

Abstract
Autoimmunity affects multiple glands in the endocrine system. Animal models and human studies highlight the importance of alleles in HLA (human leukocyte antigen)-like molecules determining tissue specific targeting that with the loss of tolerance leads to organ specific autoimmunity. Disorders such as type 1A diabetes, Grave's disease, Hashimoto's thyroiditis, Addison's disease, and many others result from autoimmune mediated tissue destruction. Each of these disorders can be divided into stages beginning with genetic susceptibility, environmental triggers, active autoimmunity, and finally metabolic derangements with overt symptoms of disease. With an increased understanding of the immunogenetics and immunopathogenesis of endocrine autoimmune disorders, immunotherapies are becoming prevalent, especially in type 1A diabetes. Immunotherapies are being used more in multiple subspecialty fields to halt disease progression. While therapies for autoimmune disorders stop the progress of an immune response, immunomodulatory therapies for cancer and chronic infections can also provoke an unwanted immune response. As a result, there are now iatrogenic autoimmune disorders arising from the treatment of chronic viral infections and malignancies.
Keywords: Type 1 diabetes, HLA, autoantibodies, immunotherapy, Addison's disease, APS-1, APS-2, Grave's disease, polyendocrine autoimmunity, iatrogenic autoimmunity
Introduction
Multiple endocrine diseases are immune mediated and now predictable. Autoimmune disorders can cluster in individuals and their relatives. A family history of autoimmunity and screening for autoantibodies can identify at risk individuals. Knowledge of these disorders along with disease associations can lead to earlier diagnosis and management resulting in less morbidity and in some cases mortality. We will review endocrine organ specific autoimmune diseases, autoimmune polyendocrine syndromes, and iatrogenic endocrine autoimmune disorders with an emphasis on immunopathogenesis hopefully leading to immunotherapy for standard and experimental clinical care.
Diabetes Mellitus
Background
By the American Diabetes Association classification, type 1A diabetes is the immune mediated form of diabetes, while type 1B represents non-immune mediated forms of diabetes with beta cell destruction leading to absolute insulin deficiency (1). There are additional forms of insulin dependent diabetes with defined etiologies. Type 2 diabetes is overall the most common form of diabetes and is characterized by insulin resistance and less beta cell loss. In the United States with a population of approximately 300 million people, there are about 1.5 million individuals with type 1A diabetes and of these approximately 170,000 are less than age 20. Type 1A diabetes incidence, similar to other immune mediated diseases such as asthma, is doubling approximately every 20 years (2). Diabetes almost always develops in the setting of genetic susceptibility best defined by polymorphisms of HLA alleles (3). Currently there is no known cure for type 1A diabetes and treatment for the disease consists of lifelong insulin administration. Immunotherapies aimed at preventing beta cell destruction at the time of clinical onset are actively being studied.
Genetic Susceptibility
There are monogenic and polygenic forms of both immune and non-immune mediated diabetes. Monogenic non-immune diabetes includes permanent neonatal diabetes mellitus (PND), transient neonatal diabetes (TND), and maturity-onset diabetes of the young (MODY). In general children with these disorders lack all anti-islet autoantibodies and therefore autoantibody assays can aid in identifying children to consider for genetic analysis. It is important to identify individuals who do not have type 1A diabetes with estimates that approximately 1.5% of children presenting with diabetes have monogenic forms of diabetes. Several monogenic forms of diabetes are reported to be better treated with sulfonylurea therapy than insulin (e.g. mutations of the ATP-sensitive beta cell selective potassium channels and HNF-1alpha mutations) (4) and diabetes due to glucokinase mutations require no therapy at all. Approximately one half of permanent neonatal diabetes is due to mutations of the proinsulin gene that leads to beta cell loss. Two monogenic syndromes with immune mediated diabetes are APS-1 and IPEX syndromes that will be discussed subsequently. The rest of this section will focus on the more common polygenic form of diabetes, type 1A diabetes.
Approximately 1/300 individuals from the general population develop type 1A diabetes compared to 1/20 siblings of patients with type 1A diabetes. The concordance rate for monozygotic twins with type 1A diabetes is greater than 60% (Figure 1), and a recent analysis of long term twin data indicates that there is no age that an initially discordant monozygotic twin is no longer at risk (5). Compared to monozygotic twins, initially discordant dizygotic twins are less often positive for anti-islet autoantibodies than non-twin siblings (6). Offspring of a father with type 1A diabetes have a greater risk compared to offspring of a mother (7).
Figure 1.
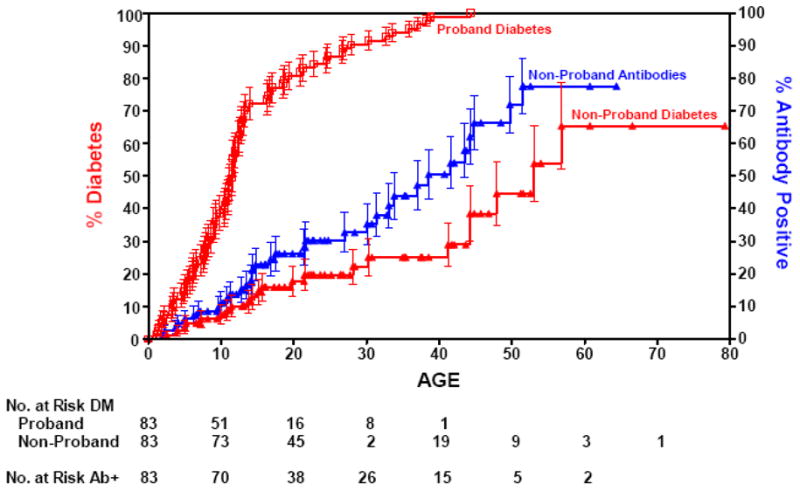
Progression to diabetes of initially discordant monozygotic twin siblings of patients with type 1 diabetes, illustrating progressive conversion to diabetes. Approximately 80% become concordant for expression of anti-islet autoantibodies. From Redondo et al Concordance for Islet Autoimmunity among Monozygotic Twins, New England Journal of Medicine, December 25, 2008.
The major determinant of genetic susceptibility to type 1A diabetes is conferred by genes in the HLA complex, which is divided into three regions: class I, II, and III. Alleles of the class II genes, DQ and DR (and to a lesser extent DP), are the most important determinants of type 1A diabetes. These class II molecules are expressed on antigen presenting cells (macrophages, dendritic cells, and B cells) and present antigens to CD4+ T lymphocytes. DR3 and DR4 haplotypes are strongly associated with type 1A diabetes with more than 90% of people with type 1A diabetes possessing one or both of these haplotypes versus 40% of the US population (8). Each unique amino acid sequence of DR and DQ is given a number. Since DRA does not vary, haplotypes can be defined by specific DRB, DQA, and DQB alleles. The highest risk DR4 haplotypes vary at both DR and DQ with DRB1*0401, DRB1*0402, DRB1*0405 and DQA1*0301, DQB1*0302. DR3 haplotypes are almost always conserved with DRB1*03 combined with DQA1*0501, DQB1*0201. The highest risk genotype has both DR3 DQB1*0201/DR4 DQB1*0302. This genotype occurs in 30 to 50% of children developing type 1A diabetes; approximately 50% of children developing type 1A diabetes before the age of five are DR3/4 heterozygotes versus 30% of young adults presenting with type 1A diabetes and 2.4% of the general population in Denver, Colorado. The excess risk for heterozygous haplotypes may be related to the trans-encoded DQ molecule (DQA and DQB encoded by different chromosomes) that can form in DR3/4 heterozygous individuals, namely DQA1*0501/DQB1*0302 (3).
In addition to HLA genes, many genetic loci contributing to diabetes risk have been implicated through genome wide association studies (Figure 2) (9), which involves analyzing thousands of single nucleotide polymorphisms from large populations to find alleles associated with a particular disease. These alleles can increase risk, i.e. high risk alleles, or protect against a certain disease. While HLA alleles confer the highest risk, multiple non-HLA genetic polymorphisms modify disease risk. The group of longer variable number of tandem nucleotide repeats (VNTR) 5′ of the insulin gene protects against diabetes. The decreased diabetes risk is associated with greater insulin message in the thymus and resultant deletion of autoreactive T cells in the thymus (10). Alleles of other identified genes primarily influence immune function such as the protein tyrosine phosphatase, non-receptor 22 (PTPN22) that regulates T cell receptor signaling. The R620W single amino acid change of PTPN22 decreases T cell receptor signaling (gain of function) and increases the risk of many autoimmune disorders including type 1A diabetes, Addison's disease, Grave's disease, rheumatoid arthritis, and others (11). Recently, a further GWAS analysis identified two additional loci, UBASH3A and BACH2, associated with type 1A diabetes; loci having OR = 1.16 and 1.13, respectively. Both of these loci were validated from two separate populations, the Wellcome Trust Case-Control Consortium and the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort (12).
Figure 2.
