

Abstract
To evaluate Flory's isolated-pair hypothesis in the context of helical peptides, we explore equilibrium conformations of α-helix-forming polypeptides as a function of temperature by means of replica exchange molecular dynamics in conjunction with the CHARMM/GB implicit solvent force field and the weighted histogram analysis method. From these simulations, Zimm–Bragg parameters, s and σ, of Ac-Alan-NMe are computed as a function of temperature. The values obtained for s(T) and σ(T) remain unchanged along the length of the polypeptide except for very short chains and yield results consistent with measurements based on short helix-forming peptides but suggest larger s values than anticipated from polymer-based measurements. From direct estimates of the density of states for Ac-Alan-NMe (n = 3–20) and peptide constructs based on the C peptide from RNase A, the conformational entropy is calculated versus temperature. The calculated S(T) shows a clear proportionality to the chain length over a wide range of temperature. This is observed in polypeptides with both significantly branched and simple methyl (alanine) side chains. These results provide evidence for the validity of Flory's isolated pair hypothesis, at least in the context of helical peptides and helix-to-coil transitions in these peptides.
Flory suggested that it is safely assumed that the state of each pair of rotation angles, (ϕ, ψ), in a polypeptide is independent of the adjoining pair (1). This means that the total number of possible conformations of a polypeptide chain can be obtained as the product of the number of possible conformations of a N- and C-terminally blocked peptide consisting of each residue in the full peptide. This “isolated-pair hypothesis” has been widely accepted because of its simplicity in relating local conformational properties to the number of possible conformations of longer peptides. However, this hypothesis also implies that the number of conformations grows exponentially with the increase of chain length, which leads to a long-asked question of how a protein can fold to its native conformation from the vast majority of nonnative states on the time scale of minutes or less (2). One answer to this “paradox,” for the folding of protein molecules, comes from the theory of funneled energy landscapes (3), which suggests that the folding problem is a directed search on a sloped energy landscape. Support for this idea, as well as the notion that protein molecules adopt native-like topologies, even in their denatured states (4–6), has become apparent from both experimental and theoretical studies during the past several years. The situation for peptides, however, remains unclear, although it has been suggested that the number of conformations is overestimated and that the isolated-pair hypothesis is not valid (7–9).
In one recent study, Rose and coworkers (8) evaluated the isolated-pair hypothesis by means of Monte Carlo exhaustive sampling. The results from their findings suggest a failure of the isolated-pair hypothesis and that the entropy cost on folding is much smaller than presently believed. Similar conclusions were reached by Sosnick and his colleagues (7) in studies of short peptides of length 1–3 aa. These results appear to be in accord with the findings of van Gunsteren and coworkers (9), who showed that a limited number of conformers were observed for peptides of various lengths during long molecular dynamics simulations. While Rose and coworkers argue that the failure of the hypothesis is caused by non-nearest-neighbor, local steric clashes that occur mainly between residues i and i + 3, i + 4, or i + 5, their results are based on polypeptides of <10 residues. Their suggestions seem quite reasonable, because it is obvious that within this range (<10 residues) there are fewer residues that might clash for the smaller peptides (e.g., there is no possible clash between i and i + 3 in a tripeptide, and there are only two in a pentapeptide and four in a septapeptide). This “end effect” is expected, however, to decrease and die away as the length of the peptide increases. This leads us to ask: Will the isolated-pair hypothesis hold for longer chains, even if it appears to fail for short ones, or will nonlocal remote clashes also play a vital role in diminishing the available conformations such that Flory's ideas fail for peptides of any chain length? We can formulate these questions more quantitatively in terms of the entropy of a peptide chain, because the number of available conformations and entropy are intimately coupled.
For a set of capped polyalanine peptides, Ac-Alan-NMe, at a certain temperature, the isolated-pair hypothesis may be formulated as
 |
[1] |
with the equality denoting that the hypothesis holds. Sn is the entropy of Ac-Alan-NMe, and Sn,i is that of the residue i of Ac-Alan-NMe (e.g., S1,1 is the entropy of residue 1 of Ac-Ala-NMe; in this case, S1,1 = S1, because there is only one residue). If non-nearest-neighbor, local steric clashes are the only factor that lead to the inequality in Eq. 1, we would still anticipate proportionality between n and Sn for some larger n:
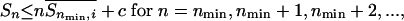 |
[2] |
where nmin is the minimum chain length at which the “end effect” becomes insignificant, c is a positive constant, 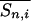 is the average of Sn,i as Sn/n, and
is the average of Sn,i as Sn/n, and 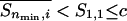 . If nonlocal steric clashes (i.e., any clash between residue i and i + 6 or larger) are not negligible, the lefthand side of Eq. 2 falls more and more short of the equality as n increases. Rose and coworkers (8) have suggested that in their systems the equality in Eq. 1 does not hold. Our question here is whether the hypothesis holds perfectly (Eq. 1), partially (Eq. 2), or not at all, and if it holds partially, for what length of peptide, nmin?
