

Abstract
Lipopolysaccharides (LPS), which are structural components of the outer surface membrane of Gram-negative bacteria, trigger innate immune responses through activation of Toll-like receptor 4 (TLR4). Such responses may be exploited for the development of adjuvants and in particular monophosphoryl lipid A (MPLA) obtained by controlled hydrolysis of LPS of Salmonella minnesota, exhibits low toxicity yet possesses beneficial immuno-stimulatory properties. We have developed an efficient synthetic approach for the preparation of a major component of MPLA (1), which has as a key feature the use of allyloxycarbonates (Alloc) as permanent protecting groups for the C-3 and C-4 hydroxyls of the proximal glucosamine unit. The latter protecting groups greatly facilitated deprotection of the fully assembled compound. Furthermore, the amino functions were protected as N-2,2,2-trichloroethoxycarbamates (Troc), which performed efficient neighboring group participation to give selectively 1,2-trans-glycosides and could easily be removed under mild conditions without affecting the permanent Alloc carbonates and anomeric dimethylthexylsilyl (TDS) ether. The synthetic methodology was also employed for the preparation of a monophosphoryl lipid A (2) derivative that has the anomeric center of the proximal sugar modified as a methyl glycoside. Compound 1 was not able to induce cytokine production in mouse macrophages whereas methyl glycoside 2 displayed activity, however it has a lower potency and efficacy than lipid A obtained by controlled hydrolysis S. minnesota. This indicates compound 2 is an attractive candidate for adjuvant development and that 1 is not the active substance of MPLA obtained by controlled hydrolysis of LPS.
Keywords: Adjuvant, Lipopolysaccharides, Tumor necrosis factor, Monophosphoryl lipid A
Introduction
Adjuvants are compounds or macromolecular complexes that boost the potency and longevity of immune response to vaccine antigens without causing toxicity.[1] They can affect the migration, maturation and antigen presentation of dendritic cells (DC), which in turn improves immune responses to antigen. Adjuvants can also affect the nature of CD4 and CD8 T-helper responses with some adjuvants promoting Th1-related responses and others preferentially inducing Th2-biased effects. Furthermore, some adjuvants enhance cross-presentation by DCs of MHC I-restricted antigens to CD8+ T cells. Adjuvants may also directly act on T- or B-cells by enhancing their proliferation and/or conversion into memory cells.
Recent studies have indicated that adjuvant activity arises in large part from activation of the innate immune system.[2] The innate immune system is an evolutionarily ancient system designed to detect the presence of microbial invaders and activate protective reactions.[3] It responds rapidly to compounds that are integral parts of pathogens that are perceived as danger signals by the host. Recognition of these molecular patterns is mediated by sets of highly conserved receptors,[4] whose activation results in acute inflammatory responses. These responses include the production of a diverse set of cytokines and chemokines, direct local attack against the invading pathogen, and initiation of responses that activate and regulate the adaptive component of the immune system.[5]
Toll-like receptor (TLR) ligands have attracted considerable attention as lead compounds for adjuvant development. However, a concern of the use of these compounds is that they can over-activate innate immune responses leading to the clinical symptoms of septic shock.[6] Thus, an important issue for the design of safe immune modulators is a detailed knowledge of structure-activity relationships to harness beneficial effects without causing toxicity.
Lipopolysaccharides (LPS), which are structural components of the outer surface membrane of Gram-negative bacteria, trigger innate immune responses through TLR4, a member of the TLR family that participates in pathogen recognition. LPS consists of a hydrophobic domain known as lipid A, a non-repeating core oligosaccharide and a distal (non-reducing end) polysaccharide (or O-antigen).[7] The lipid A moiety of E. coli consists of a hexaacylated bis-1,4′-phosphorylated glucosamine disaccharide, which has (R)-3-hydroxymyristyl residues at C-2, C-2′, C-3, and C-3′. Furthermore, both of the (3)-hydroxyacyl chains in the distal glucosamine moiety are esterified by lauric and myristic acids, and the primary hydroxyl at the C-6 position is linked to the polysaccharide through a di-KDO carbohydrate moiety (Figure 1). It has been demonstrated unequivocally that lipid A is the inflammation-inducing moiety of LPS.[8]
Figure 1.

Chemical structures of E. coli lipid A, a major component of MPLA (1) and a methyl glycoside analog (2).
Monophosphoryl lipid A, obtained by controlled hydrolysis of LPS of Salmonella minnesota (MPLA), has a much reduced toxicity yet possesses useful immunostimulatory properties.[9] It is among the first of a new generation of TLR agonists likely to be approved as a vaccine adjuvant for humans. In this respect, numerous preclinical and clinical studies have demonstrated the favorable adjuvant properties of MPLA and in general it has been found that it elicits a Th-1 or mixed Th1/Th2 response.
It has been suggested that the low toxicity of MPLA is associated with a bias toward Toll-interleukin-1 receptor (TIR)-domain-containing adapter inducing IFN-β (TRIF) signaling, due to active suppression rather than passive loss of pro-inflammatory activity of the LPS derivative.[10] Alternatively, it has been proposed that MPLA’s lack of pro-inflammatory activity is due to its inability to activate caspase-1.[11] This protease is involved in the maturation of several pro-inflammatory mediators such as IL-1β and IL-18 and the reduced production of these cytokines may explain the low toxicity of MPLA. Yet another explanation for MPLA’s low toxicity adjuvant functions is that it stimulates higher levels of the anti-inflammatory cytokine IL-10.[12]
A major compound of MPLA is a lipid A derivative with only one phosphate at C-4 of the distal GlcNAc moiety and acyloxyacyl side chains at C-2, C-2′ and C-3′ (e.g. Compound 1, Figure 1).[13]
As part of a program to develop a fully synthetic vaccine against cancer,[14] we required an efficient synthetic approach for the preparation of well-defined derivatives of MPLA.[15] During previous efforts to prepare well-defined lipid A derivatives, we observed that removal of a benzyl ether at C-4 of the proximal (reducing end) sugar by catalytic hydrogenation was very slow causing complications.[16, 17] Furthermore, sugar lactols are known to react with a wide range of reagents compromising their stability. Also, these compounds are difficult to analyze by NMR due to mixtures of alpha- and beta-lactols. To address these issues, we have developed an efficient approach for the chemical synthesis of monophosphoryl lipid A and derivatives thereof by using common disaccharide intermediates, which are convenient starting material for the preparation of a wide range of acylated derivatives. Importantly, an allyloxycarbonate (Alloc)[18] was employed as a permanent protecting group for the C-3 and C-4 hydroxyls of the proximal glucosamine moiety to avoid difficulties during deprotection. This protecting group can easily be removed by treatment with Pd(PPh3)4, without effecting acyloxyacyl- or phosphate esters. Furthermore, we have prepared a monophosphoryl lipid A derivative that has the anomeric center of the proximal sugar modified as a methyl glycoside. Previous studies have shown that replacement of an anomeric phosphate by a glycine moiety did not compromise biological activity of E. coli lipid A,[19] and thus it was hoped that methyl glycoside 2 would have comparable immunological properties to MPLA but be more stable and easier to characterize. The pro-inflammatory properties of compounds 1 and 2 have been studied by exposing mouse macrophages to the compounds followed by the measurement of the production of several cytokines, including tumor necrosis factor α (TNF-α).
Results and Discussion
Compounds 1 and 2 have been prepared by an efficient approach using the monosaccharide building blocks 3, 4 and 5 and fatty acid 6[20] (Figure 2). Our strategy for the preparation of compound 1 and 2 is based on glycosylations of trichloroacetimidate 3 with glycosyl acceptors 4 or 5, to give disaccharides that can easily be acylated and phosphorylated to give the required lipid A derivatives. The C-3 hydroxyl of 3 is already modified by a (R)-3-dodecanoyl-tetradecanoic ester because previous studies had shown that installation of this lipid at a late stage of the synthesis leads to unwanted site reaction such as phosphate migration. The amino groups of 3 is protected as N-2,2,2-trichloroethoxycarbamates (Troc), which can perform efficient neighboring group participation to provide selectively 1,2-trans-glycosides. However, this protecting group can easily be removed under mild conditions using activated Zn in acetic acid to give a free amine, which can then be acylated with 6. The benzylidene acetal of 3 can be regioselectively opened to give a C-4 hydroxyl, which can then be converted into a phosphate ester. The C-3 and C-4 hydroxyls of 4 are permanently protected as Alloc carbonates because previous studies had shown that protection by common benzyl ethers gave difficulties during deprotection. Similarly, the anomeric hydroxyl of 5 was permanently protected with dimethylthexylsilyl group (TDS), which could be removed prior to the final global reduction to yield the requisite lactol.
Figure 2.
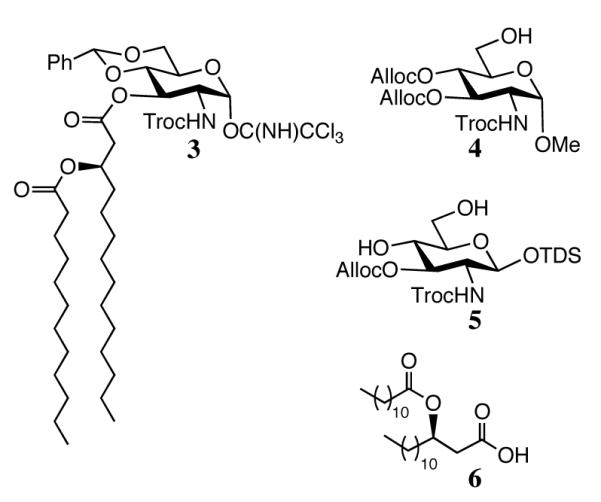
Building blocks for the synthesis of compounds 1 and 2.
Glycosyl acceptor 4 was prepared by a three-step procedure starting from compound 7[21] (Scheme 1). Thus, C-6 hydroxyl of 7 was selectively protected as TDS ether by reaction with chloro(dimethyl)thexylsilane (TDSCl) in the presence of imidazole in DMF to give 8 in a yield of 90%. The free hydroxyls of compound 8 were protected as Alloc carbonates by reaction with allyl chloroformate, in the presence of N,N,N’,N’-tetramethylethylenediamine in DCM to give fully protected 9. Removal of the TDS ether of 9 by treatment with HF-pyridine gave the target glycosyl acceptor 4.
Scheme 1.

Chemical synthesis monosaccharide building blocks. Reagent and conditions: a) TDSCl, imidazole, DMF; b) allylchloroformate, N,N,N’,N’-tetramethylethylenediamine, CH2Cl2; c) HF-pyridine, THF; d) guanidine.HCl, CH2Cl2, then dimethoxylethylbenzene, CSA, CH3CN; e) AcOH in toluene, 80 °C; f) (R)-3-dodecanoyltetradecanoic acid, EDC, DMAP, CH2Cl2; g) HF-pyridine, THF then CCl3CN, DBU, CH2Cl2.
Compounds 3 and 5 could conveniently be prepared from common building block 10.[22] Thus, the acetyl esters of 10 were removed by treatment with guanidine-HCl in DCM[23] and the 4,6-diol of the resulting compound was protected as a benzylidene acetal by treatment with dimethoxybenzaldehyde and a catalytic amount of camphorsulfonic acid (CSA) in acetonitrile to give compound 11 (Scheme 1). Acylation of 11 with (R)-3-dodecanoyltetradecanoic acid (6)[20] using the activator EDC as the activating reagent afforded 13 in a yield of 80%. Removal of the anomeric TDS moiety of 13 by treatment with HF-pyridine followed by reaction of the resulting lactol with trichloroacetonitrile in the presence of catalytic amount of DBU gave trichloroacetimidate 3 (α/β~1:1).