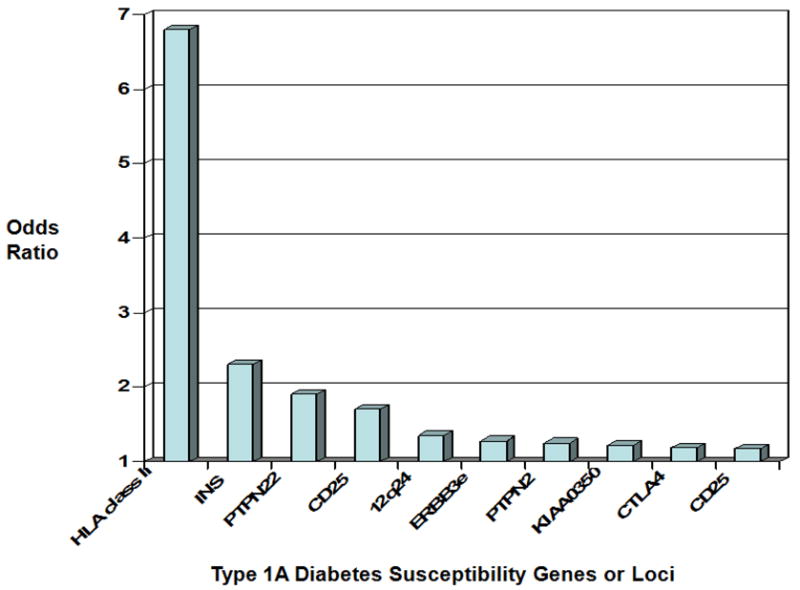
Summary of subsets of confirmed loci from whole genome screens associated with type 1A diabetes and their odds ratio (from Teaching Slides at www.barbaradaviscenter.org). Modified from Todd et al. Robust Association of four new chromosomes regions from genome-wide analyses of type 1 diabetes, Nature Genetics, July 6, 2007. HLA = human leukocyte antigen; INS = insulin, PTPN22 = protein tyrosine phosphatase non-receptor 22; CD 25 is also known as interleukin-2 receptor alpha chain; ERBB3e = an unidentified gene at 12q; PTPN2 = protein tyrosine phosphatase non-receptor 2; KIAA 0350 = a lectin-like gene; CTLA 4 = cytotoxic T lymphocyte associated antigen 4.
Environmental Factors
The incidence of type 1A diabetes is increasing dramatically at a rate of 3-5% per year for the last 50 years and this rapid increase cannot be explained by genetics. There is no evidence that the epidemic of type 1A diabetes has leveled off in Finland, one of the countries with the highest incidence. The increase in incidence of diabetes is most marked in children less than 5 years of age (13). These observations suggest that environmental factors increasing diabetes risk have been introduced or factors decreasing risk have been removed. The Diabetes Autoimmunity Study of the Young (DAISY) found no evidence that bovine milk products, vaccinations, or enteroviral infections contribute to diabetes risk but has implicated decreased omega-3 fatty acid intake (14) and early cereal introduction. There appears to be a window from the ages of 4 to 6 months where initial cereal introduction is not associated with an increased risk of islet autoimmunity, as children that received initial cereal exposure less than 3 months or after 7 months of age had a higher risk for developing islet autoantibodies (15). Omega-3 fatty acid supplementation in children with an increased genetic risk of diabetes was associated with a reduced risk of islet autoimmunity (16). There have been several studies showing an association between vitamin D supplementation during the first year of life and a reduced risk of diabetes. A large prospective study of islet autoimmunity failed to confirm an association between type 1A diabetes risk and serum alpha and gamma-tocopherol concentrations, the principal forms of vitamin E in the diet and human tissues (17). Epigenetic influences are likely to be evaluated in future studies for diabetes risk. Hypermethylation has been associated with dietary supplements (18), and there is discordance of methylation between monozygotic twins increasing with age (19).
Pathogenesis
Type 1A diabetes is a T cell mediated disease in which T cells infiltrate the islets causing insulitis and ultimately β-cell death, decreased insulin production, and insulin dependent diabetes. In a genetically susceptible individual the development of diabetes occurs in stages (Figure 3). The presence of autoantibodies against islet cell antigens is the first indication for the development of diabetes and individuals retain sufficient β-cell mass initially for euglycemia. There are currently four autoantibodies used to predict the development of type 1A diabetes: antibodies against glutamic acid decarboxylase (GAD65), a tyrosine phosphatase-like protein (ICA512 also termed IA-2), insulin, and the recently discovered zinc T8 transporter (ZnT8) (20). Following autoantibody development there is progressive loss of insulin release as the autoimmune response progresses. During later stages patients progressively develop subclinical hyperglycemia. In the final stages of development, decreased C-peptide levels cause patients to present with overt signs of diabetes.
Figure 3.

Hypothetical stages and loss of beta cells in an individual progressing to type 1A diabetes (from Teaching Slides at www.barbaradaviscenter.org). Reproduced with permission from Eisenbarth, GS.
Much of what we know about the autoimmune process in diabetes comes from the study of animal models. The nonobese diabetic (NOD) mouse is a model in which type 1A diabetes and sialitis develop spontaneously and the biobreeding (BB) rat develops both diabetes and thyroiditis. As in humans, both models have alleles of genes within the MHC complex that influence antigen presentation to T lymphocytes and development of autoimmunity. One self epitope in NOD mice has been shown to be a peptide of the insulin B chain, amino acids 9-23, that is recognized by autoreactive T lymphocytes (21). During disease progression, activated T cells invade the pancreas and destroy β-cells, resulting in insulin deficiency. Once β-cell destruction is initiated, other antigens become targets for the immune response including islet glucose-related phosphatase which is β-cell specific (22). Adoptive transfer of T cells from a diabetic mouse to an unaffected mouse results in diabetes. These animal models highlight the importance of having a genetic predisposition resulting in impaired immune regulation for autoimmunity to develop.
In humans, a recent study examining post mortem pancreas specimens from recent onset type 1A diabetics showed a temporal pattern of immune cell infiltration. Initially the inflammatory infiltrate consisted of CD8+ cytotoxic T cells and macrophages (23). CD20+ B cells were not present in early insulitis but appeared in larger numbers as beta cell death progressed. CD4+ helper T cells were present throughout insulitis but not as prevalent as cytotoxic T cells and macrophages. The exact mechanism of β-cell death remains to be elucidated but likely involves cytokines, Fas-Fas ligand induced cell death, and CD8+ T cell mediated cytotoxicity.
Diagnosis and Prediction
The hallmark of type 1A diabetes is the autoimmune destruction of the pancreatic β-cells by T cells. However, diagnosis is not made with T cell assays as they are not as well developed or standardized compared to autoantibody assays. The lack of dependable assays for autoreactive T cells leads to the reliance of autoantibodies as the initial laboratory evaluation to detect an immune response against pancreatic β-cells and distinguishing type 1 from type 2 diabetes. There are several clinical scenarios where the determination of autoantibodies is relevant. Children with transient hyperglycemia and adults presenting with hyperglycemia can present diagnostic dilemmas. Most children with transient hyperglycemia will remain normal but a subset develop type 1A diabetes; those with autoantibodies almost always progress to diabetes. Adults are much more likely to have type 2 diabetes; but 5-10% express islet autoantibodies and these individuals progress more rapidly to insulin dependence. Independent of autoantibodies, routine monitoring of blood glucose is important to prevent metabolic decompensation that can occur with many forms of diabetes.
Type 1A diabetes is a predictable disease. Autoantibodies against GAD65, insulin, IA-2, and the recently identified ZnT8 are current markers for type 1A diabetes. Relatives of patients with type 1A diabetes have been studied in detail. Expression of two or more autoantibodies (insulin, GAD65, or IA-2) has a positive predictive value of greater than 90% among relatives of patient with type 1 diabetes (Figure 4); this holds true for the general population as well (24). A single autoantibody carries a risk of approximately 20% (25;26). With the addition of a fourth autoantibody, ZnT8, prediction will only improve as 26% of patients with autoantibody negative type 1A diabetes in the DAISY study were found to have the ZnT8 autoantibodies (27).
Figure 4.

Progression to diabetes versus number of autoantibodies (GAD, ICA5112, Insulin). From Verge, CF et al. Prediction of type 1 diabetes in first-degree relatives using a combination of insulin, GAD, ICA512bdc/IA-2 autoantibodies, Diabetes, July 1996.
Treatment
The mainstay of type 1A diabetes treatment is insulin therapy. Over the last several years multiple advances in insulin preparation, insulin delivery, and glucose monitoring have considerably improved treatment. Multiple analog insulins provide either a faster onset of action or longer duration and decrease the variability of insulin absorption. Insulin pumps allow for a more physiologic administration of insulin throughout the day. Continuous glucose monitoring systems (CGMS) have been developed and measure interstial fluid glucose levels. CGMS assess blood glucose trends and provide alarms for high and low blood glucose levels. There is still a need to both calibrate the monitors and confirm low blood glucose values with fingerstick glucose determination. There is research underway with CGMS monitors controlling insulin delivery from insulin pumps.
Despite treatment with insulin therapy long-term complications, including nephropathy, retinopathy, neuropathy, and cardiovascular disease, can result. While the progress to complete insulin dependence occurs quickly after clinical onset, initially after diagnosis the pancreas is able to produce a significant amount of insulin (28); at this time immunologic intervention can save beta cell function and reduce reliance on insulin. Two international networks conducting immunotherapy trials, the Immune Tolerance Network and TrialNet, have been established. Immunotherapies in type 1A diabetes are aimed at altering the underlying immune process that results in beta cell loss. These therapies consist of agents that are non-antigen specific and those that are antigen specific. Non-antigen specific therapies target various components of the immune system and include those directed against T cells (anti-CD3 monoclonal antibodies, anti-thymocyte globulin, and cyclosporine), B cells (anti-CD20 monoclonal antibodies), and other components of the immune system (Table I). Antigen based therapies are believed to mediate immune tolerance to antigens that result in autoimmunity to beta cells. These therapies include vaccines with GAD, the B chain of insulin, and other insulin peptides (Table II). Many of these therapies have reversed hyperglycemia in the NOD mouse and several therapies show promise in altering the underlying immune process in humans (29).