. If nonlocal steric clashes (i.e., any clash between residue i and i + 6 or larger) are not negligible, the lefthand side of Eq. 2 falls more and more short of the equality as n increases. Rose and coworkers (8) have suggested that in their systems the equality in Eq. 1 does not hold. Our question here is whether the hypothesis holds perfectly (Eq. 1), partially (Eq. 2), or not at all, and if it holds partially, for what length of peptide, nmin?
By employing one of the currently most efficient computational search methods [replica exchange molecular dynamics, or REMD (10)] and an implicit solvent force field that is efficient to compute (CHARMM/GB) (11), we explore the validity of Flory's hypothesis for polypeptides up to 26 residues in length, which are long enough to observe non-nearest-neighbor steric effects.
To examine the general quality of our force field modeling and sampling methodology, we calculate (the average) thermostatistical properties of the helix-forming polypeptides and compare their features, as given by the CHARMM/GB force field, with those from experiment. To do this, we employed the standard (μ = 1) Zimm–Bragg (ZB) model (12) to estimate the helix nucleation and propagation parameters σ and s, respectively, for peptides Ac-Alan-NMe of different chain length. The standard ZB model is isomorphic with the one-dimensional Ising model, because of its assumption of local interaction between adjacent peptide units only. If the isolated-pair hypothesis holds and non-nearest-neighbor steric effects are not significant, the parameters σ and s should be independent of the chain length.
We continue our analysis by calculating the entropy of equilibrated conformations of each polypeptide as a function of temperature. To examine the adherence to Eq. 1 and Eq. 2, the density of states of the peptide is estimated by combining REMD sampling at different temperatures by using the WHAM method (13, 14). If the isolated-pair hypothesis is valid, the calculated S(T) should increase with increasing chain length (Eq. 1). Otherwise, S(T) is expected to fall short of the proportionality at larger chain length.
Materials and Methods
Capped Polyalanine and C-Peptide Homologues. N-acetyl-(l-alanil)n-N′-methylamide, or Ac-Alan-NMe, where n = 6–15 and 20, were generated using CHARMM (15) with the param19 polar hydrogen force field (16). The peptide related to the C-peptide from ribonuclease A (KETAAAKFLRQHM, Cp13) was prepared in the same way. Three homologues of Cp13, the tandem (Cp26), the N-terminal seven residue peptide fragment (CpN7), and the C-terminal six residue peptide (CpC6) were prepared similarly. Cp26 has the same composition of side chains as Cp13, but CpN7 and CpC6 do not. Nevertheless, the average of the thermostatistical properties of CpN7 and CpC6 can be regarded as the properties of a hypothetical peptide of 6.5 residues whose side chain composition is the same as that of Cp13.
The Definition of a Hydrogen Bond. We use the criteria of Ravishanker et al. (17) to define an α-helical hydrogen bond (i.e., between residues i and i + 4). Specifically we employ the definition: 1.5 Å ≤ rO—H ≤ 3.0 Å and 120° ≤ ∠ C—O—H ≤ 180°.
