

Abstract
While developing novel catalysts for carbon-carbon or carbon-heteroatom coupling (N, O, or F), we were able to introduce tridentate NHC-amidate-alkoxide palladium(II) complexes. In aqueous solution, these NHC-Pd(II) complexes showed high ability for C-H activation of various hydrocarbons (cyclohexane, cyclopentane, dimethyl ether, THF, acetone, and toluene) under mild conditions.
Keywords: NHC ligand, C-H activation, H/D exchange, Palladium complex, Intermolecular reaction
Carbon-carbon or carbon-heteroatom cross-coupling reactions catalyzed by transition metal catalysts have been widely investigated and established because of their significance in organic synthesis. In particular, homogeneous Pd catalysis has been well studied in the most powerful and versatile synthetic process such as Heck[1], Suzuki[2], Stille[3], Sonogashira[4], Negishi[5], and Buchwald-Hartwig reactions[6]. However, these reactions have required alkenyl/aryl halide substrates. Alternatively, the more desirable direct functionalization of hydrocarbons via C-H bond activation has still remained a challenge in cross-coupling reactions.
In recent years, a substantial number of metal complexes that are able to selectively activate C-H bonds under mild conditions have been discovered.[7] In spite of these advances, practical catalysts for the C-H bond functionalization remain elusive, due to the requirement of high energy to break C-H bond and subsequent vulnerability of the metal-carbon bond. Additionally, the C-H bond activation is often inhibited by water or by the product eliminated from the metal complex during the reaction.[8]
Recently, N-Heterocyclic carbene (NHC) ligands, which are known to enhance σ-donor coordination, have shown improved behaviour toward C-H bond activation during the past several years.[9] A number of organometallic species at various oxidation states (Pd, Pt, Ru, and Ir) are stabilized by the use of NHC ligands, increasing the capability to obtain efficient catalysts for C-H activation.[10] However, most examples refer to intramolecular processes, while intermolecular or catalytic examples being rare. With these challenges in mind, we report herein the efficient C-H bond activation of hydrocarbons in aqueous solution containing catalytic amounts of tridentate NHC ligand-Pd complex via H/D exchange as a probe for direct C-H functionalization. Such reactions have been studied in heterogeneous Pd catalytic systems but are rarely observed in homogeneous Pd systems.[11]
| (1) |
 |
(2) |
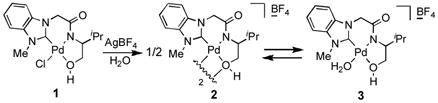
Recently, we have succeeded in preparing air-stable tridentate NHC-amidate-alkoxide ligand/palladium complex 1 from an amino alcohol.[12] We found that H/D exchange on benzene using D2O as both the solvent and deuterium source, occurred via C-H bond activation in the presence of Pd complex 1 and silver tetrafluoroborate. As shown in equation (1), the H/D exchange reaction was demonstrated with benzene (20 μL), palladium complex 1 (5 mol %) and AgBF4 in deuterium oxide (700 μL) for 22 hours at 55 °C and 100 °C. The efficiency of the H/D exchange giving deuterated isotopomers showed significant enhancement, when temperature was increased from 55 °C to 100 °C. While evaluating H/D exchange with palladium complex 1 and AgBF4, we found that a dimeric structure 2 was present in the solution as indicated by 1H-NMR spectroscopy.[13] At an elevated temperature, however, the concentration of monomeric species 3 was observed to be higher than complex 2. Consequently, we confirmed that the H/D displacement via C-H bond activation worked well only in the presence of the monomeric species 3. As shown in equation (2), NHC’s ligand/palladium complex 1 was converted to the palladium complex 2 by treating with aqueous AgBF4 for the C-H bond activation, and subsequently the monomer/dimer equilibrium process (2 ⇆ 3) took place depending on the temperature in the aqueous solution.[14] The higher efficiency of the H/D exchange process at 100 °C compared to 55 °C is attributable to the conversion of the catalyst into active mode 3 from complex 2 at the elevated temperature.