Glycosyl acceptor 5 was obtained by protection of the C-3 hydroxyl of 11 as an Alloc carbonate followed by selective removal of the benzylidene acetal of the resulting compound 12 using acetic acid.
Having glycosyl donor 3 and acceptors 4 and 5 at hand, attention was focused on glycosylation, acylation and phosphorylation. A triflic acid (TfOH) mediated glycosylation[24] of glycosyl donor 3 with acceptor 4 in the presence of molecular sieves in DCM at −40 °C gave the disaccharide 14 as exclusively the β-anomer in a yield of 82% (Scheme 2). Regioselective reductive ring opening of the benzylidene acetal of 14 using Et3SiH and of TfOH at −78 °C gave 15.[24, 25] The alcohol of 15 was phosphorylated to give 16 by reaction with N,N-diethyl-1,5-dihydro-2,3,4-benzodioxaphosphepin-3-amine in the presence of 1H-tetrazole followed by in-situ oxidation with m-chloroperoxybenzoic acid (m-CPBA).[26] The N-Troc groups of 16 was removed using Zn-dust in acetic acid and the resulting amino groups were immediately acylated with (R)-3-dodecanoyl-tetradecanoic acid (6)[20] using EDC and DMAP as the coupling reagents to afford 17. It was observed that the first acylation proceeded with a high reaction rate whereas the second amine reacted sluggishly. The relatively low overall yield of the two-step procedure is due to incomplete acylations. Removal of the Alloc groups of 17 could easily be accomplished by treatment with Pd(PPh3)4 in the presence of BuNH2 buffered with HCOOH[18] in THF to give compound 18 after purification by size exclusion chromatography using LH-20. Finally, hydrogenolysis over Pd-black resulted in the deprotection of the phosphotriester and removal of the benzyl ethers to provide monophosphoryl lipid A derivative 2. The structure of 2 was confirmed by NMR.
Scheme 2.

Reagents and conditions: a) TfOH, CH2Cl2, −40 °C, MS; b) TfOH, Et3SiH, CH2Cl2, −78 °C; c) N,N-diethyl-1,5-dihydro-2,4,3-benzodioxa-phosphepin-3-amine, tetrazole, m-CPBA, CH2Cl2; d) Zn-dust, AcOH, CH2Cl2; e) (R)-3-dodecanoyl-tetradecanoic acid, EDC, Et3N, CH2Cl2; f) Pd(PPh3)4, HCO2H, n-BuNH2, THF; g) 10% Pd on charcoal (Degussa), H2 (1 Atm.), THF, tBuOH.
The synthesis of MPLA 1 commenced with a regioselective glycosylation of trichloroacetimidate 3 with acceptor 5 to give formation of disaccharide 19 in high yield (Scheme 3). The C-4 hydroxyl of 19 was permanently protected as an Alloc carbonate by employing standard reaction conditions to give fully protected 20, the structure of which was supported by a down field shift of H-4 and the appearance of a multiplet at 4.53-4.45 corresponding to the vinyl protons of the Alloc carbonate. Next, regioselective reductive opening of the benzylidene acetal of 20 using Et3SiH and TfOH at −78 °C[25] gave 21, which after phosphorylation with N,N-diethyl-1,5-dihydro-2,3,4,-benzodioxaphosphepin-3-amine in the presence of 1H-tetrazole followed by in-situ oxidation with m-chloroperoxybenzoic acid (m-CPBA),[26] provided the phosphotriester 22. Removal of the Troc protecting groups of 22 using activated Zn followed by immediate acylation of the resulting amino group with (R)-3-dodecanoyl-tetradecanoic acid (6) in the presence of EDC gave compound 23. The latter derivative was deprotected to give MPLA 1 by removal of the Alloc carbonates using Pd(PPh3)4, cleavage of the anomeric TDS moiety employing HF in pyridine, and finally hydrogenation over Pd-black to remove the phosphate protecting group and benzyl ethers.
Scheme 3.

Reagents and conditions: a) TfOH, CH2Cl2, −40 °C; b) allylchloroformate, N,N,N’,N’-tetramethyl-ethylenediamine, CH2Cl2 ; c) TfOH, Et3SiH, CH2Cl2, −78 °C; d) N,N-diethyl-1,5-dihydro-2,4,3-benzodioxa-phosphepin-3-amine, tetrazole, m-CPBA, CH2Cl2; e) Zn-dust, AcOH, CH2Cl2; f) (R)-3-dodecanoyl-tetradecanoic acid, EDC, Et3N, CH2Cl2; g) Pd(PPh3)4, HCO2H, n-BuNH2, THF; h) HF-pyridine, THF; i) 10% Pd on charcoal (Degussa), THF, tBuOH, H2 (1 Atm.).
To examine the pro-inflammatory properties of the compounds, RAW 264.7 γNO(-) mouse macrophages were exposed over a wide range of concentrations to compounds 1 and 2, a commercial lipid A derivative obtained by controlled hydrolysis of the LPS of S. minnesota (MPLA), a synthetic lipid A derived from E. coli[16], and E. coli 055:B5 LPS. After 5.5 hours, the supernatants were harvested and examined for mouse TNF-α using a commercial capture ELISA. Potencies (EC50, concentration producing 50% activity) and efficacies (maximal level of production) were determined by fitting the dose-response curves to a logistic equation using PRISM software. As can be seen in Figure 3 and Table 1, compound 2, the synthetic lipid A derived from E. coli and MPLA yielded clear dose response curves. Surprisingly, compound 1 gave only a marginal response at the highest concentration tested. As expected, LPS had a greater potency than the lipid As because previous studies had shown that the KDO moiety of the core region of LPS contributes significantly to potency.[17] Furthermore, MPLA and lipid A from E. coli had similar potencies whereas methyl glycoside 2 was significantly less active.
Figure 3.

TNF-α production by murine macrophages after stimulation with E. coli LPS, E. coli lipid A, MPLA and synthetic compounds 1 and 2. Murine RAW 264.7 γNO(−) cells were incubated for 5.5 h with increasing concentrations of E. coli LPS, E. coli lipid A, MPLA and compounds 1 and 2 as indicated. TNF-α in cell supernatants was measured using an ELISA. Treatment with E. coli LPS, E. coli lipid A, MPLA, 1 and 2 did not affect cell viability, as judged by cellular exclusion of trypan blue.
Table 1.
EC50 values[a] (nM) of E. coli LPS and lipid A, MPLA and compound 2.
| E. coli LPS | E. coli lipid A | MPLA | Lipid A 2 | |
|---|---|---|---|---|
| TNF-α | 0.0091 (0.0074 – 0.011) |
35 (26 – 48) |
11 (9 – 14) |
184 (112 – 302) |
| IFN-β | 0.011 (0.088 – 0.013) |
229 (158 – 332) |
30 (20 – 46) |
299 (225 – 397) |
| IP-10 | 0.010 (0.087 – 0.012) |
161 (115 – 226) |
33 (26 – 43) |
139 (42 – 458) |
| IL-6 | 0.039 (0.029 – 0.053) |
184 (115 – 292) |
66 (56 – 77) |
3287 (2552 – 4237) |
Values of EC50 are reported as best-fit values and as minimum-maximum range (best-fit value ±std. error).
Importantly, the various compounds displayed differences in maximum response (efficacy) of TNF-α production with compound 2 having a lower efficacy than MPLA, which in turn had a lower maximum response than E. coli lipid A and LPS (Table 2). The supernatants were also investigated for the presence of IFN-β, IP-10 and IL-6 and similar trends for both potencies and efficacies were observed as for the presence of TNF-α.
Table 2.
Cytokine top values[a] (pg/mL) of dose-response curves of E. coli LPS and lipid A, MPLA and compound 2.
| E. coli LPS | E. coli lipid A | MPLA | Lipid A 2 | |
|---|---|---|---|---|
| TNF-α | 5782 ± 169 | 6197 ± 362 | 4783 ±185 | 2216 ± 177 |
| IFN-β | 654 ± 19 | 550 ± 37 | 418 ± 30 | 134 ± 6 |
| IP-10 | 11847 ± 367 | 11879 ± 692 | 7898 ± 353 | 2498 ± 474 |
| IL-6 | 2103 ± 77 | 1873 ± 152 | 1643 ± 51 | 622 ± 33 |
Top values are reported as best-fit values ± std. error.
The observation that synthetic methyl glycoside 2 has a significantly higher pro-inflammatory activity than its synthetic hydroxyl counter part 1 indicates that the anomeric methyl moiety can make favorable interactions with the TLR4/MD2 complex leading to cellular activation. The efficacy of cytokine induction by compound 2 was significantly lower than that of E. coli lipid A and LPS. The reduced potency and efficacy of 2 are expected to be beneficial for adjuvant development because sufficient levels of cytokines will be elicited for the induction of relevant responses, yet toxicity will be minimized.
The commercial lipid A from S. minnesota obtained by controlled hydrolysis of isolated material showed activity at a surprisingly low concentration indicating that it contains compounds other than 1 that induces pro-inflammatory responses. Recently, we found that shortening the fatty acids of monophosphoryl lipid A derivative derived from S. thyphimurium increased proinflammatory properties by as much as a thousand fold.[17] It is possible that the sample obtained by controlled hydrolysis may contain lipid A derivatives that have shorter fatty acids or contaminants that may account for the high activity. It is important to note that therapeutic grade MPLA may not contain these components and hence may exhibit a different proinflammatory profile.
Methyl glycoside 2 will be an attractive compound for incorporation into fully synthetic vaccines comprised of an inbuilt adjuvant, a helper T-epitope and a cancer-associated glycopeptide. Our previous studies have shown that such compound can elicit robust antigenic responses against tumor-associated carbohydrates.[14] It is to be expected that the methyl glycoside of 2 will protect the anomeric center making it possible to employ a variety of conjugation reactions for attachment to a glycopeptide antigen using an appropriate linker.
Conclusions
In conclusion, we have developed an efficient chemical synthesis of a monophosphoryl lipid A derivative from S. minnesota. A key feature of the approach was the permanent protection of the C-3 and C-4 hydroxyls of the proximal glucosamine moiety as Alloc carbonates, which greatly facilitated the final deprotection steps. The methyl glycoside 2 displayed lower pro-inflammatory activity than a lipid A derivative obtained by controlled hydrolysis of LPS of S. minnesota and LPS and lipid A derived from E. coli. The adjuvant properties of the synthesis derivatives are under investigations.
Experimental Section
Chemical Synthesis
General Synthetic Methods
Column chromatography was performed on silica gel 60 (EM Science, 70-230 mesh). Reactions were monitored by thin-layer chromatography (TLC) on Kieselgel 60 F254 (EM Science), and the compounds were detected by examination under UV light and by charring with 10% sulfuric acid in MeOH. Solvents were removed under reduced pressure at <40 °C. DCM was distilled from NaH and stored over molecular sieves (3 Å). THF was distilled from sodium directly prior to the application. MeOH was dried by refluxing with magnesium methoxide and then was distilled and stored under argon. Pyridine was dried by refluxing with CaH2 and then was distilled and stored over molecular sieves (3 Å). Molecular sieves (3 and 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2-3 h at 390 °C directly prior to application. Optical rotations were measured with a Jasco model P-1020 polarimeter. 1H NMR and 13C NMR spectra were recorded with Varian spectrometers (models Inova300 and Inova500) equipped with Sun workstations. NMR spectra were recorded in CDCl3 and referenced to residual CHCl3 at 7.24 ppm, and 13C NMR spectra were referenced to the central peak of CDCl3 at 77.0 ppm. Assignments were made by standard gCOSY and gHSQC. High-resolution mass spectra were obtained on a Bruker model Ultraflex MALDI-TOF mass spectrometer. Signals marked with a subscript L symbol belong to the biantennary lipids, whereas signals marked with a subscript L’ symbol belongs to their side chain.