Data Generation and Collection: REMD. The replica-exchange method (18) is one of the generalized-ensemble algorithms that yield simulations with non-Boltzmann weight factors and enable a random-walk search throughout a specific phase space. Sugita and Okamoto (10) have combined this method with molecular dynamics as REMD to provide sampling within the canonical ensemble over a wide range of temperatures for protein folding studies. In the present study, we also use REMD, with only a few minor changes, to generate the ensembles of conformations for the polypeptides mentioned above. We assign a replica of each peptide to one of sixteen temperature windows, which are exponentially distributed between 200 and 600 K (200.0 K, 215.2 K, 231.6 K, 249.1 K, 268.1 K, 288.4 K, 310.4 K, 334.0 K, 359.3 K, 386.6 K, 416.0 K, 447.6 K, 481.6 K, 518.2 K, 557.6 K, and 600.0 K). To ensure sufficiently complete conformational sampling in the molecular dynamics, a continuum solvent-based generalized Born force field, CHARMM/GB (11), is used. Furthermore, the SHAKE algorithm (19) is used to constrain hydrogen-heavy atom bonds, and the time step for dynamics integration is set to 2.0 fs. Every one thousand steps (2.0 ps), a conformation is recorded in each temperature window and a conformational exchange is attempted. Each simulation was started with replicas in a fully extended conformation that was generated with all the ϕ, ψ, and ω set to 180° and then energy-minimized. Ten thousand conformations from each window, between 20 and 40 ns, were used for analysis. Temperature adjustment was implemented by way of assigning velocities from a Gaussian distribution appropriate for the temperature window. This also applies to the end of each cycle, where the replicas are exchanged. We believe random reassignment of velocities from a Gaussian-distributed sample may be a better scheme than uniform scaling of the atomic velocities. Reassignment enables the search to move more rapidly into a different hyper-plane of the phase space while maintaining correct coupling to the canonical temperature bath. Thus, equation 12 in ref. 10 is not adopted here. All simulations were carried out using the MMTSB Tool Set (20).
ZB Model. We employ the standard (μ = 1) ZB model (12) without the infinite chain length (large-n) approximation, because the polypeptides used here are not very long. We outline the equations in this limit below. The details of the model have been well described elsewhere (12, 21, 22). The partition function, qn, by means of the standard (μ = 1) ZB model is
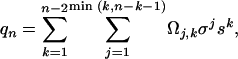 |
[3] |
where Ωj,k is the number of ways to put k α-helical hydrogen bonds into j segments, σ is the helix nucleation parameter, and s is the helix propagation parameter. Note that n – 2 is the maximum possible number of α-helical hydrogen bonds in a peptide with both termini capped, such as Ac-Alan-NMe. Because of the assumption of μ = 1 (local interaction between adjacent peptide units only), qn can also be described in a format that is more suitable for computation,
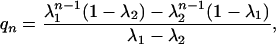 |
[4] |
where 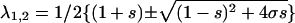 ; λ1 ≥ λ2.
; λ1 ≥ λ2.
The average number of α-helical hydrogen bonds, 〈k〉, and the average number of α-helical segments, 〈j〉, are
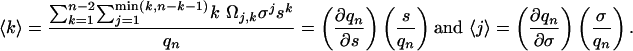 |
[5] |
The fractional helicity, θ, is simply 〈k〉/(n – 2),
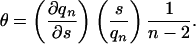 |
[6] |
The average length of a helical segment, 〈k〉/〈j〉, is
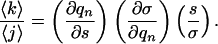 |
[7] |
Because the coordinates of the molecules at each temperature are recorded during the REMD simulation, every α-helical hydrogen bond in each molecule is located. The availability of this microscopic information enables us to obtain not only the fractional helicity but also the average length of helical segments directly, in order to fit s and σ at each temperature by Eqs. 6 and 7. We note that such is not the case in most experiment and therefore simultaneous determination of the parameters σ and s as a function of temperature are generally precluded (23–25). We obtain optimal values for s and σ by brute-force exploration of the (s, σ) space for the pair of s and σ that minimizes the sum of the squared relative errors of θ and 〈k〉/〈j〉:
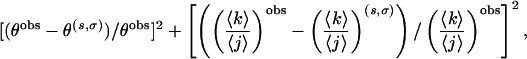 |
where θobs and (〈k〉/〈j〉)obs are from our REMD results. θ(s,σ) and (〈k〉/〈j〉)(s,σ) are those from Eqs. 6 and 7, given a set of s and σ. Typical values of the error function after optimization were on the order of 10–8.