Table 1. Non-antigen Specific Immunotherapy Trials for New Onset Type 1A Diabetes.
| Agent | Stage of Development | Comments | References and Links |
|---|---|---|---|
| Anti-CD3 Monoclonal Antibodies | Phase II/III | Reduced insulin requirements out to 18 months | (85;86) |
| Anti-CD20 Monoclonal Antibody (Rituximab) | Phase II | Ongoing | www.clinicaltrials.gov/ct2/show/NCT00279305 |
| Anti-thymocyte globulin | Phase I/II | Ongoing | www.clinicaltrials.gov/ct2/show/NCT00515099 |
| Cyclosporine | Multiple Trials | Successful remission but unacceptable side effects | (87) |
| Nicotinamide | Pilot | No effect | (88) |
| Bacille Calmette-Guerin (BCG) | Pilot | No effect | (89) |
| Anti-CD52 (Campath-1H) | Phase I | Withdrawn secondary to adverse events | www.clinicaltrials.gov/ct2/show/NCT00214214 |
| CTLA-4 Ig (Abatacept) | Phase I | Ongoing | www.clinicaltrials.gov/ct2/show/NCT00505375 |
| Mycophenolate and Daclizumab | Phase I | No effect | www.clinicaltrials.gov/ct2/show/NCT00100178 |
Table II. Selected Antigen Specific Immunotherapy Trials for Type 1A Diabetes.
| Agent | Stage of Development | Comments | References and Links |
|---|---|---|---|
| GAD65 | Phase II/III | C-peptide preserved at 18 months | (90) |
| Insulin B Chain in Incomplete Freund's Adjuvant | Phase I | Ongoing | www.clinicaltrials.gov/ct2/show/NCT00057499 |
| Proinsulin-based DNA vaccine (BHT-3021) | Phase I | C-peptide preserved at 12 months | www.bayhilltx.com |
| Oral Insulin | Prevention Trial | Subset with insulin autoantibodies having a potential response | www.clinicaltrials.gov/ct2/show/NCT00419562 |
Insulin Autoimmune Syndrome
The insulin autoimmune syndrome, also known as Hirata disease, results from autoantibodies reacting with insulin. The diagnostic criteria include fasting hypoglycemia without evidence of exogenous insulin administration, high levels of serum immunoreactive insulin, and the presence of high titer insulin autoantibodies. Patients have recurrent and spontaneous hypoglycemia. The insulin autoantibodies can be monoclonal, from a B cell lymphoma, or polyclonal. The polyclonal disorder is strongly associated with the DRB1*0406 haplotype and usually follows therapy with a sulfhydryl-containing medication such as methimizole (an antithyroid drug used to treat Grave's disease) (30).
Autoimmune Thyroid Disease
Background
Autoimmune thyroid disease consists of Grave's disease and Hashimoto's thyroiditis. It is very common, with a prevalence of 5-10% in the general population. Autoantibodies to various enzymes and proteins in the thyroid gland, thyroid peroxidase (TPO) and thyroglobulin (Tg), are the hallmark of autoimmune thyroid disease.
Grave's disease
Background
Grave's disease was first described by Robert Graves in 1835 as being associated with a goiter, palpitations, and exophthalamos. It is now know that the thyroid hormone receptor (TSHR) is stimulated by autoantibodies, thyroid stimulating immunoglobulins (TSI), and thyroid cells are activated resulting in signs and symptoms of hyperthyroidism. The clinical manifestations of hyperthyroidism include a constellation of symptoms comprised of palpitations, tremor, heat intolerance, sweating, anxiety, emotional lability, and weight loss despite a normal to increased appetite. Extrathyroidal manifestations of Grave's disease include Grave's ophthalmopathy and dermatopathy (pretibial myxedema) with little understanding of the cause of these disease components.
Pathogenesis
Grave's disease occurs in genetically susceptible individuals with the HLA alleles contributing the greatest increase in risk, similar to type 1A diabetes. In Caucasians, HLA DR3 (HLA DRB1*03) and DQA1*0501 confer the highest risk (31), while HLA DRB1*0701 is protective (32). For monozygotic twins, the concordance rate is 20% and much lower for dizygotic twins, indicating other susceptibility factors for disease development. Female sex is the main risk factor with smoking, lithium treatment, and low iodine consumption also associated with disease.
Patient with Grave's disease have diffuse lymphocytic infiltration of the thyroid gland and lose tolerance to multiple thyroid antigens, TSHR, thyroglobulin, TPO, and the sodium-iodine cotransporter. Autoantibodies develop when T cells recognize multiple epitopes of the TSHR (33) The autoantibodies can either stimulate or inhibit thyroid hormone secretion. It is a balance of these autoantibodies towards thyroid cell activation that results in hyperthyroidism. Because of these various autoantibodies with differing functions, autoantibody concentrations cannot be correlated to thyroid hormone levels in Graves' patients. Fluctuating antibody titers can result in a Thyroid Yo-Yo syndrome with alternating hyper and hypothyroidism (34). Although TSI cause Grave's disease, the serum antibody concentration can be low or undetectable in some patients. This could be due to assay insensitivity, misdiagnosis of the cause of hyperthyroidism, or intrathyroidal production of autoantibodies (35).
Grave's ophthalmopathy (GO) is associated with Grave's hyperthyroidism but the two diseases can exist independent of one another. GO is clinically evident in 25-50% of patients with hyperthyroidism, and of these patients 3-5% experience severe symptoms. GO results from increased orbital fat and muscle volume within the orbit. Histological analysis of orbital tissue reveals lymphocytic infiltration and inflammatory cytokines IL-4 and IL-10. Smoking is a strong risk factor for GO and worsens the symptoms of eye disease.
The association between Grave's hyperthyroidism and ophthalmopathy suggests that the two disorders result from an autoimmune process to one or more antigens from the thyroid and orbit. Orbital fibroblasts are thought to be the antigenic target in GO. TSHR mRNA and protein expression in orbital fibroblasts has been documented in both normal individuals and GO patients (36). It is possible that a form of TSHR or similar protein is expressed in the orbit and may serve as a cross reactive target for TSI.
Diagnosis
Grave's disease is the most common cause of hyperthyroidism. Diagnosis is made with clinical and biochemical manifestations of hyperthyroidism. Thyroid function tests show a low to suppressed TSH and an elevated thyroxine and triiodothyronine levels. Diagnosis is confirmed with a radioactive iodine uptake and scan (only tested in non-pregnant, non breastfeeding patients) showing increased homogenous uptake. TSI levels aid in the diagnosis but are not confirmatory as patients can have Grave's disease without autoantibodies present. TSI autoantibodies measured in the third trimester of pregnancy are a good predictor of neonatal Grave's disease. During pregnancy thyroid autoantibodies generally decrease, presumably due to secretion of trophoblast factors which are immunosuppressive.
Treatment
Treatment of Grave's disease has changed little over the last 50 years. Treatment options include antithyroid drugs, radioactive iodine, and surgery. Antithyroid drugs block thyroid hormone synthesis but the majority of patients relapse with discontinuation of therapy. Radioactive iodine ablation is the preferred treatment method in the United States. Ablation generally results in iatrogenic hypothyroidism, requiring life long thyroid hormone replacement. Anti-CD20 monoclonal antibody has been tried in a small number of Grave's patients. 20 patients received methimazole for Grave's disease and were rendered euthyroid. 10 patients received anti-CD20 monoclonal antibody infusions during the final three weeks of methimazole treatment. Fewer patients receiving anti-CD20 antibody treatment relapsed at one month (6/10) than those who did not (8/10) (37).
Immunotherapy trials for GO show more promise than for Grave's disease alone. Agents such as anti-CD20 monoclonal antibodies and anti-TNFα monoclonal antibodies have been used. In a pilot study, anti-CD20 monoclonal antibodies improved proptosis, soft tissue changes, and eye motility in 7 patients with moderate to severe GO. None of the treated patients followed to one year had a relapse. This was compared to 15/20 patients responding to methylprednisolone therapy; 10% had relapsed at the conclusion of the study (38). Larger randomized control trials are needed to confirm these results.
Hashimoto's Thyroiditis
Background
Hashimoto's thyroiditis (HT) is the most common endocrine autoimmune condition, affecting up to 10% of the general population. It is characterized by a gradual loss of thyroid function, goiter, and T cell infiltration on histology. HT affects women more frequently than men with a sex ratio of 7:1.
Pathogenesis
HT occurs in genetically susceptible populations but lacks a strong association with HLA. Mutations in the thyroglobulin gene (39) and CTLA-4 are associated with disease (40). T cells play a crucial role in disease pathogenesis by reacting with thyroid antigens and secreting inflammatory cytokines. Autoantibodies develop in HT to thyroid peroxidase, thyroglobulin, and to the TSHR. It is believed that these autoantibodies are secondary to thyroid follicular cell damage induced by T cells. Thyroid peroxidase is the major autoantigen and autoantibodies to TPO are closely associated with disease activity.
Diagnosis and treatment
The diagnosis and treatment of HT has changed very little over the last several decades. Diagnosis is made with clinical (fatigue, weakness, cold intolerance, weight gain, constipation, dry skin, depression, and growth failure or delayed puberty in children) and biochemical manifestations of hypothyroidism. Thyroid function tests show an elevated TSH and a low thyroxine and triiodothyronine levels. Other causes of thyroiditis (postpartum, acute, subacute, and silent) need to be excluded. Treatment is with life long thyroxine replacement with a goal to normalize the TSH. Continuous monitoring of thyroid function is needed to avoid over replacement, which can lead to premature osteoporosis and cardiac arrhythmias. Fine needle aspiration of thyroid nodules is recommended to rule out thyroid cancer, as differentiated thyroid cancer is associated with a favorable prognosis and low recurrence once detected.
Addison's Disease
Background
Thomas Addison described a group of patients affected with anemia and diseased adrenal glands in 1849. Addison's disease is a chronic disorder of the adrenal cortex resulting in decreased production of glucocorticoids, mineralocorticoids, and androgens. There is increased secretion of ACTH from the pituitary gland. Histological examination of adrenal glands from patients with autoimmune adrenal insufficiency reveals fibrosis with a mononuclear cell infiltrate, plasma cells, and rare germinal centers (41). The most common cause of primary adrenal insufficiency in developed countries is autoimmunity (70 to 90%) with tuberculosis the second most common cause (10 to 20%). Addison's disease can be present in three clinical forms: part of syndromes termed APS-1 and APS-2, and as an isolated disease.