Methyl 6-O-dimethylthexylsilyl-2-(2,2,2-trichloromethoxycarbonylamino)-2-deoxy-α-D-glucopyranoside (8)
Imidazole (6.05 g, 88.8 mmol) was added to a solution of compound 7 (31.2 g, 88.8 mmol) in DMF (25 mL). After stirring the mixture for 10 min, chloro(dimethyl)thexylsilane (17.4 g, 97.7 mmol) was added. The reaction mixture was stirred for 18 h and then the reaction was quenched by the addition of a saturated solution of Na2CO3 (50 mL). The crude material was extracted with DCM (200 mL) and washed with water (3 × 100 mL) and brine (2 × 100 mL). The organic phase was dried (MgSO4), filtered and the filtrate concentrated in vacuo. The crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 5:1, v/v) to give compound 8 as a clear oil (39.5 g, 90%). 1H NMR (300 MHz, acetone-d6): δ 6.49 (d, J = 8.1 Hz, 1H, NH), 4.78-4.67 (m, 2H, OCH2CCl3) 4.65 (d, J1-2 = 3.6 Hz, 1H, H-1), 4.13 (dd, J3- 4 = 5.1 Hz, J4-5 = 4.5 Hz, 1H, H-4), 3.89-3.71 (m, 2H, H-6) 3.65-3.54 (m, 1H, H-3), 3.45 (dd, J1-2 = 3.6 Hz, J2-3 = 2.7 Hz, 1H, H-2), 3.38-3.34 (m, 1H, H-5), 3.28 (s, 3H, OCH3) 1.49-1.43 (m, 1H, CH3CHCH3), 0.733-0.633 (m, 12H, CH3), 0.007-0.00 (m, 6H, CH3Si). 13C NMR (75 MHz, acetone-d8): 205.88 (C=O), 99.12 (C-1), 76.10 (C-4), 72.13 (C-3), 71.49 C-6), 66.63 (C-2), 65.28 (C-5), 34.2, 24.85, 19.96, 18.47, 18.45 (CH3). HR-MALDI-ToF/MS: m/z: calcd for C18H34Cl3NO7Si 509.1170; found 532.1175 [M + Na]+.
Methyl 6-O-dimethylthexylsilyl-3,4-O-allyloxycarbonyl-2-(2,2,2-trichloromethoxy carbonylamino)-2-deoxy-α-D-glucopyranoside (9)
Compound 8 (36.0 g, 70.7 mmol) was dissolved in DCM (20 mL) followed by the addition of N,N,N’,N’-tetramethylethylenediamine (8.16 g, 70.4 mmol). The reaction temperature was dropped to 0 °C followed by the addition of allylchloroformate (8.52 g, 70.7 mmol). Next, the reaction was warmed up to room temperature over a period of 24 h. The crude material was extracted with DCM (250 mL) and the organic phase washed with water (2 × 100 mL) and brine (2 × 100 mL). The organic phase was dried (MgSO4), filtered and the filtrate concentrated in vacuo. The crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 3:1, v/v) to yield compound 9 as a light yellow oil (41 g, 87%). 1H NMR (300 MHz, CDCl3): δ 5.88-5.72 (m, 2H, OCH2CH=CH2) 5.28-5.17 (m, 4H, OCH2CH=CH2) 5.15 (d, J1-2 = 6.6 Hz, 1H, H-1), 5.05 (dd, J2-3 = 9.6 Hz, J3-4 = 10.2 Hz, 1H, H-3), 4.84 (dd, J3-4 = 9.6 Hz, J4-5 = 9.6 Hz, 1H, H-4), 4.55-4.49 (m, 2H, OCH2CCl3), 4.00-3.94 (m, 2H, H-2, H-6) 3.75-3.71 (m, 2H, H-5, H-6), 3.64-3.62 (m, 1H, H-6) 3.32(s, 3H, OCH3). 13C NMR (75 MHz, CDCl3): δ 158.54, 157.69, 157.33 (C=O), 134.81(OCH2CH=CH2), 122.55 (OCH2CH=CH2), 122.34 (OCH2CH=CH2) 101.61(C-1), 73.00 (C-3), 78.12 (C-4), 76.05 (C-6), 73.80 (C-2), 72.42 (C-5), 32.96 (O-CH3), 24.85, 19.96, 18.47, 18.45 (CH3). HR-MALDI-ToF/MS: m/z calcd for C26H42Cl3NO11Si 677.1593; found 700.1597 [M + Na]+.
Methyl 3,4-O-allyloxycarbonyl-2-(2,2,2-trichloromethoxycarbonylamino)-2-deoxy-α-D-glucopyranoside (4)
Compound 9 (1.5 g, 2.21 mmol) was dissolved in THF followed by the addition of HF/pyridine (0.5 mL). The resulting reaction mixture was stirred for 8 h at room temperature. The reaction was quenched with NaHCO3 and extracted with DCM (250 mL). The organic phase was washed with water (2 × 100 mL) and brine (2 × 100 mL). The organic phase was dried (MgSO4), filtered and the filtrate concentrated in vacuo. The crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 1:2, v/v) to yield compound 4 as a white amorphous solid (1.02 g, 86%). 1H NMR (300 MHz, acetone-d6): δ 6.79 (d, J = 9.9 Hz, 1H, NH) 5.98-5.84 (m, 2H, OCH2CH=CH2) 5.60-5.23 (m, 4H, OCH2CH=CH2) 5.22 (d, J1-2 = 4.8 Hz, 1H, H-1), 5.20 (dd, J2- 3 = 4.8 Hz, J3-4 = 9.6 Hz, 1H, H-3), 4.96 (dd, J3-4 = 9.6 Hz, J4-5 = 9.7 Hz, 1H, H-4), 4.83 (brs., 2H, OCH2CCl3), 4.62-4.55 (m, 4H, OCH2CH=CH2) 4.10-4.02 (m, 2H, H-2, H-6) 3.86-3.81 (m, 2H, H-5, H-6), 3.74-3.62 (m, 1H, H-6) 3.47(s, 3H, OCH3). 13C NMR (75 MHz, acetone-d6): δ 154.81 (C=O), 154.73, 154.26 (OCH2CH=CH2), 132.22 (OCH2CH=CH2), 117.94,117.84 (OCH2CH=CH2) 98.52 (C-1), 75.57 (CH2-O) 73.19(C-3), 74.28(C-4), 73.19(C-6), 70.31(C-2), 68.60(C-5), 32.96 (O-CH3). HR-MALDI-ToF/MS: m/z calcd for C18H24Cl3NO11 536.0415; found 559.0419 [M + Na]+.
Dimethylthexylsilyl 4,6-O-benzylidine-3-O-allyloxycarbonyl-2-(2,2,2-trichloromethoxy carbonylamino)-β-D-glucopyranoside (11)
Compound 1 (25 g, 50.3 mmol) was dissolved in acetonitrile (100 mL) and dimethoxy benzaldehyde (15.3 g, 100.6 mmol) and camphor sulfonic acid were added (1.17 g, 5.03 mmol) subsequently. The reaction mixture was stirred at room temperature for 8 h. The solvent was removed under vacuo and the crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 3:1, v/v) to give 11 as a white amorphous solid material (28.3 g, 96%). 1H NMR (300 MHz, acetone-d8): δ 7.34-7.15 (m 5H, aromatic), 6.72 (d, J = 9.6 Hz, 1H, NH), 5.41 (s >CHPh) 4.72 (d, J1-2 = 5.7 Hz, 1H, H-1), 4.61-4.49 (m, 3H, H-2, COOCH2, Troc), 4.05 (dd, J3-4 = 10.5 Hz, J4-5 = 5.2 Hz, 1H, H-4), 3.71 (m, 1H, H-3), 3.58 (dd, J1-2 = 5.2 Hz, J2-3 = 10.5 Hz, 1H, H-2), 3.34-3.33 (m, 2H, H-6), 3.29-3.21 (m, 1H, H-5), 1.49-1.43 (m, 1H, CH3CHCH3), 0.733-0.633 (m, 12H, CH3), 0.007-0.00 (m, 6H, CH3Si). 13C NMR (300 MHz, acetone-d8): 205.88(C=O), 154.89, 138.46, 128.96, 128.18, 126.72 (Ar), 101.52(CHPh), 97.18(C-1), 82.10(C-3), 74.61(C-4), 71.31(C-6), 68.63(C-2), 66.68(C-5), 34.2, 24.85, 19.96, 18.47, 18.45 (CH3). HR MS (m/z) calcd for C24H36Cl3NO7Si 583.1301; found 603.1306 [M + Na]+.
Dimethylthexylsilyl-3,4-O-Allyloxycarbonyl-2-(2,2,2-trichloromethoxycarbonylamino)-β-D-glucopyranoside (12)
A solution of 11 (2.5g, 4.27 mmol) in DCM (12 mL) and N,N,N’,N’-tetramethylethylenediamine (0.45 mL, 2.99 mmol) was cooled (0 °C) and allyl chloroformate (0.55 mL, 5.12 mmol) was added drop-wise. Stirring was continued at room temperature for 18 h. The reaction mixture was quenched with saturated solution of NaHCO3 (3.0 mL) and diluted with DCM (30 mL). The organic layer was washed with water (2 × 15 mL) and brine (1 × 10 mL), dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel column chromatography (hexane:ethylacetate, 5:1, v/v → 4:1, v/v) to yield 12 as a white foam (2.1g, 76%). Rf = 3.9 (hexane/ethylacetate, 1:1, v/v); 1H-NMR (300 MHz, CDCl3): δ 7.41-7.22 (m, 5H, aromatic), 5.82-5.69 (m, 1H, OCH2CH=CH2), 5.38 (s, 1H, >CHPh), 5.22-5.06 (m, H-3, NH, OCH2CH=CH2), 4.74 (d, J = 8.8 Hz, 1H, H-1), 4.64 (d, J = 9.5 Hz, 1H, NH), 4.51-4.46 (m, 4H, OCH2CCl3, OCH2CH=CH2), 4.16 (dd, J4,5 = 8.8 Hz, J5,6a = 6.8 Hz, 1H, H-5), 3.70-3.27 (m, 4H, H-4, H-6ab, H-2), 1.54-1.46 (m, 1H, OSi(CH3)3C(CH3)3CH(CH3)3), 0.75-0.71 (m, 12H, 4CH3), 0.02 (s, 3H, SiCH3), −0.03 (s, 3H, SiCH3); 13C NMR (75 MHz, CDCl3): δ = 158.35 (C=O), 157.39 (C=O), 140.34, 134.53, 132.52, 131.61, 129.64, 122.48, 104.88, 99.96, 82.25, 78.53, 78.10, 72.36, 72.00, 69.66, 62.36, 37.33, 28.19, 23.34, 23.31, 21.91, 1.50 (SiCH3), −0.00 (SiCH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C28H40Cl3NO9 SiNa 692.0630; found 692.7123.
