though the increase in BCKDH activity following HTx was only 14.36% of normal, it was sufficient to lower the circulating levels of BCAA before killing. Analysis by tandem mass spectrometry revealed that HTx resulted in a 75% decrease in the levels of these amino acids (expressed as BCAA/ala, Figure 3a) in the blood. These results are of major significance even though levels were not normalized. Elevated levels of BCAA, particularly leucine, are the major culprit for the neurological changes seen in MSUD.4,7
Ion-exchange high-performance liquid chromatography facilitated quantitation of alloisoleucine and individual BCAAs (stereoisomeric leucine and isoleucine are not separated using tandem mass spectrometry methods), as well as examination of amino acid trends that might suggest altered physiological states (Figure 4). Comprehensive amino acid studies were also important because of the different ages of treated/untreated iMSUD mice (necessitated by our need to gauge survival time of transplanted iMSUD subjects). From these data, we hypothesize that HTx-treated animals were likely catabolic at euthanasia (35–37 days), with turnover of muscle protein resulting in increased branched chain amino acids.35 This hypothesis is supported by two observations: (i) average alloisoleucine was reduced in iMSUD–HTx animals (with 1 of 4 showing complete correction) compared to untreated iMSUD, though due to low n and high calculated error this was not a significant change. Although alloisoleucine was not normalized in all transplanted animals (Figure 4b), we hypothesize that this preliminary trend toward improvement may become significant with the addition of more animals and that these parameters reflect increased dietary BCAA tolerance. Furthermore, alloisoleucine would not be released from protein turnover in muscle as is the case for leucine, isoleucine, and valine (Figure 4a,b). (ii) iMSUD–HTx animals were hyperglycinemic at both weaning (~21 days of age, data not shown) and euthanasia (35–37 days of age; Figure 4c) consistent with a catabolic state, though due to a low n and high calculated error this was also not a significant change.
Therefore, although HTx greatly improved BCAA profiles in iMSUD mice before killing (Figure 3a), this approach was unable to compensate for fluctuations possibly due to catabolism (i.e., fed versus unfed state and a host of other metabolic parameters, Figures 3b and 4). Our hypothesis concerning the hypercatabolic state of iMSUD–HTx at euthanasia is not without flaws, however. If correct, we might expect the PBS-treated animals to be equally catabolic and the BCAA levels even higher than those observed for iMSUD–HTx, which was not the case (Figure 4). Another potential explanation is that there were insufficient viable transplanted cells at killing to have a significant effect on BCAA levels. We clearly observed a physiologic improvement near weaning, but the BCAA data at euthanasia, coupled to the fact that some transplanted mice had died just before euthanasia, suggests that the HTx effect was transient. However, any reduction in BCAA levels following HTx is expected to minimize or prevent deleterious neurological changes, particularly at the early stages before and around weaning when treatment is more difficult. With improved biochemical parameters at this early stage, animals are likely to withstand stressful situations such as infection and other metabolic stressors often encountered by MSUD patients. An extended effect of HTx might be gained in future studies with immunosuppression, lower dietary protein, and/or thiamin supplementation. A thiamin-responsive form of MSUD has been described,4 and this vitamin may help gain the extended effect of HTx in the iMSUD model.
Fourth, HTx resulted in increased body weight and prolonged survival of animals (Figure 5a,b), both very important physiological markers. In addition, subjective analysis of the mice at weaning was also striking. At the time of weaning (~21 days of age) PBS-treated and untreated iMSUD mice typically sat hunched and unmoving with unkempt fur (data not shown). Conversely, HTx treated animals behaved similar to healthy control animals, with active exploring and a smooth coat. HTx-treated animals found dead before 37 days of age perished without prodrome. However, it should be noted that survival of untreated iMSUD mice reported here (Figure 5b) was reduced compared to results previously published.30 Many generations of mice separate this study from the previous study reported by Homanics et al.30 Therefore, this difference in survival may be attributed to random genetic drift.