Estimation of Conformational Entropy. The WHAM method was utilized to build an estimate of the density of states, Ωn(iHb, Ej), for the peptides as a function of the number of α-helical hydrogen bonds iHb and potential energy level Ej. The partition function, qn, of Ac-Alan-NMe at a given temperature, therefore, in terms of the estimated Ωn (iHb, Ej) is simply
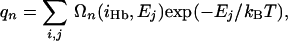 |
[8] |
where iHb corresponds to i hydrogen bonds formed, Ej is the jth energy level, T is the temperature, and kB is the Boltzmann constant. From the partition function the remaining thermodynamic properties follow simply. The total entropy of the peptide at T is given as Stotal n (T) = (En(T) – An(T))/T, where the energy is En(T) = kBT2 (∂ ln qn/∂T) and the free energy is An(T) = –kBT ln(qn). The conformational entropy of the peptide at T is
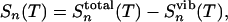 |
[9] |
with the vibrational entropy
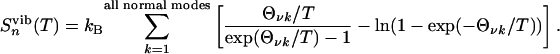 |
Θνk = hνk/kB is the vibrational temperature and h is Planck's constant (26). Each normal mode, νk, is obtained from an energy-minimized, fully α-helical conformation of the molecule. Alternatively, one could consider the ensemble of peptide conformations at each temperature and carry out vibrational analysis for each member of these ensembles to remove the vibrational contribution to the entropy. Such a calculation is very intensive, and if (as we verified in the present case) little difference is seen between the vibrational entropy of helical and extended conformations, this additional computational effort is unnecessary.
It should be noted that the estimated Ωn(iHb, Ej), and therefore En(T), An(T), and Sn(T), are independent of the bin size used in i and j, as long as i and j are not too small and the bins span the entire reaction coordinate and energy range. We use n – 1 for i binning as a natural choice, because the possible number of α-helical hydrogen bonds in Ac-Alan-NMe ranges from 0 to n – 2. For j, we trace the lowest and highest energies among the REMD trajectory and use fifty equally spaced bins between these two values.
Results and Discussion
Analysis of ZB Parameters. In Fig. 1, we show the temperature dependence of the helicity of Ac-Ala15-NMe. The results from our model for the fraction of α-helical hydrogen bonds, θ (Fig. 1a), is slightly higher than experiment and the “folding transition” is shifted to a higher temperature (≈350 K). However, the overall shape of this temperature-dependent curve mimics experiment. The average length of helical segments is shown in Fig. 1b and suggests that as the temperature is lowered below 300 K, the length of contiguous helix increases significantly. The number of α-helical hydrogen bond segments is shown in Fig. 1c. This property is defined as those hydrogen-bonded segments separated by a series of non-hydrogen-bonded peptide units of three or more, because at least three broken hydrogen bonds are needed to break a helix completely into two segments. Although there is an overall temperature shift, the CHARMM/GB model yields a reasonable distribution of polypeptide conformations compared to experiments on similar polypeptides (27, 28).
Fig. 1.

The temperature dependent helicity observed in REMD simulations of Ac-Ala15-NMe. (a) The fractional helicity, θ. (b) The average length of a helical segment, 〈k〉/〈j〉. (c) The average number of helical segments, 〈j〉.
The fitted values of the ZB parameter s(T) for the peptide Ac-Ala15-NMe from sixteen temperature windows are plotted in Fig. 2 against θ, 〈k〉/〈j〉, and 〈j〉. Eqs. 5–7 are also plotted for fixed σ as solid lines for guidance. The red curves are those from the commonly used large-n approximation, in which θ and 〈k〉/〈j〉 are n-independent. In this approximation 〈k〉/〈j〉 shoots up beyond n – 2 (= 13 in this case), the longest possible length of a helix in the peptide, whereas the “exact” 〈k〉/〈j〉 (Eq. 7) with small σ quickly converges to n – 2 as s increases. Also, the approximate 〈j〉 does not converge to 1 along s as it is supposed to do. It is clear that for the peptides used here the large-n approximation is not appropriate or applicable.
Fig. 2.

The ZB helix propagation-parameter, s(T), for Ac-Ala15-NMe plotted against: θ (a), 〈k〉/〈j〉 (b), and 〈j〉 (c). The solid thin lines are from Eqs. 6 (a), 7 (b), and 5 (c) with fixed σ of 0.001, 0.01, 0.1, 0.5, and 1.0. The thin red lines are obtained in the same way, except when using the large-n approximation instead (equations not shown here). The thick lines with filled circles are from our simulations.
In Fig. 3 a and b, the fitted values of the s(T) and σ(T) of Ac-Alan-NMe are plotted against temperature. Both s(T) and σ(T) are basically unchanged for different n, consistent with the isolated-pair hypothesis, although some deviations are observed for n = 6 and7in s(T) and 6–9in σ(T). The monotonic decrease of s(T) with temperature and the specific value of s = 1.5 at room temperature agrees well with experiments on short peptides (27, 29) but is counter to the findings for s of alanine by polymer-based experiments (30) and data-mining methods (31). The σ(T) increases until the transition temperature has a bump there, and then reaches a plateau. At lower temperatures σ(T) is of the same order of magnitude as the fixed σ determined by other groups (29, 30) but is an order of magnitude too large at other temperatures.