Dimethylhexylsilyl 3-O-[(R)-3-dodecanoyloxy-tetradecanoyl]-4,6-O-benzylidine-2-(2,2,2-trichloromethoxycarbonylamino)-β-D-glucopyranoside (13)
(R)-3-dodecanoyl-tetradecanoic acid (0.64 g, 0.150 mmol) was added to a solution of compound 11 (0.80 g, 0.137 mmol) in DCM (5 mL). EDC (0.29 g, 0.150 mmol) and DMAP (8 mg) were added sequentially and the resulting reaction mixture was stirred for 8 h at room temperature. The mixture was diluted with DCM (20 mL) and washed with 1N HCl (2 × 100 mL), water (2 × 100 mL) and brine (2 × 100 mL). The organic phase was dried (MgSO4), filtered and the filtrate concentrated in vacuo. The crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 15:1, v/v → 6:1, v/v) to yield compound 13 as a clear oil (1.08 g, 80%). 1H NMR (300 MHz, CDCl3): δ 7.37-7.14 (m 5H, aromatic), 6.77 (d, J =9.6 Hz, 1H, NH), 5.46 (s, 1H, >CHPh), 5.22. (t, J = 9.2 Hz, 1H, H-3), 5.14-5.11 (m, 1H, 3(R) CH of dodecanoyl moiety), 4.78 (d, J = 9.7 Hz, 1H, H-1), 4.64 (d, J = 13.4 Hz, 1H, OCHHCCl3), 4.48 (d, J = 14.1 Hz, 1H, OCHHCCl3), 4.19 -4.16 (m, 1H, H-2), 3.68 (t, J = 10.4 Hz, 1H, H-4), 3.58-3.36 (m, 3H, H-6ab, H-5), 2.50 (dd, J2Sa, 2Sb = 16.2 Hz, J 2Sa, 3S = 6.1 Hz, 1H, H-2Sa), 2.43 (dd, J2Sa, 2Sb = 13.4 Hz, J 2Sa, 3S = 6.7 Hz, 1H, H-2Sb), 2.07 (t, J = 6.9 Hz, 2H, CH2 of lipids), 1.40-1.45 (m, 4H), 1.20-1.10 (m, 34H, CH2 lipids), 0.82-0.76 (m, 12H, 4CH3), 0.06 (s, 3H, SiCH3), 0.02 (s, 3H, SiCH3); 13C NMR (75 MHz, CDCl3): 172.49, 169.53, 154.60 (C=O), 138.46, 128.93, 128.14, 126.49, 101.52(CHPh), 96.88(C-1), 79.19(C-3), 74.38(C-4), 72.00(C-6), 68.48(C-2), 66.59(C-5), 38.79, 34.17, 34.05, 33.69, 32.00, 29.96, (CH2-lipids), 34.2, 24.85, 19.96, 18.47, 18.45 (CH3). HR MS (m/z) calcd for C50H84Cl3NO10Si 991.4921; found 1014.4929 [M+Na]+.
Dimethylhexylsilyl 3-O-allyloxycarbonyl-2-(2,2,2-trichloromethoxycarbonylamino)-β-D-glucopyranoside (5)
Glacial acetic acid in toluene (90%, 10 mL) was added to a solution of compound 12 (1.5 g, 2.24 mmol) in toluene (5.5 mL), and the reaction mixture was heated to 80 °C for 15 h. The reaction mixture was concentrated in vacuo and the residue co-evaporated three times from toluene to remove residues of acetic acid. The residue was recrystallized from ethyl acetate:hexane (2:1, v/v) to give 5 as a white solid (1.1 g, 82%). Rf = 2.1 (hexane/ethyl acetate, 1:1, v/v); mp 115 °C; 1H-NMR (300 MHz, CDCl3): δ 5.92-5.81 (m, 1H, OCH2CH=CH2), 5.33-5.21 (m, 3H, NH, OCH2CH=CH2), 4.91 (d, J = 9.2 Hz, 1H, H-3), 4.80 (d, J = 9.5 Hz, 1H, H-1), 4.73 (d, J = 16.2 Hz, 1H, OCH HCCl3), 4.59-4.56 (m, 3H, OCH HCCl3, OCH2CH=CH2), 3.83-3.71 (m, 3H, H-6ab, H-4), 3.55-3.39 (m, 2H, H-2, H-5), 1.60-1.51 (m, 1H, OSi(CH3)3C(CH3)3CH(CH3)3), 0.82-0.76 (m, 12H, 4CH3), 0.10 (s, 3H, SiCH3), 0.09 (s, 3H, SiCH3); 13C NMR (75 MHz, CDCl3): δ = 159.00 (C=O), 157.43 (C=O), 134.40, 122.73, 99.21, 98.69, 82.24, 78.35, 78.00, 72.97, 72.51, 65.73, 61.51, 37.28, 28.15, 23.31, 23.29, 21.86, 1.53 (SiCH3), −0.09 (SiCH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C21H36Cl3NO9 SiNa 603.9563; found 603.0630.
3-O-[(R)-3-dodecanoyloxy-tetradecanoyl]-4,6-O-benzylidine-2-(2,2,2-trichloromethoxy carbonylamino)-α-D-glucopyranosyl trichloroacetimidate (3)
HF/pyridine (0.5 mL) was added to a solution of compound 13 (1.5g, 2.21 mmol) in THF. After stirring the reaction mixture for 8 h at room temperature, it was quenched with NaHCO3 and extracted with DCM (250 mL). The organic phase was washed with water (2 × 100 mL) and brine (2 × 100 mL), dried (MgSO4), filtered and the filtrate was concentrated in vacuo. The crude product was purified by flash silica gel column chromatography (hexane:ethyl acetate, 1:2, v/v) to yield a lactol as a white amorphous solid (1.02 g, 86%). 1H NMR (300 MHz, CDCl3): δ 7.48-7.31 (m 5H, aromatic), 5.88 (d, J = 8.4 Hz, 1H, NH), 5.48-5.46 (m, 2H, >CHPh, H-1), 5.42 (t, J = 10.6 Hz, 1H, H-3), 5.29 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 4.74 (d, J = 17.3 Hz, 1H, OCHHCCl3), 4.62 (d, J = 18.4 Hz, 1H, OCHHCCl3), 4.24-3.88 (m, 3H, H-4, H-2, H-5), 3.73-3.68 (m, 2H, H-6ab), 2.54 (dd, J2Sa, 2Sb = 16.2 Hz, J 2Sa, 3S = 8.1 Hz, 1H, H-2Sa), 2.47 (dd, J2Sa, 2Sb = 17.4 Hz, J 2Sa, 3S = 7.8 Hz, 1H, H-2Sb), 2.12 (t, J = 9.5 Hz, 2H), 1.49 (br.s., 4H CH2 lipids), 1.27-1.16 (br.s., 34H, CH2 lipids) 0.83 (t, J = 7.3 Hz, 6H, 2x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 173.94 (C=O), 170.65 (C=O), 154.73, 137.86, 129.31, 128.45, 128.43, 126.36, 79.63, 74.83, 70.32, 62.98, 57.63, 34.58, 32.14, 29.74, 25.71, 22.91; HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C42H66Cl3NO10Na 874.3331; found 874.2731. The resulting lactol (0.70 g, 0.824 mmol) was diluted in dry DCM (1.0 mL) under a blanket of argon. Trichloroacetonitrile (0.82 mL) and DBU (0.02 mL, 0.164 mmol) were added to the reaction mixture and the mixture was stirred for 3 h at room temperature. The crude was purified directly using flash silica gel column chromatography (hexane:ethyl acetate:Et3N, 7:1:0.01, v/v/v) to give 3 as a light yellow oil material (0.792 g, 97%). 1H NMR (300 MHz, CDCl3): δ 8.89 (s, 1H, OC(CCl3)NH), 7.58-7.38 (m 5H, aromatic), 6.48 (d, J = 6.4 Hz, 1H, NH), 5.55 (s, 1H, >CHPh), 5.53 (d, J1-2= 4.2 Hz, 1H, H-1), 5.46 (t, J = 9.8 Hz, 1H, H-3), 5.18 (t, J = 7.2 Hz, 1H, 3(R) CH of dodecanoyl moiety), 4.81 (d, J = 16.3 Hz, 1H, OCHHCCl3), 4.65 (d, J = 15.6 Hz, 1H, OCHHCCl3), 4.45-4.22 (m, 2H, H-4, H-2), 4.10-4.01 (m, 1H, H-5), 3.84-3.79 (m, 2H, H-6ab), 2.62 (dd, J2Sa, 2Sb = 14.2 Hz, J 2Sa, 3S = 7.1 Hz, 1H, H-2Sa), 2.59 (dd, J2Sa, 2Sb = 14.4 Hz, J 2Sa, 3S = 6.8 Hz, 1H, H-2Sb), 2.22 (t, J = 8.5 Hz, 2H), 1.63-1.45 (m, 4H, CH2 lipids), 1.27-1.16 (br.s., 34H, CH2 lipids) 0.85 (t, J = 7.3 Hz, 6H, 2x CH3, lipid). 13C NMR (75 MHz, CDCl3): δ 172.12 (C=O), 170.43 (C=O), 161.21, 156.21, 138.86, 131.32, 128.85, 126.19, 101.64, 78.36, 76.22, 67.73, 66.87, 64.76, 58.63, 38.08, 34.86, 32.54, 30.37, 29.62, 23.71, 22.27; HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C45H68Cl6N2O9Na 1016.7474; found 1016.9106.
Methyl [4′,6′-O-benzylidine-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-[{(R)-3-dodecanoyloxy-tetradecanoyl-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxy carbonylamino)-3,4-O-Allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (14)
A mixture of glycosyl acceptor 4 (228 mg, 0.424 mmol), trichloroacetamidate donor 3 (470 mg, 0.472 mmol) and molecular sieves (4 A, 700 mg) in DCM (2.5 mL) was placed under an atmosphere of Ar and cooled to −40 °C. TfOH (8.35 μl, 0.094 mmol, in 0.1 mL DCM) was added and stirring was continued for 20 min until the temperature had reached 0 °C. The reaction was quenched by the addition of triethylamine (35 μL). Next, the mixture was diluted with DCM (30 mL) and washed with sat. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate, 9:1, v/v → 5:1, v/v) to yield disaccharide 14 (480 mg, 82 %) as a white foamy solid. Rf = 0.46 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = −8.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.43-7.42 (m, 2H, aromatic), 7.35-7.32 (m, 3H, aromatic), 6.76 (br.s., 1H, NH), 6.42 (br.s., 1H, NH’), 5.94-5.87 (m, 2H, OCH2CH=CH2), 5.50 (s, 1H, PhCH), 5.38-5.33 (m, 4H, OCH2CH=CH2), 5.19 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 5.14 (t, J = 8.4 Hz, 1H, H-3′), 5.10 (t, J = 8.9 Hz, 1H, H-3), 4.49 (t, J = 9.1 Hz, 1H, H-4), 4.84 (t, J = 8.9 Hz, partially merged with H-1′, H-4′), 4.82 (d, J = 9.8 Hz, 1H, H-1′), 4.74 (d, J = 3.8 Hz, 1H, H-1), 4.64-4.56 (m, 8H, 2x OCH2CH=CH2, OCH2CCl3), 4.37-4.34 (m, 1H), 4.11-4.07 (m, 2H, H-2, H-2′), 3.94-3.92 (m, 2H, H-5, H-5′), 3.77 (dd, J5′,6′a = 4.9 Hz, J6′a,6′b = 16 Hz, 2H, H-6′ab), 3.67-3.58 (m, 2H, H-6ab), 3.53 (s, 3H, OCH3), 2.62 (dd, J2Sa, 2Sb = 15.2 Hz, J 2Sa, 3S = 7.5 Hz, 1H, H-2Sa), 2.51 (dd, J2Sa, 2Sb = 15.0 Hz, J 2Sa, 3S = 6.4 Hz, 1H, H-2Sb), 2.15 (t, J = 7.5 Hz, 2H), 1.53-1.52 (m, 5H), 1.31-1.29 (m, 1H, CH(CH3)2), 1.28-1.20 (m, 33H), 0.88(t, J = 7.0 Hz, 6H, 2CH3); 13C NMR (75 MHz, CDCl3): δ = 174.34 (C=O), 170.11 (C=O), 164.21 (C=O), 156.31(C=O), 155.87 (C=O), 138.92 (OCH2CH=CH2), 132.86 (OCH2CH=CH2), 128.86, 126.12 (OCH2CCl3), 103.32 (C-1′), 98.65 (C-1), 78.12 (H-3), 76.19 (C-3′), 72.34 (C-4), 71.12 (PhCH), 68.30 (OCH2CCl3), 67.26(C-5), 66.71 (C-5′), 64.23 (C-6ab), 63.19 (C-6′ab), 59.23 (C-2′), 58.21 (C-2), 56.72 (OCH3), 38.23, 34.12, 32.10, 28.43, 23.19 (CH2 of lipid chains), 15.13 (terminal CH3 of lipid chain), 14.89 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C60H88Cl6N2O20Na 1393.9021; found 1393.9182.