Pathogenesis and Genetics
Similar to type 1A diabetes Addison's disease also can be divided into stages of disease progression. Genetically predisposed individuals develop autoantibodies to the 21-hydroxylase enzyme and eventually lose the ability to produce cortisol (Figure 5) (42). Autoantibodies against 21-hydroxylase are present in more than 90% of recent onset patients. Susceptibility is conferred through the genes encoding the class II MHC, and as is the case with type 1A diabetes, there is a strong association with the DR3 haplotype. The highest risk genotype, occurring in 30% of patients with Addison's disease, consists of DR3/4, DQ2/DQ8 (43) and in this case the DRB1*0404 DR4 subtype, confers highest risk on DR4 haplotypes. The MIC-A 5.1 allele, an atypical HLA molecule (MHC class I-related gene A), is also associated with genetic risk (44). Polymorphisms of the MIC-A gene are based on the number of triplicate GCT repeats in exon 5. The translated protein interacts with the NKG2D receptor, which is important for thymic maturation of T cells (45). NKG2D can also regulate the priming of human naive CD8+ T cells (46). The allele, designated 5.1, is associated with the insertion of a base pair, which results in a premature stop codon and loss of the membrane binding region of the protein.
Figure 5.

Stages in the development of Addion's disease from Eisenbarth GS and Gottlieb PA, Autoimmune Polyendocrine Syndromes, New England Journal of Medicine, May 13, 2004. Adrenocortical function is lost over a period of years. In the first stage, genetic predisposition is conferred by a patient's HLA genotype. In the second stage, events that precipitate anti-adrenal autoimmunity occur, but are currently unknown. In the third stage, which involves presymptomatic disease, 21-hydroxylase autoantibodies predict future disease. Finally, in the fourth stage, overt Addison's disease develops. An increased plasma renin level is one of the first metabolic abnormalities to occur and is followed by the sequential development of other metabolic abnormalities (a decreased cortisol level after cosyntropin stimulation, an elevated corticotropin level, and a decreased basal cortisol level). Finally, there are severe symptoms of adrenal insufficiency, such as hypotension.
Diagnosis and Treatment
Diagnosis of Addison's disease is made in symptomatic patients with high levels of ACTH and a deficiency of cortisol or when serum cortisol levels fail to rise after an ACTH stimulation test in the presence of elevated basal ACTH levels; 21-hydroxylase autoantibodies are usually (>90%) present. The clinical manifestations are subtle (weakness, fatigue, anorexia, orthostasis, nausea, myalgias, and salt craving), and a high index of suspicion is necessary to diagnose adrenal insufficiency before an adrenal crisis. We recommend screening patients with type 1A diabetes, hypoparathyroidism, and polyendocrine autoimmunity for 21-hydroxylase autoantibodies. If present, yearly monitoring with an ACTH stimulation test is performed to allow early diagnosis and prevent an adrenal crisis. Treatment is with lifelong glucocorticoids and mineralocorticoids with counseling about the need for stress dose steroids for illnesses and prior to surgical procedures. Forty to 50% of individuals with Addison's disease will develop another autoimmune disease, necessitating lifelong monitoring for associated autoimmune conditions.
Idiopathic Hypoparathyroidism
Background
Idiopathic hypoparathyroidism (IH) results from deficiency of parathyroid hormone (PTH), which regulates serum calcium concentration, and does not have an identifiable cause. This disease is a common component of APS-1 in infants and young children. It also occurs sporadically in adults, most often affecting females with Hashimoto's thyroiditis. An autoimmune basis for IH has been suggested because of its association with other autoimmune conditions.
Pathogenesis
Hypocalcemia results from parathyroid hormone deficiency. Recent work by Kampe and colleagues identified a parathyroid autoantigen, NACHT leucine-rich-repeat protein 5 (NALP5), in individuals with APS-1 (47). NALP5 autoantibodies were identified in APS-1 patients with hypoparathyroidism and not in healthy individuals or individuals with other autoimmune disorders. Autoantibodies to the calcium sensing receptor on parathyroid glands have been described as well and can activate the receptor thereby causing a decreased production of PTH (48).
Diagnosis and treatment
Idiopathic hypoparathyroidism is diagnosed when no other causes of hypocalcemia and hypoparathyroidism can be identified. Treatment is with calcium and magnesium supplementation. To absorb calcium, active 1,25 dihydroxyvitamin D needs to be administered with calcium, and frequent monitoring of serum calcium levels are required.
Premature Ovarian Failure
Background
Premature ovarian failure (POF) is defined as amenorrhea, elevated gonadotropin levels, and hypoestrogenism before age 40. POF can occur before or after puberty. Girls should begin puberty by age 13 and menstruate within 5 years after the onset of puberty. Two distinct clinical scenarios have been identified.
Idiopathic POF with adrenal autoimmunity
Approximately 10% of females with Addison's disease will have POF. Steroid cell autoantibodies, directed against the enzymes 21 hydroxylase or 17 hydroxylase, cross react with theca interna/granulosa layers of ovarian follicles. Presence of these autoantibodies correlates with the histological diagnosis of autoimmune oophoritis (49). MHC class II is expressed on granulosa cells of patients with POF and may potentiate a local T cell autoimmune response (50).
Idiopathic POF with exclusive manifestations of ovarian autoimmunity
The vast majority (>90%) of women with POF do not have Addison's disease or steroid cell autoantibodies, calling into question the autoimmune component to disease. Thyroid autoimmunity is present in about 14% of these individuals. Approximately 10% of patients with isolated POF and without Addison's disease will have numerous ovarian follicles intact. These patients are categorized as having resistant ovary syndrome that is insensitive to ovulation induction with exogenous gonadotropins.
Treatment
Currently there is no treatment available to induce ovarian function or stop progression of autoimmune ovarian destruction. Treatment is focused on treating symptoms of estrogen deficiency and maintaining bone health to prevent osteoporosis. Infertility can be treated with in vitro fertilization with donor eggs. However, there is a relapsing and remitting component to the underlying autoimmunity and occasionally conceptions can be achieved. Screening for associated autoimmune conditions (type 1A diabetes, Addison's disease, and thyroid autoimmunity) should be considered in patients with idiopathic POF.
Lymphocytic Hypophysitis
Background
Lymphocytic hypophysitis is a rare inflammatory lesion of the pituitary gland. Approximately 500 cases have been reported in the literature since the initial report in 1962 (51). This condition is more common in females and affects women during later pregnancy and the postpartum period (e.g., postpartum hypophysitis). It is strongly associated with other autoimmune disorders. Of note ipilimumab, a monoclonal antibody that blocks CTLA-4, is an immunologic therapy used in oncology clinical trials and has induced hypophysitis (52).
Pathogenesis
The morphologic features of hypophysitis resemble those of other autoimmune endocrinopathies. The absence of granulomas on histology distinguishes this condition from glranulomatous hypophysitis seen in association with sarcoidosis, tuberculosis, and syphilis. Antipituitary antibodies have been isolated in a minority of patients with disease.
Diagnosis and treatment
Presenting symptoms include fatigue, headache, and visual field deficits. Diagnosis is confirmed by histological examination of a pituitary biopsy. Anterior pituitary hormone deficits are common and hormone replacement is indicated. High dose glucocorticoid pulse therapy has been used for treatment (53).
Autoimmune Polyendocrine Syndromes
Background
The autoimmune polyendocrine syndromes are a constellation of disorders characterized by multiple autoimmune disorders including endocrine gland failure or hyperactivity (Grave's disease). Some of the components of the syndromes have been described previously in the review. The syndromes include: APS-1, APS-2, IPEX syndrome, POEMS syndrome, non-organ specific autoimmunity (e.g., lupus erythematosus) associated with anti-insulin receptor antibodies, thymic tumors with associated endocrinopathy, and Grave's disease associated with insulin autoimmune syndrome. APS-1, APS-2, IPEX, POEMS syndrome, and diabetes associated autoimmune disorders will be discussed in further detail.
Autoimmune Polyendocrine Syndrome type 1
Background
APS-1/APECED (Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy) is a rare disorder generally seen in infants and the diagnosis is made when a child has two or three of the following: mucocutaneous candidiasis, hypoparathyroidism, or Addison's disease. Mucocutaneous candidiasis involving the mouth and nails is usually the first manifestation followed by the development of hypotension or fatigue from Addison's disease or hypocalcemia from hypoparathyroidism. APS-1 is associated with other autoimmune disorders (type 1A diabetes, vitiligo, alopecia, hepatitis, pernicious anemia, and primary hypothyroidism) and asplenism.
Pathogenesis/Genetics
APS-1 is due to a mutation in the AutoImmune REgulator (AIRE) gene which is transmitted in an autosomal recessive manner. The AIRE gene encodes a transcription factor needed for the expression and presentation of self antigens to developing lymphocytes in the thymus (54). Over 40 mutations in AIRE have been described (55), and when mutations are present, tolerance is lost to multiple self antigens. The resulting autoreactive T cells that escape deletion in the thymus have the ability to destroy multiple specific tissues, producing a phenotype of multiple autoimmune disorders. Animal models with a knockout of the AIRE gene result in widespread autoimmunity, although the phenotype is mild with lymphocytic infiltration of the liver and atrophy of the adrenal and thyroid glands. The majority of mice also exhibit autoantibodies to the pancreas, adrenal glands, testes, and liver (56). Human studies of isolated autoimmune disorders, such as Addison's disease occurring without evidence of APS-1, have not found mutations in the AIRE gene (57).
Diagnosis
Diagnosis is based upon the presence of specific autoimmune disorders and mucocutaneous candidiasis. The known AIRE gene mutations can now be screened. Meager and coworkers recently reported patients with APS-1 have multiple anti-interferon antibodies, with interferon omega reactive autoantibodies present in 100% of patients (58); assays for such autoantibodies may aid in rapid diagnosis.
Treatment
Hormone replacement is the mainstay of treatment for the endocrinopathies present in APS-1. Mucocutaneous candidiasis needs to be treated aggressively and monitored for recurrence as it can occur anywhere along the gastrointestinal tract. Untreated disease can lead to the development of epithelial cancers. Asplenism needs to be identified and vaccinations against pneumococcus, menigococcus, and hemophilus influenza need to be administered.