Our findings of partial metabolic correction in a mouse model of iMSUD by HTx were very encouraging, especially because all the above improvements were brought on by the transplantation of hepatocytes representing a tiny fraction of the whole liver at a very early stage. Although BCAAs were elevated at killing, body weight, survival, and BCKDH activity were all significantly increased at this time point (Figures 2,3–5). Liver BCKDH E2 mRNA and LacZ DNA at killing were also detectable at a small but significant measure in iMSUD–HTx mice (Figure 1). Further studies are needed and are in progress to optimize the use of hepatocytes in prolonged studies. Though it is clear that these animals are not corrected at this stage, these results indicate that HTx can be useful to improve health thus extending survival until a time point when more aggressive treatments could be applied. For example, the efficiency of hepatocyte engraftment can be substantially improved by additional HTx injections into the spleen or portal vein,36,37,38 partial hepatectomy,39 and treatment with retrorsine and/or CCl4 (refs. 40,41). Data reported here suggest that gene therapy approaches may also be beneficial, as a small amount of enzyme activity resulted in a significant effect in phenotype. For instance, ex vivo hepatocyte-directed gene therapy could be used to overexpress BCKDH in healthy donor cells42 while in vivo liver-targeted gene therapy could introduce the E2 gene to mutant hepatocytes.43 Furthermore, in this study, immunosuppression was not used despite the fact that donor hepatocytes and recipients were not immunologically matched. Future studies using immunosuppression or matched donors and recipients should also enhance outcomes.
In conclusion, this report strongly supports the feasibility of HTx for the management of MSUD, a highly complex metabolic disorder.
Materials and Methods
All studies involving animals were reviewed and approved by the University of Pittsburgh's Institutional Animal Care and Use Committee.
Mice. iMSUD mice are homozygous for LAP-tTA and TRE-E2 transgenes, which express C-terminal myc tagged human BCKDH E2 on a mouse E2 knockout (−/−) background.30 The iMSUD mouse line used in these studies was the “A” line and are on a mixed genetic background.30 Mating pairs consisted of mice homozygous for both transgenes and heterozygous for the mouse E2 knockout resulting in a 1:2:1 ratio of +/+:+/−:−/− mouse E2. Thus, all offspring from these matings were homozygous for both transgenes and expressed human E2. iMSUD mice were genotyped for the E2 knockout by Southern blot analysis as previously described.30 Mice were fed a standard mouse chow consisting of 22% protein.
Donor hepatocytes were derived from ROSA26 [B6.129S7-Gt(ROSA)26Sor/J; Jackson Laboratory (Bar Harbor, ME) stock no. 2192 (ref. 44)] mice. Heterozygous (+/−) ROSA26 mice were mated to wild type (+/+) C57Bl/6J mice (Jackson Laboratory stock no. 664) mice. This breeding strategy resulted in ~50% of offspring being ROSA26+/− (LacZ positive). ROSA26+/− mice were genotyped with PCR at birth from genomic DNA isolated from a small amount of blood using Instagene (BioRad, Hercules, CA). PCR primers and amplification parameters for this mouse strain were as described by the Jackson Laboratories;45 reactions used PCR Supermix HiFi (Invitrogen, Carlsbad, CA). Genotyping results were confirmed using a β-galactosidase Staining Kit (Mirus, Madison, WI) of a small tail clip (~0.1–0.2 mm) at birth.
Donor hepatocyte isolation. Hepatocytes from 1.5- to 2.5-week-old ROSA26+/− mice44 were isolated by collagenase perfusion.46 In brief, donor mice were sedated with 100 mg/kg ketamine (Fort Dodge Animal Health, Ft. Dodge, IA) + 10mg/kg xylazene (Vedco, St. Joseph, MO) and a midline laparotomy performed. The inferior vena cava was then perfused and the portal vein sectioned within 5–10 seconds of the start of perfusion to allow the solution to flow through the liver. The liver was sequentially perfused at a rate of 2.5 ml/min with ethylene glycol tetraacetic acid solution (Hank's buffered salt solution without calcium or magnesium, ethylene glycol tetraacetic acid 0.5 mmol/l, HEPES 10 mmol/l, pH 7.5 supplemented with 1% penicillin/streptomycin) for 5 minutes followed by a collagenase solution for 10 minutes (Eagle's minimal essential medium with calcium/magnesium plus collagenase 0.1 mg/ml, HEPES 10 mmol/l, pH 7.5 and 1% penicillin/streptomycin). Hepatocytes were removed by mechanical disassociation and filtered through sterile 150-µm nylon mesh. Cells were washed three times in Eagle's minimal essential medium by centrifugation at 50 g for 5 minutes and finally resuspended at a concentration of 2 × 106 cells/ml in PBS. The viability of donor hepatocytes was estimated by Trypan blue exclusion; only suspensions with viability >80% were used for transplants. One liver was sufficient for 30+ transplant recipients.