Methyl [6′-O-benzyl-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-[{(R)-3-dodecanoyloxy-tetradecanoyl-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxy carbonylamino)-3,4-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (15)
A mixture of disaccharide 14 (212 mg, 0.154 mmol) and molecular sieves (4A, 200 mg) in DCM (3.0 mL) was placed under an atmosphere of Ar and then cooled to −78 °C. TfOH (27.5 μl, 0.471 mmol) and triethylsilane (62 μl, 0.554 mmol) were added and the reaction mixture was stirred at −78 °C for 1 h. The reaction was quenched by the addition of triethylamine (50 μL) and methanol (0.2 mL). Next, the mixture was diluted with DCM (35 mL), and washed with sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate, 9:1, v/v→ 4:1, v/v) to yield the disaccharide 15 (148 mg, 69 %) as colorless syrup. Rf = 0.38 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = +6.1° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.43-7.31 (m, 5H, aromatic), 5.88-5.83 (m, 2H, OCH2CH=CH2), 5.58 (d, J = 10.2 Hz, 1H, NH), 5.32 (s, 2H, PhCH2), 5.30-5.22 (m, 4H, OCH2CH=CH2), 5.13 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 5.12 (t, J = 9.1 Hz, 1H, H-3′), 5.11 (t, 1H, H-3 merged with H-3′), 5.02 (t, J = 10.1 Hz, 1H, H-4), 4.98 (t, J = 9.1 Hz, 1H, H-4′), 4.84 (d, J = 9.9 Hz, 1H, H-1′), 4.75 (d, J = 3.5 Hz, 1H, H-1), 4.66-4.58 (m, 8H, 2x OCH2CH=CH2, OCH2CCl3), 4.42 (d, J = 8.0 Hz, 1H, NH), 4.10-4.02 (m, 2H, H-2, H-2′), 3.81-3.75 (m, 2H, H-5, H-5′), 3.67 (dd, J5′,6′a = 4.9 Hz, J6′a,6′b = 16 Hz, 2H, H-6′ab), 3.57-3.52 (m, 2H, H-6ab), 3.39 (s, 3H, OCH3), 2.59 (dd, J2Sa, 2Sb = 15.2 Hz, J 2Sa, 3S = 7.5 Hz, 1H, H-2Sa), 2.48 (dd, J2Sa, 2Sb = 15.0 Hz, J 2Sa, 3S = 6.4 Hz, 1H, H-2Sb), 2.27 (t, J = 7.5 Hz, 2H), 1.59-1.55 (m, 5H), 1.31-1.25 (m, 33H), 0.88(t, J = 5.9 Hz, 6H, 2CH3); 13C NMR (gHSQC, CDCl3): δ 129.56 (OCH2CH=CH2), 119.81, 118.54 (OCH2CCl3), 98.08 (C-1′), 96.75 (C-1), 75.32 (C-3), 74.19 (C-3′), 73.81 (OCH2CH=CH2), 71.84 (C-4), 69.81 (OCH2CCl3), 66.36(C-6ab), 65.71 (C-5′), 64.09 (C-6′ab), 59.13 (C-2′), 56.06 (C-2), 39.13, 38.82, 36.10, 27.83, 21.19 (CH2 of lipid chains), 15.13 (terminal CH3 of lipid chain), 14.89 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C60H90Cl6N2O20Na 1395.0760; found 1395.6084.
Methyl [6′-O-benzyl-4-O-(1,5-dihydro-3-oxo-3λ5-3H-2,4,3-benzodioxaphosphepin-3yl)-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-[{(R)-3-dodecanoyloxy-tetradecanoyl-2′- deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxycarbonylamino)-3,4-O-allyloxy carbonyl-2-deoxy-α-D-glucopyranoside (16)
Disaccharide 15 (120 mg, 0.087 mmol) in DCM (2.5 mL) was placed under an atmosphere of Ar, charged with 3% tetrazole solution in acetonitrile (0.8 mL, 0.145 mmol), and N,N-diethyl-1,5-dihydro-2,4,3-benzodioxa-phosphepin-3-amine (55 μL, 0.11 mmol) was added. After 45 min, the reaction mixture was cooled to −20 °C and m-CPBA (103 mg, 0.255 mmol) in DCM (2 mL) was added. The reaction mixture was stirred at 0 °C for another 1 h and then diluted with DCM (25 mL), washed with a sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate, 4:1, v/v → 1:1, v/v) to give 16 (110 mg, 83 %) as white foamy solid. Rf = 0.39 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = +9.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.38-7.17 (m, 9H, aromatic), 5.95-5.82 (m, 2H, OCH2CH=CH2), 5.65 (d, J = 7.5 Hz, 1H, NH), 5.44(t, J = 9.5 Hz, 1H, H-4′), 5.32-5.23 (m, 4H, OCH2CH=CH2), 5.12 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 5.10 (s, 2H, PhCH2), 5.08-5.05 (m, 2H), 4.92 (t, J = 9.6 Hz, 1H, H-3′), 4.78 (d, J = 10.1 Hz, 1H, H-1′), 4.76 (d, J = 3.0 Hz, 1H, H-1), 4.71 (t, J = 9.8 Hz, 1H, H-4), 4.63-4.58 (m, 8H, 2x OCH2CH=CH2, OCH2CCl3), 4.10-4.03 (m, 2H, H-2, H-2′), 3.93-3.89 (m, 2H, H-5, H-5′), 3.86-3.83 (m, 2H, H-6′ab), 3.73-3.70(m, 2H, H-6ab), 3.39 (s, 3H, OCH3), 2.68 (dd, J2Sa, 2Sb = 15.2 Hz, J 2Sa, 3S = 7.5 Hz, 1H, H-2Sa), 2.48 (dd, J2Sa, 2Sb = 15.0 Hz, J 2Sa, 3S = 6.4 Hz, 1H, H-2Sb), 2.26 (t, J = 7.5 Hz, 2H), 1.58-1.42 (m, 5H), 1.28-1.25 (m, 33H), 0.87(t, J = 5.9 Hz, 6H, 2CH3); 13C NMR (gHSQC, CDCl3): δ = 133.16 (OCH2CH=CH2), 120.41, 119.36 (OCH2CCl3), 98.93 (C-1′), 97.05 (C-1), 76.33 (C-3), 74.03 (OCH2CH=CH2), 71.92 (C-4), 69.01 (OCH2CCl3), 67.40 (C-6ab), 65.31 (C-5′), 64.29 (C-6′ab), 58.83 (C-2′), 56.24 (C-2), 39.33, 38.04, 36.75, 27.65, 24.30 (CH2 of lipid chains), 15.22 (terminal CH3 of lipid chain), 14.98 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C68H97Cl6N2O23P Na 1574.4321; found 1574.6469.
Methyl [6′-O-benzyl-3′-O-(R)-3-dodecanoyloxy-tetradecanoyl-4-O-(1,5-dihydro-3-oxo-3λ5-3H-2,4,3-benzodioxaphosphepin-3yl)-2′-[{(R)-3-dodecanoyloxy-tetradecanoylamino–{2′-deoxy-β-D-glucopyranosyl}]-2-(R)-3-dodecanoyloxy-tetradecanoylamino-3,4-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (17)
A solution of disaccharide 16 (105 mg, 0.029 mmol) in DCM (2 mL) was placed under an atmosphere of Ar, and Zn dust (134 mg, 0.868 mmol) and acetic acid (0.05 mL) were added. After stirring at room temperature for 1.5 h, the reaction mixture was diluted with DCM (20 mL), and filtered through a bed of celite. The filtrate was concentrated and the residue was co-evaporated three times with toluene, and then dried in vacuo. The removal of the Troc protecting group was confirmed by MALDI-TOF MS analysis. The crude free amine (80 mg) was diluted in dry DCM (1.5 mL) followed by the addition of EDC (50.9 mg, 0.027 mmol) and (R)-3-dodecanoyl-tetradecanoic acid (85.2 mg, 0.02 mmol) in DCM (2 mL) at room temperature under an atmosphere of Ar. After 24 h of stirring at room temperature, the reaction mixture was diluted with DCM (30 mL) and washed with sat. aq. NaHCO3 solution (3 × 10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate, 5:1, v/v → 2:1, v/v) to give 17 (61 mg, 45 %) as a white foamy solid. Rf = 0.41 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = −7.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.39-7.19 (m, 9H, aromatic), 6.02 (d, J = 8.0 Hz, 1H, NH), 5.92 (d, J = 9.9 Hz, 1H, NH), 5.88-5.79 (m, 2H, OCH2CH=CH2), 5.32 (t, J =10.0 Hz, 1H, H-4′), 5.25-5.13 (m, 4H, OCH2CH=CH2), 5.22 (br.s., 1H, 3(R) CH of dodecanoyl moiety partially merged with OCH2CH=CH2 ), 5.03 (s, 2H, PhCH2), 5.00-4.89 (m, 2H, H-3, H-3′), 4.89 (t, J = 10.6 Hz, 1H, H-4), 4.68 (d, J = 10.6 Hz, 1H, H-1′), 4.64 (d, J = 3.1 Hz, 1H, H-1), 4.55-4.51 (m, 4H, OCH2CH=CH2), 4.31-4.24 (m, 2H, H-2, H-2′), 3.82-3.76 (m, 2H, H-6′ab), 3.69-3.58 (m, 2H, H-6ab), 3.47-3.38 (m, 2H, H-5, H-5′), 3.28 (s, 3H, OCH3), 2.37 (dd, J2Sa, 2Sb = 14.2 Hz, J 2Sa, 3S = 8.5 Hz, 1H, H-2Sa), 2.30 (dd, J2Sa, 2Sb = 15.0 Hz, J 2Sa, 3S = 6.4 Hz, 1H, H-2Sb), 1.73-1.52 (m, 8H), 1.33-1.18 (m, 122H), 0.91(t, J = 7.0 Hz, 18H, 6CH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C114H191N2O25P Na 2042.3507; found 2042.1571.
Methyl [6′-O-benzyl-3′-O-(R)-3-dodecanoyloxy-tetradecanoyl-4-O-(1,5-dihydro-3-oxo-3λ5- 3H-2,4,3-benzodioxaphosphepin-3yl)-2′-[{(R)-3-dodecanoyloxy-tetradecanoylamino–{2′- deoxy-β-D-glucopyranosyl}]-2-(R)-3-dodecanoyloxy-tetradecanoylamino-2-deoxy-α-D-glucopyranoside (18)
A mixture of butylamine (7 μL, 0.065 mmol) and formic acid (4 μL, 0.030 mmol) was added to a solution of compound 17 (40 mg, 0.0198 mmol) in THF (1.5 mL) under Ar atmosphere. Tetrakis (triphenyl-phosphine) palladium (0) (8 mg) was added to the stirring solution. After 1 h, the reaction mixture was concentrated and the residue was purified by flash silica gel column chromatography (hexane:ethylacetate, 3:1, v/v → 1:1, v/v) to afford 18 as an amorphous solid (24 mg, 66%). Rf = 0.24 (hexane:ethyl acetate, 1:1, v/v); [α]D 24 = −18.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.38-7.17 (m, 9H, aromatic), 6.26 (d, J = 6.5 Hz, 1H, NH), 5.95 (d, J = 8.5 Hz, 1H, NH), 5.35 (t, J = 9.5 Hz, 1H, H-4′), 5.15-5.11 (m, 1H), 5.09-4.92 (m, 4H, H-3, H-3′, H-4, 3(R) CH of dodecanoyl moiety), 4.57 (d, J = 10.8 Hz, 1H, H-1′), 4.51 (d, J = 3.4 Hz, 1H, H-1), 4.05-4.3.94 (m, 2H, H-2, H-2′), 3.79-3.49 (m, 6H, H-6′ab, H-6ab, H-5, H-5′), 3.26 (s, 3H, OCH3), 2.67 (dd, J2Sa, 2Sb = 15.2 Hz, J2Sa, 3S = 8.0 Hz, 1H, H-2Sa), 2.61 (dd, J2Sa, 2Sb = 15.8 Hz, J 2Sa, 3S = 7.4 Hz, 1H, H-2Sb), 2.26 (dd, J = 16.8 Hz, J = 7.0 Hz, 4H), 1.74-1.52 (m, 8H), 1.23-1.18 (m, 122H), 0.80(t, J = 7.8 Hz, 18H, 6CH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C106H183N2O21P Na 1874.5618; found 1874.1497.