A high clinical suspicion for other autoimmune disease needs to be maintained in individuals with APS-1 and their relatives. Patients with APS-1 need to be followed at a center with experience monitoring and caring for these patients. Siblings need to be followed closely and consideration should be given to screening for anti-IFN omega autoantibodies. Recommendations are to see these patients on six month intervals and screen for autoantibodies (59). If autoantibodies are present without the associated disease, functional testing is indicated. Patients with 21-hydroxylase antibodies are often followed with annual ACTH levels, 8am cortisol levels, and cosyntropin stimulation testing, unless symptoms or signs warrants more frequent monitoring. The presence of islet cell autoantibodies warrants glucose tolerance testing to detect disease prior to overt clinical symptoms and education related to the symptoms of diabetes along with home glucose monitoring.
Autoimmune Polyendocrine Syndrome type 2
Background
APS-2, also known as Schmidt's syndrome, is the most common autoimmune polyendocrine syndrome. APS-2 has Addison's disease as its defining component with either autoimmune thyroid disease or type 1A diabetes in conjunction. Women are typically affected at a higher rate than males. Other diseases less commonly associated with APS-2 include: celiac disease, vitiligo, pernicious anemia, myasthenia gravis, stiff man syndrome, and alopecia. Familial aggregation was demonstrated by a study looking at 10 families with APS-2 and one in seven relatives had an undiagnosed autoimmune disease, the most common being thyroid disease (60). Diseases may develop years to decades apart, making knowledge of the syndromes necessary to detect disease and treat prior to morbidity and mortality. Table III compares APS-1 and APS-2 (61).
Table III.
Comparison of APS-1 and APS-2
| APS-1 | APS-2 |
|---|---|
|
|
Available from the teaching slides at www.barbaradaviscenter.org.
Pathogenesis/Genetics
The genetics of APS-2 are governed by the HLA haplotypes which confer disease risk to multiple autoimmune disorders. The DR3, DQA1*0501, DQB1*0201 haplotype increases the risk for developing type 1A diabetes, Addison's disease, and celiac disease. The DR4 haplotype of patients with all three of these diseases is associated with DQA1*0301, DQB1*0302. If a patient with type 1A diabetes has the DRB1*0404 allele and express 21-hydroxylase antibodies, there is a 100 fold increase in the risk of developing Addison's disease (62). Autoimmune diseases result from a failure to develop or maintain tolerance along with a genetic predisposition, MHC alleles, controlling specific disease development. Multiple autoimmune disorders develop when tolerance is lost to a number of self antigens.
Diagnosis and treatment
Similar to APS-1, treatment of APS-2 focuses on identifying and treating the underlying autoimmune conditions. Autoimmune thyroid disease is very common. It is prudent to screen type 1A diabetics and Addison's disease patients with a yearly TSH. We recommend screening individuals with type 1A diabetes for 21-hyroxylase and tissue transglutaminase autoantibodies. The optimal screening interval is not defined; however, autoantibodies may develop at any age and repeat testing is necessary upon negative test results. Relatives of individuals with APS-2 need to be monitored closely.
IPEX Syndrome
Background
The rare IPEX syndrome (immune dysfunction, polyendocrinopathy, enteropathy, X-linked) is caused by mutations in the forkhead box protein 3 (FOXP3) gene resulting in absent or dysfunctional regulatory T cells (63). Clinically, it presents during the first few months of life with dermatitis, growth retardation, multiple endocrinopathies, and recurrent infections. Affected neonates have overwhelming autoimmunity including type 1A diabetes developing as early as 2 days of age.
Pathogenesis/Genetics
To date, 20 mutations in FOXP3 have been identified in patients with IPEX syndrome (64;65). Most of these mutations occur in the forkhead (winged-helix) domain and leucine zipper region resulting in impaired DNA binding. The inability of FOXP3 to bind DNA in regulatory T cells impairs immune suppressor function. Dysregulated T cell function leads to overwhelming autoimmunity and recurrent infections. The scurfy mouse develops a disease very similar to IPEX and has a homologous gene, scurfin, to the human FOXP3. The scurfy mouse model allows for understanding disease pathogenesis and provides a model to evaluate treatment modalities (66;67). Neonatal thymectomy in male scurfy mice ameliorates disease and increases lifespan. Transfer of peripheral CD4+ T cells, but not CD8+ T cells, from affected mice to homologous wild type mice result in disease, while bone marrow transplantation does not induce disease. Peripheral CD4+ T cells appear to be hyper-responsive to antigens and have a decreased requirement for costimulation with CD28 (68). The inability of CD4+ T cells to regulate the immune response, from mutations in scurfin or FOXP3, results in the IPEX syndrome.
Treatment
Children affected with IPEX usually die in the first 2 years of life due to sepsis or failure to thrive. Supportive care and treatment of underlying disorders is necessary. Immunosuppressive medications have been tried in case reports or small case series. High dose glucocorticoids, tacrolimus, cyclosporine, methotrexate, sirolimus, infliximab, and rituximab have been tried with varying degrees of success. The toxicity and infectious complications limit their dosing and use. In the scurfy mouse, the disease can be cured with partial T cell chimerism, and the same appears to be true in humans with normal T lymphocytes able to regulate the abnormal immune system in a dominant fashion (69). Bone marrow transplantation can reduce symptoms and prolong survival (70)}. Transplantation should be considered early in the disease to limit the autoimmune destruction to endocrine organs and possibly reduce the infectious complications from chronic immune suppression.
POEMS Syndrome
POEMS syndrome has Polyneuropathy, Organomegaly, Endocrinopathies, M-protein, and Skin manifestations (hyperpigmentation and hypertrichosis) as clinical features. The etiologic factors of this constellation of diseases are not well defined. The syndrome is associated with plasmacytomas and osteosclerotic lesions with radiation therapy to localized lesions being beneficial. Autologous hematopoietic stem cell transplantation has improved symptoms (71).
Diabetes Associated Autoimmune Disorders
Multiple autoimmune disorders are associated with type 1A diabetes. Many of the disorders have been discussed previously in the review, and this section will focus on their relationship to type 1A diabetes.
Celiac disease is an autoimmune disorder that results in T cell infiltration of the mucosa of the small intestine. Gliadin, a protein of wheat gluten, has been identified as the antigen responsible for inducing the autoimmune process. Like type 1A diabetes, a genetic predisposition is conferred through the HLA alleles DQ2 and DQ8. Symptoms of celiac disease can be mild but can also include diarrhea, abdominal pain, iron deficiency anemia, pubertal delay, growth failure, decreased bone mineralization, and vitamin D deficiency. Tissue transglutaminase (TGA) IgA autoantibodies are a sensitive and specific marker for the autoimmune process, more so than the older anti-endomesial antibody assay. TGA autoantibodies are present in up to 16% of patients with type 1 diabetes (72). A definitive diagnosis is made with a small intestine biopsy showing flattened villae and intraepithelial lymphocytic infiltrates. Treatment is with a gluten free diet, which results in reversal of the autoimmune process and normalization of the intestinal villae (73). We recommend screening with TGA autoantibodies yearly in type 1 diabetics and performing a small intestine biopsy if a repeat TGA autoantibody is positive. The biopsy should be obtained close to the time of antibody measurement as the half life of IgA antibodies are short and the titer of TGA autoantibody fluctuates with the amount of gluten in the diet. Those with a positive biopsy are counseled on a gluten free diet. These recommendations are based upon the known risks of symptomatic Celiac disease (osteoporosis, anemia, and gastrointestinal malignancy) and the rationale that the intestinal pathology is reversible with gluten avoidance.
Addison's disease is present in 1/10,000 individuals in the general population compared to 1/200 in the type 1 diabetic population. One to 2% of type 1 diabetics have 21 hydroxylase autoantibodies (74). Many patients with Addison's disease are adrenally insufficient for years prior to diagnosis. It is advisable to screen patients with type 1 diabetes for 21 hydroxylase autoantibodies and monitor those that are positive with cosyntropin (ACTH) stimulation testing.
Autoimmune thyroid disease is common in patients with type 1 diabetes. Twenty to 30% of patients with type 1 diabetes express TPO and/or Tg autoantibodies, twice that of the general population. Long term follow up has shown 30% of patients with type 1 diabetes will develop autoimmune thyroid disease (75). It is recommend that patients with type 1 diabetes be screened for thyroid dysfunction annually with a serum thyroid stimulating hormone (TSH) level (76).
Pernicious anemia results in a macrocytic anemia from autoimmune destruction of parietal cells in the fundus and body of the stomach. The frequency of pernicious anemia in type 1 diabetes has been reported to be up to 4%, with a rate of 0.12% in the general population (77).
Vitiligo, the loss of melanocytes in the skin, is associated with many autoimmune conditions including type 1A diabetes.
Iatrogenic Endocrine Autoimmune Disorders
Background
Drug induced autoimmune diseases have been recognized for years and span multiple disciplines. Iatrogenic autoimmunity is increasing in frequency as more therapies are being designed to alter immune mechanisms in autoimmune conditions and cancer.
Pharmaceutical Agents
Interferon Alpha
The interferons are a group proteins characterized by antiviral activity, growth regulatory properties, and a variety of immunomodulatory activities. IFNα is currently used to treat patients with the hepatitis C virus. IFNα has been reported to cause Hashimoto's thyroiditis and Grave's disease. It is also associated with non-autoimmune thyroiditis. Approximately 5 to 10% of individuals treated with IFNα develop thyroid autoimmunity while another 15% develop thyroid autoantibodies without clinical disease (78). The drug also induces both islet autoantibodies and rapid progression to diabetes in a subset of patients with islet autoantibodies (79). At a minimum glucose levels should be monitored in patients and if abnormal the patient evaluated for islet autoantibodies, monitored for diabetes, and the risks/benefits of therapy carefully considered.
Interleukin-2
IL-2 induces T cell proliferation, B cell growth, and natural killer cell and monocyte activation. IL-2 has antitumor activity, and it has been used in the treatment of metastatic melanoma, renal cell carcinoma, and HIV. Thyroiditis and Hashimoto's thyroiditis have been described with IL-2 treatment either alone or in conjunction with IFNα in up to 16% of patients (80).