Hepatocyte transplantation. A sterile 30 gauge needle was used to administer 50 µl of PBS alone or PBS containing 100,000 hepatocytes directly through the abdomen of neonatal mice into the liver pulp. Each mouse was injected twice. One injection was administered between the ages of 1 and 7 days and a second injection was administered ~3–7 days later; all injections were conducted on mice between 1 and 10 days of age. At this early age, the liver was clearly visible through the skin.
Killing of mouse and tissue collection. Mice were killed by cervical dislocation when animals appeared moribund, or at the designated end of the experiment (35–37 days of age). On the date of killing, the liver was removed for processing. The liver was first flash frozen in liquid N2 and then crushed using a stainless steel mortar and pestle kept cold with dry ice and liquid N2. The pulverized tissue was separated into three partitions to be used for (i) BCKDH enzyme analysis, (ii) real-time qRT-PCR, and (iii) PCR analysis. Pulverized tissue was stored at −80 °C until the day of analysis.
PCR detection of hepatocytes. Livers were removed on the date of killing and processed as described above. On the day of analysis, DNA (~20 ng/sample) was isolated for PCR analysis via the DNeasy kit (Qiagen, Germantown, MD) or using a standard phenol:chloroform purification protocol following tissue lysis. LacZ primers and PCR cycling parameters were identical to those used for genotyping.
Real-time qRT-PCR. Livers were removed on the date of killing and processed as described above. RNA was isolated from primary mouse hepatocytes (obtained from ROSA26+/−, iMSUD, iMSUD–PBS, iMSUD–HTx, and wild-type controls) and primary human hepatocytes (obtained from liver disease patient resections) using TRIzol reagent (Invitrogen) following the manufacturer's instructions. RNA was converted to complementary DNA using the High Capacity cDNA RT Kit (Applied Biosystems, Foster City, CA). Real-time qRT-PCR was performed using the TaqMan Assays-on-Demand Gene Expression Products for β-actin (ID no. Mm02619580_g1; Applied Biosystems) following the manufacturer's protocol. In short, 100 ng of complementary DNA template, 12.5 µl of 2× Taqman Universal Master Mix, and 1.25 µl of 20× Target Assay Mix were combined into 25 µl reactions. Real-time qRT-PCR was also performed using the SYBR Green system. Mouse E2 primers for SYBR Green qRT-PCR (5′-GGAAACCACTGATCGACATTGAGA-3′ Forward; 5′-ATTGCCAGCCGACGCACT-3′ Reverse) were custom designed using a Basic Local Alignment Search Tool (Figure 1b) of the classic MSUD deletion site in mouse E2 mRNA (bp 444–617; NCBI accession no. NM 010022) and the corresponding human E2 sequence (bp 415–588; NCBI accession no. NM 001918). Primers were chosen based upon the number of mismatches to human to ensure specificity to mouse only, and verified using BiosearchTech's (Novato, CA) free online RealTimeDesign software for primers. In short, 100 ng of complementary DNA template, 12.5 µl of 2× SYBR green PCR Master Mix, and 0.075 µl of each 0.1 mmol/l primer were combined into 25 µl reactions. Each sample was run in triplicate on an ABI 7000 Real-time PCR System (Applied Biosystems). Relative quantification of real-time qRT-PCR data was performed using the Comparative CT (ΔCT) Method. In summary, CT values were first converted to an arbitrary number (AN) using a standard calculation 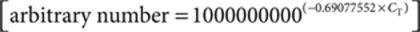 . The three arbitrary numbers (AN) for each sample were then averaged. Target gene expression levels were normalized to the corresponding actin endogenous control by calculating the ΔCT [ΔCT = aveANtarget gene/aveANactin]. Data were presented as the mean ± SEM and analyzed by ANOVA and post hoc Fisher's test.