Methyl 3′-O-[(R)-3-dodecanoyloxy-tetradecanoyl-4-O-phosphono-2′-[(R)-3-dodecanoyloxy- tetradecanoyl]-2′-deoxy-β-D-glucopyranosyl}-2-[(R)-3-dodecanoyloxy-tetradecanoyl amino]-2-deoxy-α-D-glucopyranoside (2)
A mixture of 18 (12 mg, 0.0064 mmol) and Pd-black (10% wt. Pd on charcoal, Degussa, 15.0 mg) was suspended in anhydrous THF (1.5 mL) and tBuOH (1.5 mL). The mixture was placed under an atmosphere of H2 (1atm) and the resulting reaction mixture was stirred at room temperature for 10 h, after which it was neutralized with triethylamine (0.1 mL). The catalyst was removed by filtration through a bed of celite, which was washed with THF (2 × 2 mL). The combined filtrates were concentrated in vacuo to afford 2 as a colorless film (6.8 mg, 68%). [α]D 24 = −31.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3:CD3OD, 1:1, v/v): δ 5.29-5.27 (m, 1H, H-3), 4.78 (1H, H-1, merged with CD3OD peak), 4.74 (d, J = 9.0 Hz, 1H, H-1′), 4.41 (dd, J1= 17.8 Hz, J2 = 10.1 Hz, 1H, H-4), 4.29 (dist. t, J = 9.0 Hz, 1H, H-4′), 4.14 (dd, J5,6a = 2.9 Hz, J6a,6b = 14.1 Hz, 1H, H-6a), 4.07-4.03 (m, 2H, H-2, H-2′), 3.92 (d, J6b,6a = 15.7 Hz, 1H, H-6b), 3.80 (dist. quartet, J6′a,6′b = 16.4 Hz, 1H, H-6′a), 3.76 (t, J = 10.2 Hz, 1H, H-3′), 3.49 (d, J6′b,6′a = 16.7 Hz, 1H, H-6′b), 3.44-3.41 (m, 2H, H-5, H-5′), 3.32 (br.s., 3H, OCH3), 2.82 (dd, J2Sa, 2Sb = 15.3 Hz, J 2Sa, 3S = 7.4 Hz, 1H, H-2Sa), 2.70 (dd, J2Sa, 2Sb = 16.8 Hz, J 2Sa, 3S = 7.0 Hz, 1H, H-2Sb), 2.65 (dd, J1= 15.8 Hz, J2= 8.4 Hz, 2H), 1.77-1.63 (m, 12H), 1.36-1.21 (m, 114H), 1.16 (t, J = 7.1 Hz, 18H, 6CH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C91H171N2O21P Na 1682.3060; found 1682.1757.
Dimethylthexylsilyl[4′,6′-O-benzylidine-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-[{(R)-3-dodecanoyloxy-tetradecanoyl-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxycarbonylamino)-3-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (19)
A mixture of glycosyl acceptor 3 (210 mg, 0.361 mmol), trichloroacetamidate donor 5 (300 mg, 0.301 mmol) and molecular sieves (4A, 650 mg) in DCM (2.5 mL) was placed under an atmosphere of Ar and cooled to −40 °C. TfOH (1.34 μL, 0.015 mmol, in 0.1 mL DCM) was added and the resulting reaction mixture was stirred for 15 min until the temperature had reached 0 °C. The reaction was quenched by the addition of triethylamine (30 μL). Next, the mixture was diluted with DCM (35 mL), and washed with sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate 9:1, v/v → 4:1, v/v) to yield the disaccharide 19 (349 mg, 82 %) as a white foamy solid. Rf = 0.44 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = −11.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.41-7.33 (m, 2H, aromatic), 5.95-5.86 (m, 2H, OCH2CH=CH2), 5.46 (d, J = 8.2 Hz, 1H, NH), 5.42 (s, 1H, PhCH), 5.38-5.22 (m, 4H, OCH2CH=CH2), 5.20 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 5.13-5.05 (m, 2H, H-3, H-3′), 4.79 (dist. t, J = 10.1 Hz, 1H, H-4), 4.71-4.60 (m, 3H, H-1, H-1′, H-4′, all partially merged), 4.58-4.51 (m, 8H, 2x OCH2CH=CH2, OCH2CCl3), 4.28-4.24 (m, 1H), 3.90 (d, J6b,6a = 15.7 Hz, 1H, H-6b), 3.71 (t, J6a,6b = 16.4 Hz, 1H, H-6a), 3.64-3.42 (m, 6H, H-6′ab, H-2, H-2′, H-5, H-5′), 3.94-3.92 (m, 2H, H-5, H-5′), 2.52 (dd, J2Sa, 2Sb = 14.2 Hz, J 2Sa, 3S = 7.8 Hz, 1H, H-2Sa), 2.46 (dd, J2Sa, 2Sb = 14.4 Hz, J 2Sa, 3S = 7.4 Hz, 1H, H-2Sb), 2.23 (t, J = 7.4 Hz, 2H), 1.54 (br.s., 5H), 1.37-1.29 (m, 33H), 0.94-0.85 (m, 12H, 4 CH3), 0.06 (s, 3H, SiCH3), 0.0 (s, 3H, SiCH3); 13C NMR (gHSQC, CDCl3): δ = 131.72 (OCH2CH=CH2), 122.86, 119.02 (OCH2CCl3), 98.42 (C-1′), 96.65 (C-1), 77.02 (C-3), 76.19 (C-3′), 74.91 (OCH2CH=CH2), 72.01 (PhCH), 71.34 (C-4), 68.81 (OCH2CCl3), 68.26(C-6ab), 66.70 (C-5′), 63.19 (C-6′ab), 58.13 (C-2′), 58.06 (C-2), 38.23, 38.12, 35.10, 28.83, 22.19 (CH2 of lipid chains), 14.13 (terminal CH3 of lipid chain), 13.89 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C58H98Cl6N2O18 SiNa 1375.2048; found 1375.3142.
Dimethylthexylsilyl [4,6′-O-benzyl-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-[{(R)-3-dodecanoyloxy-tetradecanoyl-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-dimethylthexylsilyl- [6′-O-benzyl-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-{(R)-3-dodecanoyloxy-tetradecanoyl-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxycarbonylamino)-3,4-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (21)
Disaccharide 19 (350 mg, 0.247 mmol) in DCM (2.5 ml) was placed under an atmosphere of Ar and charged with N,N,N’,N’-tetramethylethylenediamine (0.18 mL, 1.23 mmol). The reaction mixture was cooled to 0 °C and allyloxycarbonyl chloride (0.13 mL, 1.23 mmol) was added slowly. The progress of reaction was monitored by TLC and MALDI-TOF MS. After 12 h of stirring at room temperature, the reaction mixture was diluted with DCM (30 mL), washed with sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The structure of the compound was confirmed by MS and the compound was used in the next step without further purification. A mixture of the crude 20 (270 mg) and molecular sieves (4A, 270 mg) in DCM (3.0 mL) was placed under an atmosphere of Ar and cooled to −78 °C. TfOH (32 μL) and triethylsilane (72 μL, 0.091 mmol) were added to the reaction mixture. The reaction mixture was stirred at −78 °C for 1 h after which the reaction was quenched by the addition of triethylamine (50 μL) and methanol (0.2 mL). Next, the mixture was diluted with DCM (35 mL), and washed with sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane:ethyl acetate, 9:1, v/v → 3:1, v/v) to yield the disaccharide 21 (168 mg, 62 %) as a colorless thick syrup. Rf = 0.32 (hexane:ethyl acetate, 4:1, v/v); [α]D 24 = −10.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.32-7.18 (m, 5H, aromatic), 5.78-5.71 (m, 2H, OCH2CH=CH2), 5.31 (d, J = 9.2 Hz, 1H, NH), 5.19-5.10 (m, 4H, OCH2CH=CH2), 5.05 (br.s., 1H, 3(R) CH of dodecanoyl moiety), 4.99-4.94 (m, 2H, H-3, H-3′), 4.91 (d, J = 10.0 Hz, 1H, H-1′), 4.88 (br.s., 1H, H-1), 4.73 (t, J = 9.1 Hz, 1H, H-4), 4.71-4.69 (m, 1H, H-4′, partially merged with OCH2CH=CH2), 4.53-4.45 (m, 8H, 2x OCH2CH=CH2, OCH2CCl3), 3.98 (d, J6b,6a = 15.7 Hz, 1H, H-6b), 3.68-3.52 (m, 2H, H-2, H-2′), 3.55-3.49 (m, 3H, H-6a, H-6′ab), 3.43-3.37 (m, 2H, H-5, H-5′), 2.43 (dd, J2Sa, 2Sb = 16.2 Hz, J 2Sa, 3S = 6.8 Hz, 1H, H-2Sa), 2.39 (dd, J2Sa, 2Sb = 14.7 Hz, J 2Sa, 3S = 7.9 Hz, 1H, H-2Sb), 2.14 (t, J = 7.3 Hz, 2H), 1.53-1.55 (m, 6H), 1.12-0.88 (m, 33H), 0.69-0.58 (m, 12H, 4 CH3), 0.04 (s, 3H, SiCH3), 0.0 (s, 3H, SiCH3); 13C NMR (gHSQC, CDCl3): δ = 133.72 (OCH2CH=CH2), 119.06, 115.87 (OCH2CCl3), 96.22 (C-1′), 95.01 (C-1), 75.02 (C-3), 74.71 (C-3′), 74.01 (OCH2CH=CH2), 69.30 (C-4), 63.70 (C-5′), 63.19 (C-6′ab), 58.13 (C-2′), 58.16 (C-2), 36.03, 35.12, 30.10, 26.83, 20.85 (CH2 of lipid chains), 15.23 (terminal CH3 of lipid chain), 14.21 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C67H106Cl6N2O20 SiNa 1521.3476; found 1521.4486.