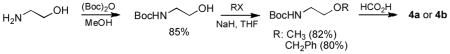
To further investigate the C-H activation of this class of Pd complexes, we extended the structural motifs of palladium to various tridentate ligands. To circumvent the dimerization of monomeric species in aqueous solution at low temperatures, we employed a methyl ether (7a) or benzyl ether (7b) group instead of the previously employed hydroxyl moiety as oxygen-site of the ligands (Scheme 1). Compounds 4a and 4b were prepared from ethanolamine and valinol, respectively.[15] Bromoacetylation of the amino ethers was followed by amide formation and N-alkylation with benzimidazole to yield 5a and 5b. The benzimidazole salts 6a and 6b were obtained by allowing 5a and 5b to react with CH3I in THF. For the coordination of NHC’s to palladium, metal exchange was carried out through a silver NHC complex.[16] N-Methyl iodide salts 6a and 6b were reacted with Ag2O to give silver NHC complexes, and subsequent treatment of the silver compounds with PdCl2(CH3CN)2 in CH3CN provided the desired complexes 7a and 7b in 79 and 72% yields, respectively. The structures of Pd/ligand complexes 7a and 7b were further confirmed by the molecular ion peaks (ESI-MS) at m/z 388.0029 [M+] (calcd: 388.0044) and m/z 506.0813 [M+] (calcd: 506.0827).
Scheme 1.

In addition, we designed an O-C-N tridentate ligand system, an example of which is the alkoxy-NHC-amidate ligand/palladium complex 11 (Scheme 2). Amide compound 9 was easily derived from aniline and bromoacetyl bromide. By means of N-alkylation with benzimidazole and compound 9, the benzimidazole-amide adduct was obtained in DMF solution, which after second N-alkylation with iodoethanol gave the iodine salt 10 in 62% yield for two steps. Subsequently, construction of alkoxy-NHC-amidate ligand/palladium complex 11 was successfully accomplished by treatment with half an equivalent of Ag2O and one equivalent of PdCl2(CH3CN)2 sequentially in CH3CN solution at room temperature.
Scheme 2.

First, to optimize the H/D exchange reaction conditions, various reaction temperatures and catalyst loadings were screened with Pd complexes 8a, 8b and 12 for benzene in D2O. The H/D exchange can be conveniently monitored by 1H NMR spectroscopy through observation of the decreasing proton signals of benzene in the aqueous phase relative to the poly(dimethylsiloxane) as an external reference standard. Before H/D exchange, AgBF4 was added into a MeCN solution of Pd complexes 7a, 7b and 11 to remove Cl anion, so that Pd complexs 8a, 8b, and 12 could be ready for the C-H activation. In addition, unreacted AgBF4 and AgCl were removed by a celite column. In D2O solution, the MeCN 1H-signal shifted from 2.06 ppm (free MeCN in D2O) to 2.16 ppm as well as broadening of the signal indicated that there was weak interaction between MeCN and Pd metal.
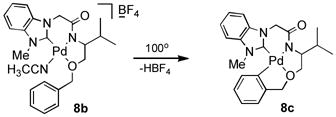
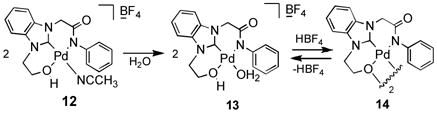
As shown in Table 1, we found that the reaction temperature could be lowered to 55 °C without causing a significant decrease in the extent of deuterium incorporation relative to that observed in reactions run at 100 °C (entry 3). Interestingly, we discovered that at 55 °C increasing the catalyst loading beyond 1.7 mol% (entry 4) did not lead to a detectable increase in deuterium incorporation. While use of less catalyst (entries 1 and 2) lead to a decrease in the extent of H/D exchange, 1.7 mole% is apparently enough to saturate the system and achieve optimum conversion. In addition, longer reaction times may not affect the catalytic cycle dramatically (entry 3). When the ligand 8b possessing a benzyl ether group was utilized, deuterium incorporation slowed down somewhat at 55 °C (entry 5), however deuterium incorporation was barely seen at an elevated temperature (100 °C) due to deactivation of 8b by coordination between palladium and benzyl aromatic C-H intramolecular activation to generate 8c (Equation 3).[17] Moreover, alkoxy-NHC-amidate ligand/palladium complex 12 gave the lowest level of deuterium incorporation into benzene at 55 °C because of the steric hindrance of the phenyl group as well as the existence of dimer/monomer equilibrium in aqueous solution (Equation 4).[14] This result wherein the efficiency of the H/D exchange showed significant enhancement at a higher temperature is in accordance with behaviour of the active catalyst derived from 1 (Equation 1).
Table 1.