Fig. 3.

The parameters s(T) and σ(T) calculated for Ac-Alan-NMe of n = 6–15 and 20. The sets relatively apart from others are linked by solid lines: (a) s(T) of n = 6 and 7 (bottom to top); (b) σ(T) of n = 6–9 (top to bottom). (c) The free energy change of the alanine residue from the manifold of coil states to α-helical states against the temperature, calculated from s(T) of Ac-Ala15-NMe.
From s(T) we can estimate thermodynamic properties of α-helix formation for alanine. In Fig. 3c, the free energy change of an alanine residue from a coil manifold of states to α-helical states, ΔG = –RTln(s(T)), is plotted against temperature. The regression line to this data over a range of temperatures gives the enthalpy change, ΔH, as the intercept and the entropy change, ΔS, as the negative slope, assuming the ΔS is constant within the temperature range. From the range between 200 and 386.6 K, ΔH and ΔS are estimated to be –0.83 kcal·mol–1 and –2.11 cal·mol–1·K–1, respectively. By calculating σ ≈ exp(3ΔS/kB), σ is found to be 0.042, which matches well with σ(T) in the same temperature range (see Fig. 3b). The ΔH by calorimetry is –0.9 ± 0.1 kcal·mol–1 (32), and as Kallenbach and coworkers (33) calculated, this leads to ΔS of –2.20 ± 0.37 cal·mol–1·K–1. Our results are in agreement with these. We note that unfolded conformations from our simulations are also observed to sample the polyproline II helical region observed by Kallenbach and coworkers; however, the population of such conformations is small. The meaning of the regression line for high temperatures is not clear. It gives ΔS of 1.05 cal·mol–1·K–1 for ≥400 K, which results in a disturbingly small σ of 1.3 × 10–7.
Similar values for s(T) and larger-than-experiment σ(T) were also observed in other simulations. Using the CHARMM force field with explicit solvent, a large equilibrium constant (equivalent to σs) was obtained for Ac-Ala3-NMe (34), and similar values of s were found for Ac-Alan-NMe (3 ≤ n ≤ 15) at ambient temperature (35). Also, Mitsutake and Okamoto (36) calculated s(T) and σ(T) by using the large-n approximation for short peptides by a force field based on ECEEP/2. Their σ(T) is even larger than that we observe, but it would be about the same if fitted without this approximation (cf. Fig. 2b). Furthermore, Garcia and coworkers (37, 38) observed a similarly larger σ value for polyalanines in both explicit and implicit (GB) water when using the AMBER force field. Because similar s and σ values were obtained from a number of different workers using different force fields, the energy landscapes representing polypeptides from these force fields are likely to be similar, furthering the suggestion that our findings regarding the isolated-pair hypothesis (see next section) are not CHARMM/GB specific.
The experimentally obtained σ itself varies by more than an order of magnitude (29, 30); consequently, it is difficult to judge the causes of the difference between the simulated σ and experimental values. One possible reason is that these force fields favor helical conformations, consistent with findings by Garcia and Sanbonmatsu (37), who modified the AMBER force field to reduce helical propensity and found σ values closer to experiment. This finding found further support when Zaman et al. (7) showed that the modified AMBER, as well as OPLS-AA, favors helical conformations less than other force fields. These findings suggest that OPLS-AA would probably yield a σ value for helix initiation closer to experiment. In addition, our observation that the current force fields underestimate the entropy cost of helix initiation could be a result of inadequate representations of the hydrophobic effect in current models with implicit (38) and explicit solvent representations (34, 35, 38).
Conformational Entropy. To examine the length and composition dependence of the peptide entropy, we calculate the conformational entropy at a given temperature T, Sn(T), by Eq. 9. This is shown in Fig. 4 for both the Ac-Alan-NMe peptides and the C-peptide homologues. The curves are shifted so that the minimum value matches zero, which is appropriate if only one (or a few) states are sampled at the lowest temperatures, as we see in our simulations. These curves should reach plateau values at low and high temperatures, and this behavior does occur. However, because of our approximate removal of vibrational entropy as described above, some additional curvature occurs at these extremes. The general feature that is quite clear is that the longer the molecule the larger the entropy, and for each molecule, the higher the temperature the larger the entropy, as expected. Fig. 5 a and b display Sn(T) versus n at 300, 400, and 500 K. For all temperatures, Sn(T) for both Ac-Alan-NMe and the C-peptide homologues is linear in n (correlation coefficient of 0.95 or higher, except 0.71 of Ac-Alan-NMe at 300 K).