. The three arbitrary numbers (AN) for each sample were then averaged. Target gene expression levels were normalized to the corresponding actin endogenous control by calculating the ΔCT [ΔCT = aveANtarget gene/aveANactin]. Data were presented as the mean ± SEM and analyzed by ANOVA and post hoc Fisher's test.
BCKDH complex enzyme activity assay. Livers were removed on the day of killing and processed as described above. On the day of analysis, livers were homogenized (1:9 wt/wt) in 0.25 sucrose, 10 mmol/l Tris–HCl and centrifuged 600 g for 10 minutes and the supernatant utilized for the enzyme assay. BCKDH activity was determined by measuring the release of 14CO2 from α-keto [1-14C] isocaproate (Amersham Biosciences, Piscataway, NJ).47 Assays were done in duplicate and carried out for 15 minutes at 37 °C; 14CO2 was trapped in hydroxide of Hyamine (Sigma-Aldrich, St Louis, MO) and radioactivity was determined by liquid scintillation spectrometry. Liver protein concentration was assessed with a Bicinchoninic acid assay. Calculation: xmol α-ketoisocaproic acid oxidized = DPM 14CO2/DPM 14C-α-ketoisocaproic acid/15 minutes/mg liver protein. Data were presented as the mean ± SEM. An F-test for equality of two standard deviations determined data were nonparametric. Therefore, differences between genotypes were compared by the nonparametric Kruskal–Wallis test and a Mann–Whitney analysis.
Whole blood and serum amino acid analysis. Whole blood samples were collected at 1 PM from the tail tip at weaning and approximately weekly until killing. Blood was spotted on a Guthrie card and evaluated for BCAA and alanine concentrations using tandem mass spectrometry (Pediatrix Analytical, Sunrise, FL31). Due to small blood volumes, it was not possible to completely soak the filter-paper spots; accordingly, data were presented as a ratio (total BCAA/ala) to correct for blood volume differences. This ratio is a sensitive indicator of MSUD phenotype.48 Data were analyzed by repeated measures ANOVA and post hoc Fisher's test.
Serum was isolated from whole blood collected by cardiac puncture in some animals at killing through centrifugation for 10 minutes at 3,000 g. Amino acids from mouse serum were quantified using standard ion-exchange high-performance liquid chromatography with postcolumn ninhydrin derivatization.32 Data were presented as the mean ± SEM and analyzed by ANOVA and post hoc Tukey's test.
Body weight and survival. Date of birth, date of treatment, weight at weaning (~21 days of age), and date of death were recorded for iMSUD–HTx, iMSUD–PBS, untreated iMSUD, and control mice. Data were analyzed via the Kaplan–Meier method and log-rank test (for survival), or ANOVA and post hoc Fisher's test (for body weight).
Acknowledgments
We thank James DiPerna and Christina Hansen (Pediatrix Analytical) for expert technical assistance. This work was supported by NIH grants DK51960, DK57386, AA10422, and NS40270. Financial support was also generously provided by the MSUD Family Support Group and the Scott C. Foster Metabolic Disease Fund.