H/D exchange of benzene in D2O by 8a, 8b, and 12.a
 | |||||
|---|---|---|---|---|---|
| H/D conversions (%)b |
|||||
| Entry | Catalyst (mol %) | 55 °C (6h) | 100 °C (6h) | 55 °C (22h) | 100 °C (22h) |
| 1 | 8a (0.4) | 48 | - | - | - |
| 2 | 8a (0.9) | 81 | - | - | - |
| 3 | 8a (1.7) | 90 | 89 | 95 | 96 |
| 4 | 8a (6.9) | 89 | 89 | - | - |
| 5 | 8b (1.7) | 69 | 8 | - | - |
| 6 | 12 (1.7) | 23 | 86 | - | - |
All reactions were carried out with 20μL of benzene in 0.7 mL of D2O solution.
The H/D conversions were determined by1H NMR.
 |
(3) |
 |
(4) |
Additionally, in an experiment designed to study the time dependence of the deuterium displacement, a solution of benzene with catalyst 8a in D2O was heated to 55 °C and monitored by 1H NMR spectroscopy. As shown in Figure 1(A), over a period of 22 hours, the resonances for the benzene (δ 6.71 at 55 °C) disappeared without any significant change in the signal intensities of the other resonances. Furthermore, to unequivocally establish C-H activation with Pd complex, we also probed deuterium-proton displacement between C6D6 and H2O at 55 °C and H/D incorporation was monitored by 2H NMR spectroscopy. As shown in Figure 1(B), the signal for benzene-d6 decreased in intensity over a period of 16 h. Overall conversion of C6D6 to C6H6 by 3.6 μmol of 8a was 75%, which is lower than the percent of conversion of C6H6 to C6D6 (89.5% in 6 h, entry 2 in table 1) as expected.
Figure 1.

(A) 1H-NMR spectra of a solution of 8a and C6H6 in D2O at 55 °C recorded over a period of 22 h. REF1 (internal standard): poly)dimethylsiloxane). (B) 2H-NMR spectra of solution of 8a and C6D6 in H2O at 55 °C recorded after 16 h REF2 (internal standard): CD3CN.
On the basis of these results, various organic substrates were investigated to explore the feasibility of the H/D exchange reaction with palladium NHC-ligand complex 8a. In general, effective multiple deuterium incorporation into the alkyl C-H was observed with D2O as both the solvent and deuterium source. Table 2 lists the organic substrates examined and their corresponding extents of deuterium incorporation. Catalyst 8a showed deuteration ability for a wide range of molecules including saturated hydrocarbons, ethers, and ketones. Remarkably, the catalyst was also capable of C-H activation of saturated hydrocarbons such as cyclohexane and cyclopentane, and excellent total deuterium incorporation (96 ~ 97%) was observed (entries 1 ~ 4). Recently, there have been a few examples for the H/D exchange of cyclohexane and cyclopentane, in which efficient thermal catalysis was carried out by Ir or Pt species using deuterated organic solvents or D2 as the deuterium source.[18] These C-H activations were developed with modest conversions, and they had additional drawbacks such as air/water sensitivity of the metal catalysts and high reaction temperature requirement. On the contrary, our palladium complex 8a appeared to be superior for C-H activation of saturated hydrocarbons with significant air/water stability.
Table 2.
H/D exchange of various organic substrates in D2O by Pd catalyst 8a.a
| Entry | 8a | Substrateb | T (°C) | D conversion (%)c/TONd |
|
|---|---|---|---|---|---|
| 6 hrs | 22 hrs | ||||
| 1 | 3.86 | 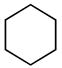 |
55 | 81/464 | 93/536 |
| 2 | 3.86 | 100 | 96/552 | 93/536 | |
| 3 | 3.86 | 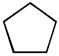 |
55 | 72/399 | 76/422 |
| 4 | 3.86 | 100 | 96/533 | 97/541 | |
| 5 | 3.86 | 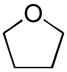 |
55 | 2/10 | 2/10 |
| 6 | 3.86 | 100 | 44/222 | 52/267 | |
| 7 | 3.86 | 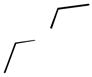 |
55 | 0.5/2 | 1/4 |
| 8 | 3.86 | 100 | 5/25 | 9/43 | |
| 9 | 15.4 | 100 | 27/27 | 46/46 | |
| 10 | 3.86 | 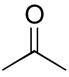 |
55 | 12/52 | 15/65 |
| 11 | 3.86 | 100 | 26/110 | 52/218 | |
| 12 | 15.4 | 100 | 55/58 | 63/66 | |
| 13 | 3.86 | 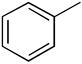 |
55 | 83/323 | 87/340 |
| 14 | 3.86 | 100 | 95/369 | 97/379 | |
All reactions were performed with Pd complex 8a (μmol) in sealed glass tubes containing D2O and substrate at 55 and 100 °C.