Fig. 4.

(a) The increase of conformational entropy of Ac-Alan-NMe with temperature, where n is 6–15 and 20. (b) The increase of conformational entropy of the C-peptide homologues (CpC6, CpN7, Cp13, and Cp26) versus the chain length and temperature.
Fig. 5.

(a) The increase of conformational entropy of Ac-Alan-NMe versus chain length n at the temperatures 300, 400, and 500 K. The regression lines are Sn(300 K) = 0.0325 + 0.00151n, Sn(400 K) = 0.0269 + 0.00494n, and Sn(500 K) = 0.0060 + 0.0731n. (b) The increase of conformational entropy of the C-peptide homologues versus chain length at the temperatures 300, 400, and 500 K. The regression lines are Sn(300 K) = 0.0125 + 0.0062n, Sn(400 K) = 0.0041 + 0.00852n, and Sn(500 K) = –0.0186 + 0.0112n. (c) The increase of conformational entropy of Ac-Alan-NMe for n = 3–5 at the temperatures 300 and 400 K. The red regression lines are of n = 3–5 for the same temperature. The black lines are identical to the regression lines in a.(d) The conformational entropy per residue versus temperature for Ac-Alan-NMe and the C-peptide homologues.
If Eq. 2, Flory's hypothesis, does not hold, or nonlocal steric clashes are not negligible, Eq. 2 should fall short of the equality as n increases. This failure should be more obvious in the C-peptide homologues than in Ac-Alan-NMe, because the larger side chains may cause clashes even if the main chain portions do not. These plots show a linear n dependency; hence, the effects of nonlocal steric clashes must be minimal. Although n is limited to 20 (Ac-Alan-NMe) or 26 (the C-peptide homologues) and is not evaluated for some of the integers in those ranges, the clear proportionality between Sn(T) and n in Fig. 5 a and b indicates that Eq. 2 holds and that nmin is not within this range of n.
What, then, is nmin? Its value must be ≤6, because nmin is the smallest n for which we observe proportionality between Sn and n. On the other hand, if non-nearest-neighbor effects are obvious, nmin should appear somewhere around 10, because nmin is the minimum length at which the “end effect” of non-nearest-neighbor local clashes becomes negligible. To further explore this question, the same calculations were carried out for n = 3–5, and Sn(T) of Ac-Alan-NMe at 300 and 400 K are displayed as the red filled and small black circles with the red regression lines in Fig. 5 c. The black lines are identical to those in Fig. 5a. All the red circles fall on or below the black regression lines; however, the points of n = 4 and 5 are closer to the regression lines than those of n = 3. Note that the red lines have steeper slopes than the black lines, just like S1,1 of Eq. 1 and  of Eq. 2. Also, the intercepts of these red lines are close to zero, whereas those of the black lines have positive intercepts (>0.02 kcal·mol–1·K–1), like c of Eq. 2. At 500 K, the regression line for n = 6–20 has a near zero intercept (Fig. 5 a), which makes sense because the majority of conformations are rather extended and local steric effects become less significant at this high temperature. These observations suggest that, basically, Eq. 1 holds for the region of n ≤ 5 and Eq. 2 holds for 6 ≤ n; thus, nmin is 6.
of Eq. 2. Also, the intercepts of these red lines are close to zero, whereas those of the black lines have positive intercepts (>0.02 kcal·mol–1·K–1), like c of Eq. 2. At 500 K, the regression line for n = 6–20 has a near zero intercept (Fig. 5 a), which makes sense because the majority of conformations are rather extended and local steric effects become less significant at this high temperature. These observations suggest that, basically, Eq. 1 holds for the region of n ≤ 5 and Eq. 2 holds for 6 ≤ n; thus, nmin is 6.
This finding is in general agreement with the results from Rose and coworkers that indicate the failure of Flory's hypothesis (Eq. 1) in Alan of n = 5 (8). However, their results suggest a growing divergence from Eq. 2 as n increases. Our findings suggest that this is not the case. The observations by van Gunstern and coworkers (9), that the number of conformers observed will not exponentially increase with the chain length, is counterintuitive and interesting. In table 1 of their article, however, there seems to be a correlation between the number of conformers and simulation length for (almost) the same chain length, possibly indicating insufficient conformational sampling.