Ipilimumab
Ipilimumab is a monoclonal antibody that blocks cytotoxic T lymphocyte associated antigen 4 (CTLA-4), a receptor on T cells; blockade of CTLA-4 results in T cell activation, proliferation, and differentiation. Ipilimumab has been used to treat patients with metastatic renal cell cancer and melanoma. Endocrinopathies, hypophysitis and hypothyroidism, as well as non-endocrine autoimmune disorders have been reported (81). Up to 59% of participants in the National Institutes of Health (NIH) studies treated with anti-CTLA-4 monoclonal antibodies have presented with autoimmune toxicities. Many of the autoimmune events are transient and some can be successfully treated with high dose glucocorticoids. Five percent (8/163) of patients treated with anti-CTLA-4 monoclonal antibodies at the NIH have developed hypophysitis (82). In the case of hypophysitis, anterior pituitary hormone deficiencies have been reported to be present for up to two years despite discontinuation of therapy with ipilimumab.
Campath-1H
Campath-1H is a humanized anti-CD52 monoclonal antibody that suppresses Th1 lymphocytes. Grave's disease has been associated with treatment in patients with multiple sclerosis and new onset type 1A diabetes. In a study of 29 multiple sclerosis patients treated with Campath-1H, 9 developed Grave's disease after 6-31 months of treatment (83).
Highly Active Antiretroviral Therapy (HAART)
HAART, used to treat HIV infection, has been associated with Grave's disease. The prevalence is rare and occurs 16-19 months after initiation of therapy. Following HAART, there is immune reconstitution and this is when autoimmunity develops, likely resulting from changes to CD4+ T cells (84).
Conclusions
Improved understanding of the immune pathogenesis of endocrine diseases has lead to the initial development of therapies that target the underlying autoimmunity. Type 1A diabetes, one of the most well studied organ specific autoimmune diseases, is now predictable in humans and therapies are emerging to augment the underlying autoimmune destruction of beta cells. With continued basic understanding of the immunologic mechanisms causing autoimmunity, better therapies can be designed to improve the quality of life for patients and their families afflicted with these disorders.
Acknowledgments
Supported by grants from the National Institutes of Health (DK32083, DK32493, DK057538) Autoimmunity Prevention Center (AI50964), Diabetes Endocrine Research Center (P30 DK57516), Clinical Research Centers (MO1 RR00069, MO1 RR00051), the Immune Tolerance Network (AI15416), the American Diabetes Association, the Juvenile Diabetes Research Foundation, the Brehm Coalition, and the Children's Diabetes Foundation.
Abbreviations
- ACTH
Adrenocorticotrophic hormone
- AIRE
Autoimmune regulator
- APS-1
Autoimmune Polyendocrine Syndrome type 1
- APS-2
Autoimmune Polyendocrine Syndrome type 2
- BB
Biobreeding
- CTLA
Cytotoxic T-lymphocyte-associated antigen
- DAISY
Diabetes Autoimmunity Study in the Young
- GAD
Glutamic acid decarboxylase
- GO
Grave's orbitopathy
- HAART
Highly Active Antiretroviral Therapy
- HT
Hashimoto's thyroiditis
- HLA
Human leukocyte antigen
- IA-2
Islet associated antigen (ICA512)
- IFN
Interferon
- IH
Idiopathic hypoparathyroidism
- IL
Interleukin
- IPEX
Immune dysfunction, polyendocrinopathy, enteropathy, X-linked
- MHC
Major histocompatability complex
- MIC-A
MHC class I-related molecule A
- MODY
Mature-onset diabetes of the young
- NOD
Nonobese diabetic
- POEMS
Polyneuropathy, organomegaly, endocrinopathy, serum monoclonal protein, and skin changes
- POF
Premature ovarian failure
- PND
Permanent neonatal diabetes
- PTH
Parathyroid hormone
- PTPN22
Protein tyrosine phosphatase, non-receptor 22
- Tg
Thyroglobulin
- TGA
Tissue transglutaminase
- TND
Transient neonatal diabetes
- TNF
Tumor necrosis factor
- TPO
Thyroid peroxidase
- TSH
Thyroid stimulating hormone
- TSI
Thyroid stimulating immunoglobulins
- TSHR
Thyroid stimulating hormone receptor
- VNTR
Variable number of tandem nucleotide repeats
- ZnT8
Zinc T8 transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Diagnosis and classification of diabetes mellitus. Diab care. 2008 January; 1:31. S55–S60. doi: 10.2337/dc08-S055. [DOI] [PubMed] [Google Scholar]
- 2.Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008 May 24;371(9626):1777–82. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 3.Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P. HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk: Analysis of the Type 1 Diabetes Genetics Consortium Families. Diabetes. 2008 February 5;57:1084–92. doi: 10.2337/db07-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008 April;4(4):200–13. doi: 10.1038/ncpendmet0778. [DOI] [PubMed] [Google Scholar]
- 5.Redondo MJ, Jeffrey J, Fain PR, Eisenbarth GS, Orban T. Concordance for islet autoimmunity among monozygotic twins. N Engl J Med. 2008 December 25;359(26):2849–50. doi: 10.1056/NEJMc0805398. [DOI] [PubMed] [Google Scholar]
- 6.Redondo MJ, Fain PR, Krischer JP, Yu L, Cuthbertson D, Winter WE, Eisenbarth GS. Expression of beta-cell autoimmunity does not differ between potential dizygotic twins and siblings of patients with type 1 diabetes. J Autoimmun. 2004 November;23(3):275–9. doi: 10.1016/j.jaut.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Bonifacio E, Pfluger M, Marienfeld S, Winkler C, Hummel M, Ziegler AG. Maternal type 1 diabetes reduces the risk of islet autoantibodies: relationships with birthweight and maternal HbA(1c) Diabetologia. 2008 July;51(7):1245–52. doi: 10.1007/s00125-008-1022-z. [DOI] [PubMed] [Google Scholar]
- 8.Lie BA, Ronningen KS, Akselsen HE, Thorsby E, Undlien DE. Application and interpretation of transmission/disequilibrium tests: transmission of HLA-DQ haplotypes to unaffected siblings in 526 families with type 1 diabetes. Am J Hum Genet. 2000 February;66(2):740–3. doi: 10.1086/302780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JH, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007 July;39(7):857–64. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pugliese A, Zeller M, Fernandez A, Jr, Zalcberg LJ, Bartlett RJ, Ricordi C, Pietropaolo M, Eisenbarth GS, Bennett ST, Patel DD. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nature Genetics. 1997;15(3):293–7. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 11.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005 November 6;37(12):1317–9. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 12.Grant SF, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Taback SP, Frackelton EC, Eckert AW, Annaiah K, Lawson ML, Otieno FG, Santa E, Shaner JL, Smith RM, Skraban R, Imielinski M, Chiavacci RM, Grundmeier RW, Stanley CA, Kirsch SE, Waggott D, Paterson AD, Monos DS, et al. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. diab. 2009 January;58(1):290–5. doi: 10.2337/db08-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008 May 24;371(9626):1777–82. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 14.Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, Orton HD, Baron AE, Clare-Salzler M, Chase HP, Szabo NJ, Erlich H, Eisenbarth GS, Rewers M. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA. 2007 September 26;298(12):1420–8. doi: 10.1001/jama.298.12.1420. [DOI] [PubMed] [Google Scholar]
- 15.Norris JM, Barriga K, Klingensmith G, Hoffman M, Eisenbarth GS, Erlich H, Rewers M. Timing of cereal exposure in infancy and risk of islet autoimmunity. The Diabetes Autoimmunity Study in the Young (DAISY) JAMA. 2003;290(13):1713–20. doi: 10.1001/jama.290.13.1713. [DOI] [PubMed] [Google Scholar]
- 16.Norris JM, Yin X, Lamb MM, Barriga K, Seifert J, Hoffman M, Orton HD, Baron AE, Clare-Salzler M, Chase HP, Szabo NJ, Erlich H, Eisenbarth GS, Rewers M. Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes. JAMA. 2007 September 26;298(12):1420–8. doi: 10.1001/jama.298.12.1420. [DOI] [PubMed] [Google Scholar]
- 17.Uusitalo L, Nevalainen J, Niinisto S, Alfthan G, Sundvall J, Korhonen T, Kenward MG, Oja H, Veijola R, Simell O, Ilonen J, Knip M, Virtanen SM. Serum alpha- and gamma-tocopherol concentrations and risk of advanced beta cell autoimmunity in children with HLA-conferred susceptibility to type 1 diabetes mellitus. Diabetologia. 2008 May;51(5):773–80. doi: 10.1007/s00125-008-0959-2. [DOI] [PubMed] [Google Scholar]
- 18.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003 August;23(15):5293–300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005 July 26;102(30):10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wenzlau JM, Juhl K, Yu L, Moua O, Sarkar SA, Gottlieb P, Rewers M, Eisenbarth GS, Jensen J, Davidson HW, Hutton JC. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A. 2007 October 23;104(43):17040–5. doi: 10.1073/pnas.0705894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci U S A. 2002 April 16;99(8):5539–44. doi: 10.1073/pnas.082120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutton JC, Eisenbarth GS. A pancreatic {beta}-cell-specific homolog of glucose-6-phosphatase emerges as a major target of cell-mediated autoimmunity in diabetes. Proc Natl Acad Sci U S A. 2003 July 14;100:8626–8. doi: 10.1073/pnas.1633447100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009 February;155(2):173–81. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Achenbach P, Bonifacio E, Ziegler AG. Predicting type 1 diabetes. Curr Diab Rep. 2005 April;5(2):98–103. doi: 10.1007/s11892-005-0035-y. [DOI] [PubMed] [Google Scholar]