20 μL.
For %D, 1H NMR spectroscopy was used for analysis.
[D conversion × mole of substrate]/[mole of Pd × 100].
Ether compounds such as THF and diethyl ether showed lower levels of D-incorporation compared to aromatic and saturated hydrocarbons, however we observed that it was also a facile process with catalyst 8a under conditions of higher temperatures and longer reaction times (entries 6 and 8) with increased catalyst loading (entry 9). To our knowledge, there are only few examples for the H/D exchange of THF, which were effected by Ir catalysis at lower levels of D-incorporation at longer reaction times and higher temperatures.[19] Although the selective C-H activation for the α- and β-positions of THF and diethyl ether was not examined under these conditions, we demonstrated H/D exchange for these ether compounds with palladium complexes. These findings were quite similar to the case of H/D exchange with ketone as the substrate. While deuterium was incorporated into acetone at higher initial conversion values than the ethers, an increase in the three aforementioned reaction variables led to a significant improvement of percent D-incorporation for this substrate (entries 10 ~12). Also, we examined the deuterium incorporation of toluene in D2O to elucidate the possibility of selective C-H activation for aryl and alkyl groups. Similarly to other examined substrates, toluene showed high H/D conversions (95 ~ 97%) at 100 °C (entry 14), however we were not able to observe selective C-H activation for CH3 (95% in 6 h) or C6H5 C-H bonds (95% in 6 h).
 |
(5) |
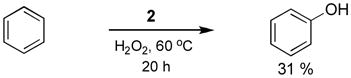
Based on these studies, which tested the possibility of direct oxidation of a sp2 C-H bond, C6H6, was added to a reaction mixture of 2 (10 mol %) in 30% H2O2 solution (Equation 5). We detected the oxidation product of phenol via C-H activation to afford phenol as a product.
In summary, we have demonstrated efficient and strong C-H activation with water soluble NHC-Pd(II) complexes that showed efficient H/D exchange of various organic substrates under D2O under mild conditions (55 °C or 100 °C). Importantly, the catalytic reaction was not inhibited by coordination of water to Pd. Strongly electron donating groups such as NHC, amide N, and O would increase the electron density of Pd, allowing a weak interaction between electrophilic Pd and water. In addition, we were able to obtain a preliminary result for the direct functionalization in aqueous media. This independent character of the developed catalysts from water and polar solvents can open up a new avenue in Pd(II) catalysis. Accordingly, utilization of this system for the incorporation of deuterium and other useful functional groups into the C-H bonds of numerous classes of organic compounds will be extensively pursued and reported in due course.
Experimental Section
Catalytic H/D exchange reaction
Pd cation compounds were prepared when required. Catalyst (7a, 7b, or 11) and 1.5 eq of AgBF4 were stirred in 2 mL of MeCN solution for 30 minutes. After passing through celite column, The filtrate was dried in a rotary evaporator. The dried compound was dissolved in 0.7 mL of D2O and placed in a J-Young NMR tube with an external standard capillary consisting of C6F6 solution with the poly(dimethylsiloxane). Then the hydrocarbon substrate (20 μL) was added in D2O solution. The resulting mixture was heated at 55 °C/100 °C. Deuteration levels were monitored by 1H NMR using an external capillary standard consisting of a solution of the poly (dimethylsiloxane).
Oxidation of Benzene (Equation 5)
Pd cationic compound 2 was prepared when required. The catalyst 1 (2 mg, 0.5 mol%) and AgBF4 (1.5 eqiv.) were stirred in 2 mL of MeCN solution for 30 minutes. Then filtrate was dried in a rotary evaporator after passing through celite column. The dried compound was dissolved in 0.2 mL of H2O. Then 0.2 mL of H2O2 and 0.1 mL of C6H6 was added to the aqua solution. The resulting mixture was heated at 60 °C for 20 hours. After the catalytic reaction, 2 μL of pyridine for the internal reference and 0.2 mL of D2O were added. Production of 16 equivalent of phenol was detected by WET1D NMR technique.