From the findings we present above, we conclude that Flory's isolated-pair hypothesis does not fail in the form of Eq. 2, at least for Ac-Alan-NMe and the C-peptide homologues with the CHARMM/GB force field. There do exist non-nearest-neighbor, local steric effects (i.e., c in Eq. 2 is not zero), but nonlocal steric effects are not significant. However, we note that calculations for Ac-Alan-NMe of even larger n or proteins with topologies other than α-helical have not been examined, and there remains the possible presence of the nonlocal, remote steric effects in these systems. Clearly, such effects will occur as a result of the finite volume of the peptide chain, as peptides long enough to achieve their persistence length [20 Å for polyalanine (39)] under specific solvent conditions are accessed.
The slope of the regression lines in Fig. 5 a and b give the average conformational entropy per residue. In Fig. 5 d, we display S(T) per residue for Ac-Alan-NMe and the C peptide. With the assumption of additivity between the entropy of the main chain and the side chain, the entropy difference between the two curves gives the average side chain entropy per residue of the C peptide. For instance, at 300 K the entropy difference of 0.00475 kcal·mol–1·K–1 corresponds to about 11 conformations per residue. Because there are a total of 31 χ angles in the C peptide, the average number of dihedral angles per residue is 2.38, leading to about 2.7 conformations per χ angle.
Conclusions
We have shown that the conformational entropy of capped polyalanine, Ac-Alan-NMe, is proportional to the chain length, n, over a wide range of temperatures and for chain lengths of n = 3–20. The proportionality between chain entropy and chain length is also observed in a set of polypeptides with side chains, the C-peptide homologues that maintain the same composition of residue types. The ZB parameters s(T) and σ(T) of Ac-Alan-NMe are basically independent to n, in accordance with the observed entropy-chain length proportionality. These results support the validity of the isolated-pair hypothesis with local steric effects.
Acknowledgments
Y.Z.O. thanks J. Karanicolas and K. V. Damodaran for insightful discussions and technical assistance. The REMD algorithms used here are based on methods developed by M. Feig and J. Karanicolas. These methods are available through the National Institutes of Health funded research resource (RR12255) Multiscale Modeling Tools in Structural Biology at http://mmtsb.scripps.edu. Financial support from the National Institute of Health (Grant GM48805) is greatly appreciated.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: REMD, replica exchange molecular dynamics; ZB, Zimm–Bragg.
References
- 1.Flory, P. J. (1969) Statistical Mechanics of Chain Molecules (Wiley, New York).
- 2.Levinthal, C. (1969) in Mossbauer Spectroscopy in Biological Systems, eds. Debrunner, P., Tsibris, J. C. M. & Muenck, E. (Univ. of Illinois Press, Urbana), pp. 22–24.
- 3.Bryngelson, J. D., Onuchic, J. N. & Wolynes, P. G. (1995) Proteins 21, 167–195. [DOI] [PubMed] [Google Scholar]
- 4.Shortle, D. & Ackerman, M. S. (2001) Science 293, 487–489. [DOI] [PubMed] [Google Scholar]
- 5.Lietzow, M. A., Jamin, M., Dyson, H. J. & Wright, P. E. (2002) J. Mol. Biol. 322, 655–662. [DOI] [PubMed] [Google Scholar]
- 6.Zagrovic, B., Snow, C. D., Khaliq, S., Shirts, M. R. & Pande, V. S. (2002) J. Mol. Biol. 323, 153–164. [DOI] [PubMed] [Google Scholar]
- 7.Zaman, M. H., Shen, M. Y., Berry, R. S., Freed, K. F. & Sosnick, T. R. (2003) J. Mol. Biol. 331, 693–711. [DOI] [PubMed] [Google Scholar]