REFERENCES
- Harris RA, Joshi M., and , Jeoung NH. Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem Biophys Res Commun. 2004;313:391–396. doi: 10.1016/j.bbrc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Yeaman S. The 2-oxo acid dehydrogenase complexes: recent advances. Biochem J. 1989;257:625–632. doi: 10.1042/bj2570625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DT., and , Shih VE.Maple syrup urine disease (branched-chain ketoaciduria) The Metabolic and Molecular Basis of Inherited Disease 2001McGraw-Hill Medical Publishing: New York; 1971–2005.In: Scriver, CR, Beaudet, AL, Sly, WS, Valle, D (eds)8th edn [Google Scholar]
- Dancis J, Hutzler J., and , Levitz M. Metabolism of the white blood cells in maple syrup urine disease. Biochem Biophys Acta. 1960;43:342–343. doi: 10.1016/0006-3002(60)90448-0. [DOI] [PubMed] [Google Scholar]
- Menkes J, Hurst P., and , Craig J. A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics. 1954;14:462–466. [PubMed] [Google Scholar]
- Cox RP., and , Chuang DT.Maple syrup urine disease: clinical and molecular genetic considerations The Molecular and Genetic Basis for Neurological Disease 1993Butterworth-Heinemann: Boston; 189–207.In: Rosenberg, R, Prusiner, S, DiMauro, R, Barchi, R and Kunkel, L (eds) [Google Scholar]
- Strauss KA, Mazariegos GV, Sindhi R, Squires R, Finegold DN, Vockley G, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557–564. doi: 10.1111/j.1600-6143.2005.01209.x. [DOI] [PubMed] [Google Scholar]
- Wendel U, Saudubray JM, Bodner A., and , Schadewaldt P. Liver transplantation in maple syrup urine disease. Eur J Pediatr. 1999;158 Suppl 2:S60–S64. doi: 10.1007/pl00014324. [DOI] [PubMed] [Google Scholar]
- Bodner-Leidecker A, Wendel U, Saudubray JM., and , Schadewaldt P. Branched-chain L-amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation. J Inherit Metab Dis. 2000;23:805–818. doi: 10.1023/a:1026708618507. [DOI] [PubMed] [Google Scholar]
- Netter JC, Cossarizza G, Narcy C, Hubert P, Ogier H, Revillon Y, et al. Mid term outcome of 2 cases with maple syrup urine disease: role of liver transplantation in the treatment. Arch Pediatr. 1994;1:730–734. [PubMed] [Google Scholar]
- Strom SC, Bruzzone P, Cai H, Ellis E, Lehmann T, Mitamura K, et al. Hepatocyte transplantation: clinical experience and potential for future use. Cell Transplant. 2006;15 Suppl 1:S105–S110. doi: 10.3727/000000006783982395. [DOI] [PubMed] [Google Scholar]
- Dhawan A, Mitry RR, Hughes RD, Lehec S, Terry C, Bansal S, et al. Hepatocyte transplantation for inherited factor VII deficiency. Transplantation. 2004;78:1812–1814. doi: 10.1097/01.tp.0000146386.77076.47. [DOI] [PubMed] [Google Scholar]
- Vroemen JP, Buurman WA, Heirwegh KP, van der Linden CJ., and , Kootstra G. Hepatocyte transplantation for enzyme deficiency disease in congenic rats. Transplantation. 1986;42:130–135. doi: 10.1097/00007890-198608000-00005. [DOI] [PubMed] [Google Scholar]
- Groth C, Arborgh B, Bjorken C, Sundberg B., and , Lundgren G. Correction of hyperbilirubinemia in the glucuronyltransferase-deficient rat by intraportal hepatocyte transplantation. Transplant Proc. 1977;9:313–316. [PubMed] [Google Scholar]
- Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, et al. Treatment of the Crigler–Najjar syndrome type I with hepatocyte transplantation. N Eng J Med. 1998;338:1422–1426. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- Matas AJ, Sutherland DE, Steffes MW, Mauer SM, Sowe A, Simmons RL, et al. Hepatocellular transplantation for metabolic deficiencies: decrease of plasma bilirubin in Gunn rats. Science. 1976;192:892–894. doi: 10.1126/science.818706. [DOI] [PubMed] [Google Scholar]