Supplementary Material
Acknowledgments
We acknowledge generous financial supports from the National Institute of General Medical Sciences of the National Institute of Health (RO1 GM 71495) as well as the Hydrocarbon Research Foundation.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.200######.
References
- 1.Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 2.a) Suzuki A. J Organomet Chem. 1999;576:147. [Google Scholar]; b) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]
- 3.Mitchell TN. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Wiley-VCH Verlag GmbH; Weinheim, Germany: 1998. pp. 167–202. [Google Scholar]
- 4.Negishi E-I, Anastasia L. Chem Rev. 2003;103:1979. doi: 10.1021/cr020377i. [DOI] [PubMed] [Google Scholar]
- 5.Negishi E-I, Hu Q, Huang ZH. Aldrichimica Acta. 2005;38:71. [Google Scholar]
- 6.a) Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805. [Google Scholar]; b) Hartwig JF. Angew Chem Int Ed. 1998;37:2046. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.a) Periana RA, Bhalla G, Tenn WJ, III, Young KJH, Liu XY, Mironov O, Jones CJ, Ziatdinov VR. J Mol Catal A. 2004;220:7. [Google Scholar]; b) Goldman AS, Goldberg KI. Activation and Functionalization of C-H Bonds. Oxford University Press; Washington D.C.: 2004. pp. 1–43. [Google Scholar]; c) Labinger JA, Bercaw JE. Nature. 2002;417:506. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; d) Crabtrss RH. J Chem Soc Dalton Trans. 2001:2437. [Google Scholar]; e) Jones WD. Science. 2000;287:1942. [Google Scholar]
- 8.Periana RA, Mironov O, Taube D, Bhalla G, Jones C. Science. 2003;301:814. doi: 10.1126/science.1086466. [DOI] [PubMed] [Google Scholar]
- 9.a) Tanabe Y, Hanasaka F, Fujita K, Yamaguchi R. Organometallics. 2007;26:4618. [Google Scholar]; b) Corberan R, Sanau M, Peris E. Organometallics. 2006;25:4002. [Google Scholar]; c) Cariou R, Fischmeister C, Toupet C, Dixneuf PH. Organometallics. 2006;25:2126. [Google Scholar]; d) Appelhans LN, Zuccaccia D, Kovacevic A, Chianese AR, Miecznikowski JR, Macchioni A, Clot E, Eisenstein O, Crabtree RH. J Am Chem Soc. 2005;127:16299. doi: 10.1021/ja055317j. [DOI] [PubMed] [Google Scholar]; e) Cabeza JA, de Rio I, Miguel D, Sanches-Vega MG. Chem Comm. 2005:3956. doi: 10.1039/b506287j. [DOI] [PubMed] [Google Scholar]; f) Scott NM, Dorta R, Stevens ED, Correa A, Cavallo L, Nolan SP. J Am Chem Soc. 2005;127:3516. doi: 10.1021/ja043249f. [DOI] [PubMed] [Google Scholar]; g) Burling S, Mahon MF, Paine BM, Whittlesey MK, Williams JMJ. Organometallics. 2004;23:4537. [Google Scholar]; h) Dorta R, Stevens ED, Nolan SP. J Am Chem Soc. 2004;126:5054. doi: 10.1021/ja049545+. [DOI] [PubMed] [Google Scholar]; i) Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi TL, Ding S, Day MW, Grubbs RH. J Am Chem Soc. 2003;125:2546. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]
- 10.a) Scott NM, Nolan SP. Eur J Org Chem. 2005;127:3516. doi: 10.1021/ja043249f. [DOI] [PubMed] [Google Scholar]; b) Scott NM, Nolan SP. Eur J Inorg Chem. 2005:1815. [Google Scholar]; c) Ahrens S, Strassner T. Inorg Chim Acta. 2006;359:4789. [Google Scholar]; d) Huynh HV, Han Y, Ho JHH, Tan GK. Organometallics. 2006;25:3267. [Google Scholar]; e) Burling S, Paine BM, Nama D, Brown VS, Mahon MF, Prior TJ, Pregosin PS, Whittlesey MK, Williams JMJ. J Am Chem Soc. 2007;129:1987. doi: 10.1021/ja065790c. [DOI] [PubMed] [Google Scholar]; f) Fructos MR, de Fremont P, Nolan SP, Diaz-Requejo MM, Perez PJ. Organometallics. 2006;25:2237. [Google Scholar]; g) Corberan R, Sanau M, Peris E. J Am Chem Soc. 2006;128:3974. doi: 10.1021/ja058253l. [DOI] [PubMed] [Google Scholar]