- 8.Pappu, R. V., Srinivasan, R. & Rose, G. D. (2000) Proc. Natl. Acad. Sci. USA 97, 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Gunsteren, W. F., Büergi, R., Peter, C. & Daura, X. (2001) Angew. Chem. Int. Ed. Engl. 40, 351–355. [PubMed] [Google Scholar]
- 10.Sugita, Y. & Okamoto, Y. (1999) Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- 11.Dominy, B. N. & Brooks, C. L., III. (1999) J. Phys. Chem. B 103, 3765–3773. [Google Scholar]
- 12.Zimm, B. H. & Bragg, J. K. (1959) J. Chem. Phys. 31, 526–535. [Google Scholar]
- 13.Ferrenberg, A. M. & Swendsen, R. H. (1989) Phys. Rev. Lett. 63, 1195–1198. [DOI] [PubMed] [Google Scholar]
- 14.Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A. & Rosenberg, J. M. (1992) J. Comp. Chem. 13, 1011–1021. [Google Scholar]
- 15.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comp. Chem. 4, 187–217. [Google Scholar]
- 16.Neria, E., Fischer, S. & Karplus, M. (1996) J. Chem. Phys. 105, 1902–1921. [Google Scholar]
- 17.Vijakumar, S., Vishveshwara, S., Ravishanker, G. & Beveridge, D. L. (1994) Am. Chem. Soc. Symp. Ser. 569, 175–193. [Google Scholar]
- 18.Swendsen, R. H. & Wang, J. S. (1986) Phys. Rev. Lett. 57, 2607–2609. [DOI] [PubMed] [Google Scholar]
- 19.Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. (1977) J. Comput. Phys. 23, 327–341. [Google Scholar]
- 20.Feig, M., Karanicolas, J. & Brooks, C. L., III (2003) J. Mol. Graph. Model, in press. [DOI] [PubMed]
- 21.Cantor, C. R. & Schimmel, P. R. (1980) Biophysical Chemistry (Freeman, New York), Vol. 3, pp. 1041–1074. [Google Scholar]
- 22.Qian, H. & Schellman, J. A. (1992) J. Phys. Chem. 96, 3987–3994. [Google Scholar]
- 23.von Dreele, P. H., Lotan, N., Ananthanarayanan, V. S., Andreatta, R. H., Poland, D. & Scheraga, H. A. (1971) Macromolecules 4, 408–424. [Google Scholar]
- 24.Scholtz, J. M., Qian, H., York, E. J., Stewart, J. M. & Baldwin, R. L. (1991) Biopolymers 31, 1463–1470. [DOI] [PubMed] [Google Scholar]
- 25.Yang, J., Zhao, K., Gong, Y., Vologodskii, A. & Kallenbach, N. R. (1998) J. Am. Chem. Soc. 120, 10646–10652. [Google Scholar]
- 26.McQuarrie, D. A. (2000) Statistical Mechanics (University Science Books, Sausalito, CA).
- 27.Marqusee, S., Robbins, V. H. & Baldwin, R. L. (1989) Proc. Natl. Acad. Sci. USA 86, 5286–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalongo, W., Dugad, L. & Stellwagen, E. (1994) J. Am. Chem. Soc. 116, 8288–8293. [Google Scholar]
- 29.Chakrabartty, A., Kortemme, T. & Baldwin, R. L. (1994) Protein Sci. 3, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Platzer, K. E. B., Ananthanarayanan, V. S., Andreatta, R. H. & Scheraga, H. A. (1972) Macromolecules 5, 177–187. [Google Scholar]
- 31.Ooi, T. & Oobatake, M. (1991) Proc. Natl. Acad. Sci. USA 88, 2859–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez, M. M., Chin, D.-H., Baldwin, R. L. & Makhatadze, G. I. (2002) Proc. Natl. Acad. Sci. USA 99, 1298–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi, Z., Olson, C. A., Rose, G. D., Baldwin, R. L. & Kallenbach, N. R. (2002) Proc. Natl. Acad. Sci. USA 99, 9190–9195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tobias, D. J. & Brooks, C. L., III (1991) Biochemistry 30, 6059–6070. [DOI] [PubMed] [Google Scholar]
- 35.Young, W. S. & Brooks, C. L., III (1996) J. Mol. Biol. 259, 560–572. [DOI] [PubMed] [Google Scholar]
- 36.Mitsutake, A. & Okamoto, Y. (2000) J. Chem. Phys. 112, 10638–10647. [Google Scholar]
- 37.Garcia, A. E. & Sanbonmatsu, K. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nymeyer, H. & Garcia, A. (2002) Prot. Sci. 11, Suppl. 1, 205. [Google Scholar]
- 39.Cantor, C. R. & Schimmel, P. R. (1980) Biophysical Chemistry (Freeman, New York), Vol. 3, pp. 1013. [Google Scholar]