Dimethylthexylsilyl [6′-O-benzyl-2′-deoxy-4-O-(1,5-dihydro-3-oxo-3λ5-3H-2,4,3-benzodioxaphosphepin-3yl)-2′-(2,2,2-trichloromethoxycarbonylamino)-3′-O-{(R)-3-dodecanoyloxy-tetradecanoyl]-2′-deoxy-β-D-glucopyranosyl}]-2-(2,2,2-trichloromethoxycarbonylamino)-3,4-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (22)
Disaccharide 21 (130 mg, 0.0866 mmol) in DCM (2.5 mL) was placed under an atmosphere of Ar and 3% tetrazole solution in acetonitrile (0.32 μl, 0.346 mmol) and N,N-diethyl-1,5-dihydro-2,4,3-benzodioxa-phosphepin-3-amine (74 μL, 0. 346 mmol) were added. After 45 min, the reaction mixture was cooled to −20 °C and m-CPBA (104 mg, 0.606 mmol) in DCM (2 mL) was added. The reaction mixture was stirred at 0 °C for another 1 h and then diluted with DCM (30 mL), washed with sat. aq. NaHCO3 solution (10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel column chromatography (hexane:ethylacetate, 4:1, v/v → 1:1, v/v) to give 22 (122 mg, 84 %) as a white foam. Rf = 0.35 (hexane:ethyl acetate, 3:1, v/v); [α]D 24 = −23.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.67 (d, J = 9.8 Hz, 2H, aromatic), 7.47 (t, J = 10.2 Hz, 2H, aromatic), 7.36-7.16 (m, 5H, aromatic), 5.88-5.77 (m, 2H, OCH2CH=CH2), 5.67 (d, J= 6.7 Hz, 1H, NH), 5.58 (t, J = 8.8 Hz, 1H, H-4′), 5.35-5.21 (m, 4H, OCH2CH=CH2), 5.21- 4.88 (m, 4H, 3(R) CH of dodecanoyl moiety, H-4, PhCH2), 4.99-4.94 (m, 2H, H-3, H-3′), 4.91 (d, J = 10.0 Hz, 1H, H-1′), 4.88 (br.s., 1H, H-1), 4.61-4.55 (m, 2H, H-3, H-3′), 4.52-4.48 (m, 4H, OCH2CCl3), 3.97 (d, J6b,6a = 10.7 Hz, 1H, H-6b), 3.88 (d, J6′b,6′a = 9.8 Hz, 1H, H-6′b), 3.73 -3.62 (m, 4H, H-6a, H-6′a, H-2, H-2′), 3.46-3.37 (m, 2H, H-5, H-5′), 2.86 (dd, J2Sa, 2Sb = 16.2 Hz, J 2Sa, 3S = 6.8 Hz, 1H, H-2Sa), 2.85 (dd, J2Sa, 2Sb = 14.7 Hz, J 2Sa, 3S = 7.9 Hz, 1H, H-2Sb), 2.41 (t, J = 7.3 Hz, 2H), 1.79-1.62 (m, 6H), 1.10-0.88 (m, 33H), 0.66-0.53 (m, 18H, 6 CH3), 0.05 (s, 3H, SiCH3), 0.0 (s, 3H, SiCH33); 13C NMR (gHSQC, CDCl3): δ = 131.20 (OCH2CH=CH2), 120.09, 118.87 (OCH2CCl3), 98.71 (C-1′), 97.64 (C-1), 88.71 (C-3), 76.75 (OCH2CH=CH2), 74.64 (C-4′), 68.06 (C-6ab′), 66.52 (C-6′ab), 63.31.13 (C-2′), 63.01 (C-2), 34.03, 32.32, 30.91, 28.32, 20.11 (CH2 of lipid chains), 15.13 (terminal CH3 of lipid chain), 12.61 (terminal CH3 of lipid chain); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C75H113Cl6N2O23 PSiNa 1705. 4766; found 1705.3152.
Dimethylthexylsilyl [6′-O-benzyl-3′-O-(R)-3-dodecanoyloxy-tetradecanoyl-4-O-(1,5-dihydro-3-oxo-3λ5-3H-2,4,3-benzodioxaphosphepin-3yl)-2′-(R)-3-dodecanoyloxy-tetradecanoylamino–{2′-deoxy-β-D-glucopyranosyl}]-2-(R)-3-dodecanoyloxy- tetradecanoylamino-3,4-O-allyloxycarbonyl-2-deoxy-α-D-glucopyranoside (23)
Disaccharide 22 (80 mg, 0.0475 mmol) in DCM (2.5 mL) was placed under an atmosphere of Ar, and Zn dust (90 mg, 1.426 mmol) and acetic acid (0.05 mL) were added. After stirring at room temperature for 1.5 h, the reaction mixture was diluted with DCM (20 mL), and filtered through a bed of celite. The filtrate was concentrated and the residue co-evaporated three times with toluene, and then dried in vacuo. The deprotection of the Troc group was confirmed by MALDI-TOF MS analysis. The crude free amine (63 mg, 0.0473 mmol), dissolved in DCM (1.5 mL) and triethylamine (25 μL), was added to a stirring solution of EDC (37 mg, 0.189 mmol) and (R)-3-dodecanoyl-tetradecanoic acid (60.5 mg, 0.142 mmol) in DCM (2 mL) at room temperature under an atmosphere of Ar. After stirring for 24 h at room temperature, the reaction mixture was diluted with DCM (30 mL) and washed with sat. aq. NaHCO3 solution (3 × 10 mL), water (2 × 10 mL) and brine (10 mL). The organic layer was dried (Na2SO4), filtered and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel column chromatography (hexane:ethyl acetate, 4:1, v/v→ 2:1, v/v) to give 23 (49 mg, 48 %) as a white foam. Rf = 0.38 (hexane:ethyl acetate, 3:1, v/v); [α]D 24 = 21.6° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.39-7.16 (m, 9H, aromatic), 5.89-5.80 (m, 2H, OCH2CH=CH2), 5.66 (d, J= 6.7 Hz, 1H, NH), 5.48 (t, J = 8.2 Hz, 1H, H-4′), 5.27-5.21 (m, 4H, OCH2CH=CH2), 5.17- 4.89 (m, 4H, 3(R) CH of dodecanoyl moiety, H-4, PhCH2), 4.80-4.76 (m, 2H, H-3, H-3′), 4.68 (d, J = 8.8 Hz, 1H, H-1′), 4.66 (br.s., 1H, H-1), 4.55-4.43 (m, 2H, H-3, H-3′), 3.85 (d, J6b,6a = 9.7 Hz, 1H, H-6b), 3.80 (d, 1H, J6′b,6′a = 8.6 Hz, H-6′b), 3.64 -3.57 (m, 4H, H-6a, H-6′a, H-2, H-2′), 3.41-3.27 (m, 2H, H-5, H-5′), 2.46-2.51 (m, 2H, H-2Sa, H-2Sb), 1.63-1.10 (m, 130H), 1.79-1.62 (m, 6H), 1.10-0.88 (m, 33H), 0.71-0.63 (m, 18H, 6 CH3), 0.05 (s, 3H, SiCH3), 0.0 (s, 3H, SiCH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C121H207N2O25 PSiNa 2171.9959; found 2171.3125.
Dimethylthexylsilyl [6′-O-benzyl-3′-O-(R)-3-dodecanoyloxy-tetradecanoyl-4-O-(1,5-dihydro-3-oxo-3λ5-3H-2,4,3-benzodioxaphosphepin-3yl)-2′-(R)-3-dodecanoyloxy-tetradecanoyl–{2′-deoxy-β-D-glucopyranosyl}]-2-(R)-3-dodecanoyloxy-tetradecanoylamino-2-deoxy-α-D-glucopyranoside (24)
A mixture of butyl amine (7.4 μL, 0.0744 mmol) and formic acid (2.8 μL, 0.0744 mmol) was added to a solution of compound 23 (40 mg, 0.0186 mmol) in THF (1.5 mL) under an atmosphere of Ar. Tetrakis(triphenyl-phosphine) palladium (0) (8 mg) was added to the resulting solution and stirring was continued for 1 h. The reaction mixture was concentrated and the residue was purified by flash silica gel column chromatography (hexane:ethyl acetate, 3:1, v/v → 1:1, v/v) to afford 24 as a sticky solid (25.7 mg, 70%). Rf = 0.28 (hexane:ethyl acetate, 2:1, v/v); [α]D 24 = 19.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.43-7.18 (m, 9H, aromatic), 5.87 (br.s., 1H, NH), 5.53 (t, J = 8.6 Hz, 1H, H-4′), 5.33-5.30 (m, 1H, H-4), 5.27- 5.00 (m, 3H, 3(R) CH of dodecanoyl moiety, PhCH2), 4.80-4.66 (m, 4H, H-1, H-1′, H-3, H-3′), 4.15 (d, J6b,6a = 9.1 Hz, 1H, H-6b), 3.88 (d, J6′b,6′a = 8.6 Hz, 1H, H-6′b), 3.74 -3.67 (m, 4H, H-6a, H-6′a, H-2, H-2′), 3.51-3.29 (m, 2H, H-5, H-5′), 2.76-2.61 (m, 2H, H-2Sa, H-2Sb), 1.63-1.10 (m, 130H), 2.24 (t, J = 9.0 Hz, 2H), 1.78-1.70 (m, 6H), 1.42-1.21 (m, 130H), 0.71-0.63 (m, 18H, 6 CH3), 0.06 (s, 3H, SiCH3), 0.0 (s, 3H, SiCH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C113H199N2O21 PSiNa 2003.8492; found 2003.5231.
3′-O-(R)-3-dodecanoyloxy-tetradecanoyl-4-O-phosphono-2′-[ (R)- 3-dodecanoyloxy-tetradecanoyl]-2′-deoxy-β-D-glucopyranosyl}-2--[(R)-3-dodecanoyloxy-tetradecanoyl amino]-2-deoxy-α-D-glucopyranose (1)
A mixture of 24 (24 mg, 0.0121 mmol) and HF-pyridine (18 μL, 0.726 mmol, 65-70%) in anhydrous THF (1.5 mL) was stirred at room temperature for 10 h. The reaction mixture was quenched by the addition of solid NaHCO3 (25 mg) and then diluted with DCM (35 mL), filtered and the filtrate was concentrated in vacuo. The residue was purified by flash silica gel column chromatography (DCM:MeOH, 15:1, v/v → 10:1, v/v) to yield a lactol (14 mg, 68%) as a colorless thick syrup. The lactol (14 mg, 0.007 mmol) was added to a suspension of Pd-black (10% wt. Pd on charcoal, Degussa, 18.0 mg) in a mixture of anhydrous THF (1.5 mL) and tBuOH (1.5 mL). The mixture was placed under an atmosphere of H2 (1 atm) and the resulting reaction mixture was stirred at room temperature for 10 h, after which it was neutralized with triethylamine (0.12 mL), and the catalyst removed by filtration through bed of celite, which was washed with THF (2 × 2 mL). The combined filtrates were concentrated in vacuo to afford 1 as a colorless flim (7.8 mg, 68%). [α]D 24 = 19.0° (c = 1.0, CHCl3); 1H NMR (500 MHz, CDCl3:CD3OD, 1:1, v/v): δ 5.34-5.08 (m, 5H), 4.78-4.44 (m, 4H, H-4′, H-3, H-1′, H-1 peaks are partially merged with CD3OD), 4.33 (br.s., 1H, 3(R) CH of dodecanoyl moiety,), 4.23-4.02 (m, 2H, H-4, H-3′), 3.82-3.31 (m, 6H, H-6ab, H-6′ab, H-2, H-2′, H-5, H-5′), 2.78-2.51 (m, 2H, H-2Sa, H-2Sb), 2.21 (br.s., 4H), 1.73-1.09 (m, 130H), 0.81-0.73 (m, 18H, 6 CH3); HR-MALDI-ToF/MS: m/z: for [M + Na]+ calcd. C90H169N2O21 PNa 1669.2794; found 1669.0132.
Biological Experiments
Reagents for Biological Experiments
E. coli 055:B5 LPS was obtained from List Biologicals and monophosphorylated (detoxified) lipid A obtained by controlled hydrolysis of the LPS of S. minnesota (MPLA) was obtained from Avanti Polar Lipids Inc. The synthesis of E. coli lipid A has been reported elsewhere[16] and was reconstituted in PBS with DMSO (10%). The synthetic compounds 1 and 2 were reconstituted in PBS containing THF (10%). Final concentrations of DMSO and THF in the biological experiments never exceeded 0.5% to avoid toxic effects.