- 11.a) Sajiki H, Hattori K, Aoki F, Yasunaga K, Hirota K. Synlett. 2002:1149. [Google Scholar]; b) Sajiki H, Aoki F, Esaki H, Maegawa T, Hirota K. Org Lett. 2004;6:1485. doi: 10.1021/ol0496374. [DOI] [PubMed] [Google Scholar]; c) Sajiki H, Esaki H, Aoki F, Maegawa T, Hirota K. Synlett. 2005:1385. [Google Scholar]; d) Esaki H, Aoki F, Maegawa T, Hirota K, Sajiki H. Heterocycles. 2005;66:361. [Google Scholar]; e) Maegawa T, Akashi A, Esaki H, Aoki F, Sajiki H, Hirota K. Synlett. 2005:845. [Google Scholar]; f) Sajiki H, Ito N, Esaki H, Maesawa T, Maegawa T, Hirota K. Tetrahedron Lett. 2005;46:6995. [Google Scholar]; g) Ito N, Watahiki T, Maesawa T, Maegawa T, Sajiki H. Adv Synth Catal. 2006;348:1025. [Google Scholar]; h) Esaki H, Ito N, Sakai S, Maegawa T, Monguchi Y, Sajiki H. Tetrahedron. 2006;62:10954. [Google Scholar]; i) Esaki H, Aoki F, Umemura M, Kato M, Maegawa T, Monguchi Y, Sajiki H. Chem Eur J. 2007;13:4052. doi: 10.1002/chem.200601615. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi S, Yoo KS, O’Neill J, Lee JH, Stewart T, Jung KW. Angew Chem Int Ed. 2008;47:9326. doi: 10.1002/anie.200803793. [DOI] [PubMed] [Google Scholar]
- 13.For the NMR analysis, see the supporting information.
- 14.a) Dervisi A, Koursarou D, Ooi L, Horton PN, Hursthouse MB. Dalton Trans. 2006:5717. doi: 10.1039/b609607g. [DOI] [PubMed] [Google Scholar]; b) de Vries JG. Dalton Trans. 2006:421. doi: 10.1039/b506276b. [DOI] [PubMed] [Google Scholar]; c) de Vries HM, Mulders JMCA, Mommers JHM, Henderickx HJW, de Vries JG. Org Lett. 2003;5:3285. doi: 10.1021/ol035184b. [DOI] [PubMed] [Google Scholar]; d) Rosner T, Le Bars J, Pfaltz A, Blackmond DG. J Am Chem Soc. 2001;123:1848. doi: 10.1021/ja003191e. [DOI] [PubMed] [Google Scholar]
-
15.Reaction routes for 4a and 4b:
- 16.a) Wang HMJ, Lin IJB. Organometallics. 1998;17:972. [Google Scholar]; b) Wang IJB, Vasam CS. Coordination Chem Rev. 2007;251:642. [Google Scholar]
- 17.For the NMR analysis, see the supporting information.
- 18.a) Golden JT, Andersen RA, Bergman RG. J Am Chem Soc. 2001;123:5837. doi: 10.1021/ja0155480. [DOI] [PubMed] [Google Scholar]; b) Santos LL, Mereiter K, Paneque M, Slugovc C, Carmona E. New J Chem. 2003;27:107. [Google Scholar]; c) Wong-Foy AG, Bhalla G, Liu XY, Periana RA. J Am Chem Soc. 2003;125:14292. doi: 10.1021/ja037849a. [DOI] [PubMed] [Google Scholar]; d) Lei GD, Sachtler WMH. J Cat. 1993;140:601. [Google Scholar]; e) Augustine RL, Wesdyk R. Langmuir. 1985;1:262. [Google Scholar]
- 19.a) Luecke HF, Arndtsen BA, Burger P, Bergman RG, Am J. Chem Soc. 1996;118:2517. [Google Scholar]; b) Gutierrez-Puebla E, Monge A, Paneque M, Poveda ML, Taboada S, Trujillo M, Carmona E. J Am Chem Soc. 1999;121:346. [Google Scholar]; c) Klei SR, Golden JT, Tilley TD, Bergman RG, Am J. Chem Soc. 2002;124:2092. doi: 10.1021/ja017219d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.