- 25.Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA, Chase HP, Eisenbarth GS. Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. diab. 1996;45(7):926–33. doi: 10.2337/diab.45.7.926. [DOI] [PubMed] [Google Scholar]
- 26.Achenbach P, Warncke K, Reiter J, Williams AJ, Ziegler AG, Bingley PJ, Bonifacio E. Type 1 diabetes risk assessment: improvement by follow-up measurements in young islet autoantibody-positive relatives. Diabetologia. 2006 December;49(12):2969–76. doi: 10.1007/s00125-006-0451-9. [DOI] [PubMed] [Google Scholar]
- 27.Wenzlau JM, Moua O, Sarkar SA, Yu L, Rewers M, Eisenbarth GS, Davidson HW, Hutton JC. SlC30A8 is a major target of humoral autoimmunity in type 1 diabetes and a predictive marker in prediabetes. Ann N Y Acad Sci. 2008 December;1150:256–9. doi: 10.1196/annals.1447.029. [DOI] [PubMed] [Google Scholar]
- 28.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329(14):977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 29.Staeva-Vieira T, Peakman M, von Herrath M. Translational Mini-Review Series on Type 1 Diabetes: Immune-based therapeutic approaches for type 1 diabetes. Clin Exp Immunol. 2007 April;148(1):17–31. doi: 10.1111/j.1365-2249.2007.03328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchigata Y, Kuwata S, Tsushima T, Tokunaga K, Miyamoto M, Tsuchikawa K, Hirata Y, Juji T, Omori Y. Patients with Graves' disease who developed insulin autoimmune syndrome (Hirata disease) possess HLA-Bw62/Cw4/DR4 carrying DRB1*0406. J Clin Endocrinol Metab. 1993;77:249–54. doi: 10.1210/jcem.77.1.8325948. [DOI] [PubMed] [Google Scholar]
- 31.Marga M, Denisova A, Sochnev A, Pirags V, Farid NR. Two HLA DRB 1 alleles confer independent genetic susceptibility to Graves disease: relevance of cross-population studies. Am J Med Genet. 2001 August 1;102(2):188–91. doi: 10.1002/ajmg.1431. [DOI] [PubMed] [Google Scholar]
- 32.Chen QY, Huang W, She JX, Baxter F, Volpe R, Maclaren NK. HLA-DRB1*08, DRB1*03/DRB3*0101, and DRB3*0202 are susceptibility genes for Graves' disease in North American Caucasians, whereas DRB1*07 is protective. J Clin Endocrinol Metab. 1999 September;84(9):3182–6. doi: 10.1210/jcem.84.9.5991. [DOI] [PubMed] [Google Scholar]
- 33.Martin A, Nakashima M, Zhou A, Aronson D, Werner AJ, Davies TF. Detection of major T cell epitopes on human thyroid stimulating hormone receptor by overriding immune heterogeneity in patients with Graves' disease. J Clin Endocrinol Metab. 1997 October;82(10):3361–6. doi: 10.1210/jcem.82.10.4299. [DOI] [PubMed] [Google Scholar]
- 34.Gillis D, Volpe R, Daneman D. A young boy with a thyroid yo-yo. J Pediatr Endocrinol Metab. 1998;11(3):467–70. doi: 10.1515/jpem.1998.11.3.467. [DOI] [PubMed] [Google Scholar]
- 35.Weetman AP. Graves' disease. N Engl J Med. 2000 October 26;343(17):1236–48. doi: 10.1056/NEJM200010263431707. [DOI] [PubMed] [Google Scholar]
- 36.Starkey KJ, Janezic A, Jones G, Jordan N, Baker G, Ludgate M. Adipose thyrotrophin receptor expression is elevated in Graves' and thyroid eye diseases ex vivo and indicates adipogenesis in progress in vivo. J Mol Endocrinol. 2003 June;30(3):369–80. doi: 10.1677/jme.0.0300369. [DOI] [PubMed] [Google Scholar]
- 37.El Fassi D, Nielsen CH, Bonnema SJ, Hasselbalch HC, Hegedus L. B lymphocyte depletion with the monoclonal antibody rituximab in Graves' disease: a controlled pilot study. J Clin Endocrinol Metab. 2007 May;92(5):1769–72. doi: 10.1210/jc.2006-2388. [DOI] [PubMed] [Google Scholar]
- 38.Salvi M, Vannucchi G, Campi I, Curro N, Dazzi D, Simonetta S, Bonara P, Rossi S, Sina C, Guastella C, Ratiglia R, Beck-Peccoz P. Treatment of Graves' disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007 January;156(1):33–40. doi: 10.1530/eje.1.02325. [DOI] [PubMed] [Google Scholar]
- 39.Ban Y, Greenberg DA, Concepcion E, Skrabanek L, Villanueva R, Tomer Y. Amino acid substitutions in the thyroglobulin gene are associated with susceptibility to human and murine autoimmune thyroid disease. Proc Natl Acad Sci U S A. 2003 December 9;100(25):15119–24. doi: 10.1073/pnas.2434175100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ban Y, Davies TF, Greenberg DA, Kissin A, Marder B, Murphy B, Concepcion ES, Villanueva RB, Barbesino G, Ling V, Tomer Y. Analysis of the CTLA-4, CD28, and inducible costimulator (ICOS) genes in autoimmune thyroid disease. Genes Immun. 2003 December;4(8):586–93. doi: 10.1038/sj.gene.6364018. [DOI] [PubMed] [Google Scholar]
- 41.Sommers SC. Adrenal glands. In: Kissane JM, editor. CV Mosby. Anderson's Pathology; St. Louis, MO: 2002. pp. 1429–50. [Google Scholar]
- 42.Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syndromes. N Engl J Med. 2004 May 13;350(20):2068–79. doi: 10.1056/NEJMra030158. [DOI] [PubMed] [Google Scholar]
- 43.Betterle C, Volpato M, Rees Smith B, Furmaniak J, Chen S, Zanchetta R, Greggio NA, Pedini B, Boscaro M, Presotto F. II. Adrenal cortex and steroid 21-hydroxylase autoantibodies in children with organ-specific autoimmune diseases: markers of high progression to clinical Addison's disease. J Clin Endocrinol Metab. 1997;82(3):939–42. doi: 10.1210/jcem.82.3.3849. [DOI] [PubMed] [Google Scholar]
- 44.Gambelunghe G, Falorni A, Ghaderi M, Laureti S, Tortoioli C, Santeusanio F, Brunetti P, Sanjeevi CB. Microsatellite polymorphism of the MHC class I chain-related (MIC-A and MIC-B) genes marks the risk for autoimmune Addison's disease. J Clin Endocrinol Metab. 1999 October;84(10):3701–7. doi: 10.1210/jcem.84.10.6069. [DOI] [PubMed] [Google Scholar]
- 45.Hue S, Monteiro RC, Berrih-Aknin S, Caillat-Zucman S. Potential role of NKG2D/MHC class I-related chain A interaction in intrathymic maturation of single-positive CD8 T cells. J Immunol. 2003 August 15;171(4):1909–17. doi: 10.4049/jimmunol.171.4.1909. [DOI] [PubMed] [Google Scholar]
- 46.Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol. 2005 April 15;174(8):4480–4. doi: 10.4049/jimmunol.174.8.4480. [DOI] [PubMed] [Google Scholar]
- 47.Alimohammadi M, Bjorklund P, Hallgren A, Pontynen N, Szinnai G, Shikama N, Keller MP, Ekwall O, Kinkel SA, Husebye ES, Gustafsson J, Rorsman F, Peltonen L, Betterle C, Perheentupa J, Akerstrom G, Westin G, Scott HS, Hollander GA, Kampe O. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. 2008 March 6;358(10):1018–28. doi: 10.1056/NEJMoa0706487. [DOI] [PubMed] [Google Scholar]
- 48.Kifor O, Moore FD, Jr, Delaney M, Garber J, Hendy GN, Butters R, Gao P, Cantor TL, Kifor I, Brown EM, Wysolmerski J. A syndrome of hypocalciuric hypercalcemia caused by autoantibodies directed at the calcium-sensing receptor. J Clin Endocrinol Metab. 2003 January;88(1):60–72. doi: 10.1210/jc.2002-020249. [DOI] [PubMed] [Google Scholar]
- 49.Bakalov VK, Anasti JN, Calis KA, Vanderhoof VH, Premkumar A, Chen S, Furmaniak J, Smith BR, Merino MJ, Nelson LM. Autoimmune oophoritis as a mechanism of follicular dysfunction in women with 46,XX spontaneous premature ovarian failure. Fertil Steril. 2005 October;84(4):958–65. doi: 10.1016/j.fertnstert.2005.04.060. [DOI] [PubMed] [Google Scholar]
- 50.Hill JA, Welch WR, Faris HM, Anderson DJ. Induction of class II major histocompatibility complex antigen expression in human granulosa cells by interferon gamma: a potential mechanism contributing to autoimmune ovarian failure. Am J Obstet Gynecol. 1990 February;162(2):534–40. doi: 10.1016/0002-9378(90)90425-7. [DOI] [PubMed] [Google Scholar]
- 51.Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008 September;7(8):631–7. doi: 10.1016/j.autrev.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, Topalian SL, Haworth LR, Levy C, Rosenberg SA, Sherry RM. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005 November;28(6):593–8. doi: 10.1097/01.cji.0000178913.41256.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kristof RA, Van RD, Klingmuller D, Springer W, Schramm J. Lymphocytic hypophysitis: non-invasive diagnosis and treatment by high dose methylprednisolone pulse therapy. J Neurol Neurosurg Psychiatry. 1999 September;67(3):398–402. doi: 10.1136/jnnp.67.3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002 November 15;298(5597):1395–401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 55.Kumar PG, Laloraya M, She JX. Population genetics and functions of the autoimmune regulator (AIRE) Endocrinol Metab Clin North Am. 2002 June;31(2):321–38. doi: 10.1016/s0889-8529(01)00011-1. vi. [DOI] [PubMed] [Google Scholar]