- Lee KW, Lee JH, Shin SW, Kim SJ, Joh JW, Lee DH, et al. Hepatocyte transplantation for glycogen storage disease type Ib. Cell Transplant. 2007;16:629–637. doi: 10.3727/000000007783465019. [DOI] [PubMed] [Google Scholar]
- Muraca M, Gerunda G, Neri D, Vilei MT, Granato A, Feltracco P, et al. Hepatocyte transplantation as a treatment for glycogen storage disease type 1a. Lancet. 2002;359:317–318. doi: 10.1016/S0140-6736(02)07529-3. [DOI] [PubMed] [Google Scholar]
- Irani AN, Malhi H, Slehria S, Gorla GR, Volenberg I, Schilsky ML, et al. Correction of liver disease following transplantation of normal rat hepatocytes into Long-Evans Cinnamon rats modeling Wilson's disease. Mol Ther. 2001;3:302–309. doi: 10.1006/mthe.2001.0271. [DOI] [PubMed] [Google Scholar]
- Horslen SP, McCowan TC, Goertzen TC, Warkentin PI, Cai HB, Strom SC, et al. Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder Pediatrics 20031111262–1267.6 Pt 1 [DOI] [PubMed] [Google Scholar]
- Strom S, Fisher RA, Rubinstein WS, Barranger JA, Towbin RB, Charron M, et al. Transplantation of human hepatocytes. Transpl Proc. 1997;29:2103–2106. doi: 10.1016/s0041-1345(97)00252-2. [DOI] [PubMed] [Google Scholar]
- Jiang J, Salido EC, Guha C, Wang X, Moitra R, Liu L, et al. Correction of hyperoxaluria by liver repopulation with hepatocytes in a mouse model of primary hyperoxaluria type-1. Transplantation. 2008;85:1253–1260. doi: 10.1097/TP.0b013e31816de49e. [DOI] [PubMed] [Google Scholar]
- Moscioni AD, Roy-Chowdhury J, Barbour R, Brown LL, Roy-Chowdhury N, Competiello LS, et al. Human liver cell transplantation. Prolonged function in athymic-Gunn and athymic-analbuminemic hybrid rats. Gastroenterology. 1989;96:1546–1551. [PubMed] [Google Scholar]
- Oren R, Dabeva MD, Petkov PM, Hurston E, Laconi E., and , Shafritz DA. Restoration of serum albumin levels in nagase analbuminemic rats by hepatocyte transplantation. Hepatology. 1999;29:75–81. doi: 10.1002/hep.510290147. [DOI] [PubMed] [Google Scholar]
- Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, Finegold M, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- Strom SC, Fisher RA, Thompson MT, Sanyal AJ, Cole PE, Ham JM, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation. 1997;63:559–569. doi: 10.1097/00007890-199702270-00014. [DOI] [PubMed] [Google Scholar]
- Strom SC, Chowdhury J., and , Fox IJ. Hepatocyte transplantation for the treatment of human disease. Semin Liver Dis. 1999;19:39–48. doi: 10.1055/s-2007-1007096. [DOI] [PubMed] [Google Scholar]
- Fisher RA, Bu D, Thompson M, Tisnado J, Prasad U, Stirling R, et al. Defining hepatocellular chimerism in a liver failure patient bridged with hepatocyte infusion. Transplantation. 2000;69:303–307. doi: 10.1097/00007890-200001270-00018. [DOI] [PubMed] [Google Scholar]
- Bilir BM, Guinette D, Karrer F, Kumpe DA, Krysl J, Stephens J, et al. Hepatocyte transplantation in acute liver failure. Liver Transpl. 2000;6:32–40. doi: 10.1002/lt.500060113. [DOI] [PubMed] [Google Scholar]
- Homanics GE, Skvorak K, Ferguson C, Watkins S., and , Paul HS. Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med Gen. 2006;7:33. doi: 10.1186/1471-2350-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chace DH, Hillman SL, Millington DS, Kahler SG, Roe CR., and , Naylor EW. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin Chem. 1995;41:62–68. [PubMed] [Google Scholar]
- Slocum RH., and , Cummings JG.Amino acid analysis of physiological samples Techniques in Diagnostic Human Biochemical Genetics 1991Wiley-Liss: New York; 87–126.In: Hommes, FA (ed)1st edn [Google Scholar]
- Skvorak KJ. Animal models of maple syrup urine disease. J Inherit Metab Dis. 2009;32:229–246. doi: 10.1007/s10545-009-1086-z. [DOI] [PubMed] [Google Scholar]