Cell Maintenance
RAW 264.7 γNO(−) cells, derived from the RAW 264.7 mouse monocyte/macrophage cell line, were obtained from ATCC. The cells were maintained in RPMI 1640 medium (ATCC) with L-glutamine (2 mM), adjusted to contain sodium bicarbonate (1.5 g L−1), glucose (4.5 g L−1), HEPES (10 mM) and sodium pyruvate (1.0 mM) and supplemented with penicillin (100 u mL−1)/streptomycin (100 μg mL−1) and fetal bovine serum (10%). Cells were maintained in a humid 5% CO2 atmosphere at 37 °C.
Cytokine Induction and ELISAs
RAW 264.7 γNO(−) cells were plated on the day of the exposure assay as 2 × 105 cells/well in 96-well tissue culture plates (Nunc) and incubated with different stimuli for 5.5 and 24 h. Culture supernatants were collected and stored frozen (−80 °C) until assayed for cytokine production. TNF-α, IFN-β and IP-10 were assayed in supernatants collected after 5.5 h incubation and IL-6 after 24 h. All cytokine ELISAs were performed in 96-well MaxiSorp plates (Nunc). Cytokine DuoSet ELISA Development Kits (R&D Systems) were used for the cytokine quantification of TNF-α, IP-10 and IL-6 according to the manufacturer’s instructions. The absorbance was measured at 450 nm with wavelength correction set to 540 nm using a microplate reader (BMG Labtech). Concentrations of IFN-β in culture supernatants were determined as follows. ELISA MaxiSorp plates were coated with rabbit polyclonal antibody against mouse IFN-β (PBL Biomedical Laboratories). IFN-β in standards and samples was allowed to bind to the immobilized antibody. Rat anti-mouse IFN-β antibody (USBiological) was then added, producing an antibody-antigen-antibody “sandwich”. Next, horseradish peroxidase (HRP) conjugated goat anti-rat IgG (H+L) antibody (Pierce) and a chromogenic substrate for HRP 3,3′,5,5′-tetramethylbenzidine (TMB; Pierce) were added. After the reaction was stopped, the absorbance was measured at 450 nm with wavelength correction set to 540 nm.
Data Analysis
Concentration-response data were analyzed using nonlinear least-squares curve fitting in Prism (GraphPad Software, Inc.). These data were fit with the following four parameter logistic equation: Y = Emax / (1 + (EC50/X)Hill slope), where Y is the cytokine response, X is the concentration of the stimulus, Emax is the maximum response and EC50 is the concentration of the stimulus producing 50% stimulation. The Hill slope was set at 1 to be able to compare the EC50 values of the different inducers. All values are presented as the means ± SD of triplicate measurements, with each experiment being repeated three times.
Supplementary Material
Acknowledgments
We thank Dr Yanghui Zhang for synthesizing the E. coli lipid A and Dr Jidnyasa Gaekwad for performing the cell activation studies. This research was supported by the Institute of General Medicine (NIGM) of the National Institutes of Health (NIH) (R01GM061761) and the National Cancer Institute (NCI) (R01CA088986). Dr Michael De Castro was supported by a Carl Storm Postdoctoral Fellowship - National Institutes of Health and Mr A. M. Abdel-Aal El-Sayed’s stay in Dr Boons’ laboratory was supported by Dr I. Toth (University of Queensland) and Francine Kroesen, Arlo D. Harris and UQ-GSRTG scholarships.
Footnotes
Supporting Information (see also the footnote in the first page of this article): NMR spectra synthetic compounds.
Supporting information for the article is available.
References
- [1] a).O’Hagan DT, MacKichan ML, Singh M. Biomol. Eng. 2001;18:69–85. doi: 10.1016/s1389-0344(01)00101-0. [DOI] [PubMed] [Google Scholar]; b) Jiang ZH, Koganty RR. Curr. Med. Chem. 2003;10:1423–1439. doi: 10.2174/0929867033457340. [DOI] [PubMed] [Google Scholar]; c) Aguilar JC, Rodriguez EG. Vaccine. 2007;25:3752–3762. doi: 10.1016/j.vaccine.2007.01.111. [DOI] [PubMed] [Google Scholar]; d) Chiarella P, Massi E, De Robertis M, Signori E, Fazio VM. Expert Opin. Biol. Ther. 2007;7:1551–1562. doi: 10.1517/14712598.7.10.1551. [DOI] [PubMed] [Google Scholar]; e) Ishizaka ST, Hawkins LD. Expert Rev. Vaccines. 2007;6:773–784. doi: 10.1586/14760584.6.5.773. [DOI] [PubMed] [Google Scholar]; f) McKee AS, Munks MW, Marrack P. Immunity. 2007;27:687–690. doi: 10.1016/j.immuni.2007.11.003. [DOI] [PubMed] [Google Scholar]; g) Alving CR, Rao M. Vaccine. 2008;26:3036–3045. doi: 10.1016/j.vaccine.2007.12.002. [DOI] [PubMed] [Google Scholar]; h) Johnson DA. Curr. Top. Med. Chem. 2008;8:64–79. doi: 10.2174/156802608783378882. [DOI] [PubMed] [Google Scholar]; i) Lahiri A, Das P, Chakravortty D. Vaccine. 2008;26:6777–6783. doi: 10.1016/j.vaccine.2008.09.045. [DOI] [PubMed] [Google Scholar]; j) Harandi AM, Davies G, Olesen OF. Expert Rev. Vaccines. 2009;8:293–298. doi: 10.1586/14760584.8.3.293. [DOI] [PubMed] [Google Scholar]; k) Reed SG, Bertholet S, Coler RN, Friede M. Trends Immunol. 2009;30:23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- [2].Kwissa M, Kasturi SP, Pulendran B. Expert Rev. Vaccines. 2007;6:673–684. doi: 10.1586/14760584.6.5.673. [DOI] [PubMed] [Google Scholar]
- [3].Beutler B. Mol. Immunol. 2004;40:845–859. doi: 10.1016/j.molimm.2003.10.005. [DOI] [PubMed] [Google Scholar]
- [4].van Amersfoort ES, van Berkel TJC, Kuiper J. Clin. Microbiol. Rev. 2003;16:379–414. doi: 10.1128/CMR.16.3.379-414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] a).Akira S, Takeda K, Kaisho T. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]; b) Dabbagh K, Lewis DB. Curr. Opin. Infect. Dis. 2003;16:199–204. doi: 10.1097/00001432-200306000-00003. [DOI] [PubMed] [Google Scholar]; c) Bevan MJ. Nat. Rev. Immunol. 2004;4:595–602. doi: 10.1038/nri1413. [DOI] [PubMed] [Google Scholar]; d) Finlay BB, Hancock REW. Nat. Rev. Microbiol. 2004;2:497–504. doi: 10.1038/nrmicro908. [DOI] [PubMed] [Google Scholar]; e) Pasare C, Medzhitov R. Semin. Immunol. 2004;16:23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]; f) Pasare C, Medzhitov R. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- [6].Munford RS. Infect. Immun. 2008;76:454–465. doi: 10.1128/IAI.00939-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] a).Darveau RP. Curr. Opin. Microbiol. 1998;1:36–42. doi: 10.1016/s1369-5274(98)80140-9. [DOI] [PubMed] [Google Scholar]; b) Caroff M, Karibian D, Cavaillon JM, Haeffner-Cavaillon N. Microbes Infect. 2002;4:915–926. doi: 10.1016/s1286-4579(02)01612-x. [DOI] [PubMed] [Google Scholar]; c) Raetz CRH, Whitfield C. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8] a).Galanos C, Luderitz O, Rietschel ET, Westphal O, Brade H, Brade L, Freudenberg M, Schade U, Imoto M, Yoshimura H, Kusumoto S, Shiba T. Eur. J. Biochem. 1985;148:1–5. doi: 10.1111/j.1432-1033.1985.tb08798.x. [DOI] [PubMed] [Google Scholar]; b) Imoto M, Yoshimura H, Sakaguchi N, Kusumoto S, Shiba T. Tetrahedron Lett. 1985;26:1545–1548. [Google Scholar]
- [9].Casella CR, Mitchell TC. Cell Mol. Life Sci. 2008;65:3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- [11].Okemoto K, Kawasaki K, Hanada K, Miura M, Nishijima M. J. Immunol. 2006;176:1203–1208. doi: 10.4049/jimmunol.176.2.1203. [DOI] [PubMed] [Google Scholar]
- [12].Salkowski CA, Detore GR, Vogel SN. Infect. Immun. 1997;65:3239–3247. doi: 10.1128/iai.65.8.3239-3247.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Evans JT, Cluff CW, Johnson DA, Lacy MJ, Persing DH, Baldridge JR. Expert Rev. Vaccines. 2003;2:219–229. doi: 10.1586/14760584.2.2.219. [DOI] [PubMed] [Google Scholar]
- [14] a).Buskas T, Ingale S, Boons GJ. Angew. Chem. Int. Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. [DOI] [PubMed] [Google Scholar]; b) Ingale S, Wolfert MA, Gaekwad J, Buskas T, Boons GJ. Nat. Chem. Biol. 2007;3:663–667. doi: 10.1038/nchembio.2007.25. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ingale S, Wolfert MA, Buskas T, Boons GJ. Chembiochem. 2009;10:455–463. doi: 10.1002/cbic.200800596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15] a).Johnson DA, Keegan DS, Sowell CG, Livesay MT, Johnson CL, Taubner LM, Harris A, Myers KR, Thompson JD, Gustafson GL, Rhodes MJ, Ulrich JT, Ward JR, Yorgensen YM, Cantrell JL, Brookshire VG. J. Med. Chem. 1999;42:4640–4649. doi: 10.1021/jm990222b. [DOI] [PubMed] [Google Scholar]; b) Jiang ZH, Budzynski WA, Qiu DX, Yalamati D, Koganty RR. Carbohydr. Res. 2007;342:784–796. doi: 10.1016/j.carres.2007.01.012. [DOI] [PubMed] [Google Scholar]
- [16].Zhang Y, Gaekwad J, Wolfert MA, Boons GJ. J. Am. Chem. Soc. 129:5200–5216. doi: 10.1021/ja068922a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang Y, Gaekwad J, Wolfert MA, Boons GJ. Chem. Eur. J. 2008;14:558–569. doi: 10.1002/chem.200701165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harada T, Yamada H, Tsukamoto H, Takahashi T. J. Carbohydr. Chem. 1995;14:165–170. [Google Scholar]
- [19].Fujimoto Y, Adachi Y, Akamatsu M, Fukase Y, Kataoka M, Suda Y, Fukase K, Kusumoto S. J. Endotoxin Res. 2005;11:341–347. doi: 10.1179/096805105X76841. [DOI] [PubMed] [Google Scholar]
- [20].Keegan DS, Hagen SR, Johnson DA. Tetrahedron: Asymmetry. 1996;7:3559–3564. [Google Scholar]
- [21].Higashi K, Nakayama K, Shioya E, Kusama T. Chem. Pharm. Bull. 1991;39:2502–2504. doi: 10.1248/cpb.39.3244. [DOI] [PubMed] [Google Scholar]
- [22].Dullenkopf W, Castro-Palomino JC, Manzoni L, Schmidt RR. Carbohydr. Res. 1996;296:135–147. doi: 10.1016/s0008-6215(96)00237-6. [DOI] [PubMed] [Google Scholar]
- [23].Ellervik U, Magnusson G. Tetrahedron Lett. 1997;38:1627–1628. [Google Scholar]
- [24].Schmidt RR, Kinzy W. Adv. Carbohydr. Chem. Biochem. 1994;50:21–123. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- [25].Sakagami M, Hamana H. Tetrahedron Lett. 2000;41:5547–5551. [Google Scholar]
- [26].Watanabe Y, Komoda Y, Ebisuya K, Ozaki S. Tetrahedron Lett. 1990;31:255–256. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.