- 56.Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kampe O, Eskelin P, Pelto-Huikko M, Peltonen L. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum Mol Genet. 2002 February 15;11(4):397–409. doi: 10.1093/hmg/11.4.397. [DOI] [PubMed] [Google Scholar]
- 57.Meyer G, Donner H, Herwig J, Bohles H, Usadel KH, Badenhoop K. Screening for an AIRE-1 mutation in patients with Addison's disease, type 1 diabetes, Graves' disease and Hashimoto's thyroiditis as well as in APECED syndrome. Clin Endocrinol (Oxf) 2001 March;54(3):335–8. doi: 10.1046/j.1365-2265.2001.01230.x. [DOI] [PubMed] [Google Scholar]
- 58.Meager A, Visvalingam K, Peterson P, Moll K, Murumagi A, Krohn K, Eskelin P, Perheentupa J, Husebye E, Kadota Y, Willcox N. Anti-Interferon Autoantibodies in Autoimmune Polyendocrinopathy Syndrome Type 1. PLoS Med. 2006 June 13;3(7):e289. doi: 10.1371/journal.pmed.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perheentupa J. APS-I/APECED: The clinical disease and therapy. In: Eisenbarth GS, editor. Autoimmune Polyendocrine Syndromes. Philadelphia: W.B. Saunders Company; 2002. pp. 295–320. [Google Scholar]
- 60.Eisenbarth GS, Wilson PW, Ward F, Buckley C, Lebovitz HE. The polyglandular failure syndrome: disease inheritance, HLA- type and immune function. Ann Intern Med. 1979;91(4):528–33. doi: 10.7326/0003-4819-91-4-528. [DOI] [PubMed] [Google Scholar]
- 61.Michels AW, Eisenbarth GS. Autoimmune polyendocrine syndrome type 1 (APS-1) as a model for understanding autoimmune polyendocrine syndrome type 2 (APS-2) J Intern Med. 2009 May;265(5):530–40. doi: 10.1111/j.1365-2796.2009.02091.x. [DOI] [PubMed] [Google Scholar]
- 62.Yu L, Brewer KW, Gates S, Wu A, Wang T, Babu S, Gottlieb P, Freed BM, Noble J, Erlich H, Rewers M, Eisenbarth G. DRB1*04 and DQ alleles: expression of 21-hydroxylase autoantibodies and risk of progression to Addison's disease. J Clin Endocrinol Metab. 1999;84(1):328–35. doi: 10.1210/jcem.84.1.5414. [DOI] [PubMed] [Google Scholar]
- 63.Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG. Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest. 2006 June;116(6):1713–22. doi: 10.1172/JCI25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–26. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 65.Le BS, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest. 2006 June;116(6):1473–5. doi: 10.1172/JCI28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature Genet. 2001;27(1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 67.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003 April;4(4):337–42. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 68.Singh N, Chandler PR, Seki Y, Baban B, Takezaki M, Kahler DJ, Munn DH, Larsen CP, Mellor AL, Iwashima M. Role of CD28 in fatal autoimmune disorder in scurfy mice. Blood. 2007 August 15;110(4):1199–206. doi: 10.1182/blood-2006-10-054585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patel DD. Escape from tolerance in the human X-linked autoimmunity-allergic disregulation syndrome and the Scurfy mouse. J Clin Invest. 2001;107(2):155–7. doi: 10.1172/JCI11966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mazzolari E, Forino C, Fontana M, D'Ippolito C, Lanfranchi A, Gambineri E, Ochs H, Badolato R, Notarangelo LD. A new case of IPEX receiving bone marrow transplantation. Bone Marrow Transplant. 2005 March 21; doi: 10.1038/sj.bmt.1704954. [DOI] [PubMed] [Google Scholar]
- 71.Creange A, Chater A, Brouet JC, Jaccard A, Rahmouni A, Lefaucheur JP, Santin A. A case of POEMS syndrome treated by autologous hematopoietic stem-cell transplantation. Nat Clin Pract Neurol. 2008 December;4(12):686–91. doi: 10.1038/ncpneuro0942. [DOI] [PubMed] [Google Scholar]
- 72.Rewers M, Liu E, Simmons J, Redondo MJ, Hoffenberg EJ. Celiac disease associated with type 1 diabetes mellitus. Endocrinol Metab Clin North Am. 2004 March;33(1):197–214. doi: 10.1016/j.ecl.2003.12.007. xi. [DOI] [PubMed] [Google Scholar]
- 73.Farrell RJ, Kelly CP. Celiac sprue. N Engl J Med. 2002 January 17;346(3):180–8. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- 74.Barker JM, Yu J, Yu L, Wang J, Miao D, Bao F, Hoffenberg E, Nelson JC, Gottlieb PA, Rewers M, Eisenbarth GS. Autoantibody “sub-specificity” in type 1 diabetes: Risk for organ specific autoimmunity clusters in distinct groups. Diab care. 2005;28:850–5. doi: 10.2337/diacare.28.4.850. [DOI] [PubMed] [Google Scholar]
- 75.Umpierrez GE, Latif KA, Murphy MB, Lambeth HC, Stentz F, Bush A, Kitabchi AE. Thyroid dysfunction in patients with type 1 diabetes: a longitudinal study. Diabetes Care. 2003 April;26(4):1181–5. doi: 10.2337/diacare.26.4.1181. [DOI] [PubMed] [Google Scholar]
- 76.Silverstein J, Klingensmith G, Copeland K, Plotnick L, Kaufman F, Laffel L, Deeb L, Grey M, Anderson B, Holzmeister LA, Clark N. Care of children and adolescents with type 1 diabetes: a statement of the American Diabetes Association. Diabetes Care. 2005 January;28(1):186–212. doi: 10.2337/diacare.28.1.186. [DOI] [PubMed] [Google Scholar]
- 77.Perros P, Singh RK, Ludlam CA, Frier BM. Prevalence of pernicious anaemia in patients with Type 1 diabetes mellitus and autoimmune thyroid disease. Diabet Med. 2000 October;17(10):749–51. doi: 10.1046/j.1464-5491.2000.00373.x. [DOI] [PubMed] [Google Scholar]
- 78.Russo MW, Fried MW. Side effects of therapy for chronic hepatitis C. Gastroenterology. 2003 May;124(6):1711–9. doi: 10.1016/s0016-5085(03)00394-9. [DOI] [PubMed] [Google Scholar]
- 79.Schreuder TC, Gelderblom HC, Weegink CJ, Hamann D, Reesink HW, Devries JH, Hoekstra JB, Jansen PL. High incidence of type 1 diabetes mellitus during or shortly after treatment with pegylated interferon alpha for chronic hepatitis C virus infection. Liver Int. 2007 November 21; doi: 10.1111/j.1478-3231.2007.01610.x. [DOI] [PubMed] [Google Scholar]
- 80.Weijl NI, Van der Harst D, Brand A, Kooy Y, Van LS, Schroder J, Lentjes E, van Rood JJ, Cleton FJ, Osanto S. Hypothyroidism during immunotherapy with interleukin-2 is associated with antithyroid antibodies and response to treatment. J Clin Oncol. 1993 July;11(7):1376–83. doi: 10.1200/JCO.1993.11.7.1376. [DOI] [PubMed] [Google Scholar]
- 81.Maker AV, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Hughes M, Yellin MJ, Haworth LR, Levy C, Allen T, Mavroukakis SA, Attia P, Rosenberg SA. Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother. 2006 July;29(4):455–63. doi: 10.1097/01.cji.0000208259.73167.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, Topalian SL, Haworth LR, Levy C, Rosenberg SA, Sherry RM. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005 November;28(6):593–8. doi: 10.1097/01.cji.0000178913.41256.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jones JL, Coles AJ. Campath-1H treatment of multiple sclerosis. Neurodegener Dis. 2008;5(1):27–31. doi: 10.1159/000109935. [DOI] [PubMed] [Google Scholar]
- 84.Chen F, Day SL, Metcalfe RA, Sethi G, Kapembwa MS, Brook MG, Churchill D, de RA, Robinson S, Lacey CJ, Weetman AP. Characteristics of autoimmune thyroid disease occurring as a late complication of immune reconstitution in patients with advanced human immunodeficiency virus (HIV) disease. Medicine (Baltimore) 2005 March;84(2):98–106. doi: 10.1097/01.md.0000159082.45703.90. [DOI] [PubMed] [Google Scholar]
- 85.Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, Rother K, Diamond B, Harlan DM, Bluestone JA. A Single Course of Anti-CD3 Monoclonal Antibody hOKT3{gamma}1(Ala-Ala) Results in Improvement in C-Peptide Responses and Clinical Parameters for at Least 2 Years after Onset of Type 1 Diabetes. Diabetes. 2005 June;54(6):1763–9. doi: 10.2337/diabetes.54.6.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin JM, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach JF, Pipeleers D, Chatenoud L. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005 June 23;352(25):2598–608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 87.Jenner M, Bradish G, Stiller C, Atkison P. Cyclosporin A treatment of young children with newly-diagnosed type 1 (insulin-dependent) diabetes mellitus. London Diabetes Study Group. Diabetologia. 1992 September;35(9):884–8. doi: 10.1007/BF00399937. [DOI] [PubMed] [Google Scholar]
- 88.Elliott RB, Chase HP. Prevention or delay of type I (insulin-dependent) diabetes mellitus in children using nicotinamide. diabetol. 1991;34(5):362–5. doi: 10.1007/BF00405010. [DOI] [PubMed] [Google Scholar]
- 89.Allen HF, Klingensmith GJ, Jensen P, Simoes E, Hayward A, Chase HP. Effect of BCG vaccination on new-onset insulin-dependent diabetes mellitus: A randomized clinical study. Diab care. 1998;22(10):1703–7. doi: 10.2337/diacare.22.10.1703. [DOI] [PubMed] [Google Scholar]
- 90.Ludvigsson J, Faresjo M, Hjorth M, Axelsson S, Cheramy M, Pihl M, Vaarala O, Forsander G, Ivarsson S, Johansson C, Lindh A, Nilsson NO, Aman J, Ortqvist E, Zerhouni P, Casas R. GAD Treatment and Insulin Secretion in Recent-Onset Type 1 Diabetes. N Engl J Med. 2008 October 8; doi: 10.1056/NEJMoa0804328. [DOI] [PubMed] [Google Scholar]