- Zinnanti WJ, Lazovic J, Griffin K, Skvorak KJ, Paul HS, Homanics GE, et al. Dual mechanisms of brain injury and novel treatment strategy in maple syrup urine disease Brain 2009132903–918.Pt 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illsinger S LT, Meyer U,, Vaske B., and , Das AM. Branched chain amino acids as a parameter for catabolism in treated phenylketonuria. Amino Acids. 2005;28:45–50. doi: 10.1007/s00726-004-0150-0. [DOI] [PubMed] [Google Scholar]
- Mito M, Ebata H, Kusano M, Onishi T, Saito T., and , Sakamoto S. Morphology and function of isolated hepatocytes transplanted into rat spleen. Transplatation. 1979;28:499–505. doi: 10.1097/00007890-197912000-00013. [DOI] [PubMed] [Google Scholar]
- Holzman MD, Rozga J, Neuzil DF, Griffin D, Moscioni AD., and , Demetriou AA. Selective intraportal hepatocyte transplantation in analbuminemic and Gunn rats. Transplantation. 1993;55:1213–1219. doi: 10.1097/00007890-199306000-00002. [DOI] [PubMed] [Google Scholar]
- Ponder KP, Gupta S, Leland F, Darlington G, Finegold M, DeMayo J, et al. Mouse hepatocytes migrate to liver parenchyma and function indefinitely after intrasplenic transplantation. Proc Natl Acad Sci USA. 1991;88:1217–1221. doi: 10.1073/pnas.88.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha C, Deb N, Sappal B, Ghosh S, Roy-Chowdhury N., and , Roy-Chowdhury J. Amplification of engrafted hepatocytes by preparative manipulation of the host liver. Artif Organs. 2001;25:522–528. doi: 10.1046/j.1525-1594.2001.025007522.x. [DOI] [PubMed] [Google Scholar]
- Laconi E, Oren R, Mukhopadhyay DK, Hurston E, Laconi S, Pani P, et al. Long-term, near-total liver replacement by transplantation of isolated hepatocytes in rats treated with retrorsine. Am J Path. 1998;153:319–329. doi: 10.1016/S0002-9440(10)65574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Fu T, Nelson JA, Superina RA., and , Soriano HE. Liver repopulation after cell transplantation in mice treated with retrorsine and carbon tetrachloride. Transplantation. 2002;73:1818–1824. doi: 10.1097/00007890-200206150-00020. [DOI] [PubMed] [Google Scholar]
- Wang X, Mani P, Sarkar DP, Roy-Chowdhury N., and , Roy-Chowdhury J. Ex vivo gene transfer into hepatocytes. Methods Mol Biol. 2009;481:117–140. doi: 10.1007/978-1-59745-201-4_11. [DOI] [PubMed] [Google Scholar]
- Ding Z, Georgiev P., and , Thöny B. Administration-route and gender-independent long-term therapeutic correction of phenylketonuria (PKU) in a mouse model by recombinant adeno-associated virus 8 pseudotyped vector-mediated gene transfer. Gene Ther. 2006;13:587–593. doi: 10.1038/sj.gt.3302684. [DOI] [PubMed] [Google Scholar]
- Friedrich G., and , Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5:1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- JAX 2008The Jackson Laboratory: Bar Harbor, ME; Gt(ROSA)26Sor, Gt(ROSA)26Sortm1Sho, Version 1 Genotyping Protocol [Google Scholar]
- Strom SC, Pisarov LA, Dorko K, Thompson MT, Schuetz JD., and , Schuetz EG. Use of human hepatocytes to study P450 gene induction. Methods Enzymol. 1996;272:388–401. doi: 10.1016/s0076-6879(96)72044-x. [DOI] [PubMed] [Google Scholar]
- Paul HS., and , Adibi SA. Mechanism of increased conversion of branched chain keto acid dehydrogenase from inactive to active form by a medium chain fatty acid (octanoate) in skeletal muscle. J Biol Chem. 1992;267:11208–11214. [PubMed] [Google Scholar]
- Morton DH, Strauss KA, Robinson DL, Puffenberger EG., and , Kelley RI. Diagnosis and treatment of maple syrup urine disease: a study of 36 patients. Pediatrics. 2002;109:999–1008. doi: 10.1542/peds.109.6.999. [DOI] [PubMed] [Google Scholar]