

Abstract
Polycystin 1 and polycystin 2 are large transmembrane proteins, which, when mutated, cause autosomal dominant polycystic kidney disease (ADPKD), a highly prevalent human genetic disease. The polycystins are thought to form a receptor-calcium channel complex in the plasma membrane of renal epithelial cells and elicit a calcium influx in response to mechanical stimulation, such as fluid flow across the apical surface of renal epithelial cells. The functional role of the polycystins in mechanosensation remains largely unknown. Here, we found that myocyte enhancer factor 2C (MEF2C) and histone deacetylase 5 (HDAC5), two key regulators of cardiac hypertrophy, are targets of polycystin-dependent fluid stress sensing in renal epithelial cells in mice. We show that fluid flow stimulation of polarized epithelial monolayers induced phosphorylation and nuclear export of HDAC5, which are crucial events in the activation of MEF2C-based transcription. Kidney-specific knockout of Mef2c, or genetrap-inactivation of a MEF2C transcriptional target, MIM, resulted in extensive renal tubule dilation and cysts, whereas Hdac5 heterozygosity or treatment with TSA, an HDAC inhibitor, reduced cyst formation in Pkd2−/− mouse embryos. These findings suggest a common signaling motif between myocardial hypertrophy and maintenance of renal epithelial architecture, and a potential therapeutic approach to treat ADPKD.
Keywords: Autosomal dominant polycystic kidney disease, Histone deacetylase 5, Myocyte enhancer factor 2C, Mouse
INTRODUCTION
During renal development, nephron formation relies on coordinated regulation of cell proliferation, cell polarity, differentiation and apoptosis. Epithelial cell organization is disrupted in autosomal dominant polycystic kidney disease (ADPKD) kidneys, where numerous fluid-filled cysts develop as a result of aberrant cell proliferation, loss of planar cell polarity and transepithelial fluid secretion and changes of epithelial cell polarity and cytoskeleton (Grantham, 2003; Wilson and Goilav, 2007). ADPKD is caused by mutations in PKD1 or PKD2, which encode polycystin 1 (PC1) and polycystin 2 (PC2), respectively (Mochizuki et al., 1996; Rossetti et al., 2001; Watnick and Germino, 1999). PC1 is a large G-protein-coupled receptor-like protein with a complex array of functions and has been shown to bind polycystin 2 (PC2), a TRP calcium channel, through a COOH-terminal coiled-coil region (Boletta and Germino, 2003).
PC1 and PC2 localize to a number of cellular compartments such as the primary cilia, microtubule-based structures that extend from the apical surface of epithelial cells into the tubule lumen (Berbari et al., 2009; Eley et al., 2005), as well as other epithelial surfaces, and PC2 associates prominently with the endoplasmic reticulum (ER) (reviewed in Wilson and Goilav, 2007). The cilia localization has gained the most attention as physical bending of the primary cilia or fluid flow across the apical surface of epithelial cells caused an increase in intracellular Ca2+ (Praetorius and Spring, 2001). Later studies showed that polycystins are required for the fluid flow-induced Ca2+ influx, suggesting that these proteins, possibly through their association with cilia, function as mechanosensors (Grimm et al., 2002; Nauli et al., 2003). However, conditional inactivation of genes required for ciliogenesis in adult mice did not lead to rapid development of cysts despite the loss of primary cilia (Davenport et al., 2007; Patel et al., 2008). These findings cast doubts on whether the loss of mechanosensory function mediated by the primary cilia is responsible for cyst formation.
Despite a lack of clarity on the functional location of the polycystins, loss of polycystins correlates with disruption of flow-dependent intracellular calcium signaling and a reduction in steady-state intracellular Ca2+ levels (Nauli et al., 2006; Yamaguchi et al., 2006). Recent studies have demonstrated a role for intracellular Ca2+ in cAMP-dependent cell proliferation. cAMP stimulates the proliferation of ADPKD cells in culture but inhibits the proliferation of normal renal cells. Incubation of normal cells with a calcium channel blocker caused a phenotypic switch such that cAMP stimulated cell proliferation. Furthermore, elevation of intracellular calcium prevented cAMP-induced hyper-proliferation of ADPKD cells (Yamaguchi et al., 2006). Interestingly, treatment of Cy/+ rats, a PKD rat model, with a calcium channel blocker increased the progression of PKD, whereas treatment with a calcium mimetic inhibited late-stage cyst growth (Gattone et al., 2009; Nagao et al., 2008). Signaling molecules or transcription factors, such as MAP kinases, STAT3 and ID2, have been implicated in the regulation of cell proliferation downstream of the polycystins (Bhunia et al., 2002; Li et al., 2005b; Nagao et al., 2003; Yamaguchi et al., 2003); however, it is unclear if these pathways are directly regulated by calcium and mechanosensory function of polycystins. Furthermore, the importance of fluid flow-induced calcium signaling through polycystins was called into question in a recent study (Köttgen et al., 2008). Amidst these unresolved questions, elucidating molecular pathways that directly respond to the fluid flow-induced calcium signal might provide new insights into the role of polycystin mechanosensory function in the control of epithelial organization and proliferation.
In order to identify downstream targets regulated by the mechanosensory function of the polycystins, we performed an expression microarray analysis designed to detect genes that are differentially expressed in response to fluid flow shear stress in a PC1-dependent manner in polarized renal epithelial cells. This analysis identified myocyte enhancer factor 2C (Mef2c) and histone deacetylase 5 (Hdac5) among the fluid flow-responsive genes. We demonstrate that the fluid flow-dependent calcium rise leads to phosphorylation of HDAC5 and its nuclear export. These events lead to activation of MEF2C target genes, one of which is found to be missing in metastasis (MIM), a regulator of the actin cytoskeleton. Using mouse models, we present evidence that MEF2C and MIM are required for the maintenance of normal epithelial organization in the kidney and that pharmacological inhibition of HDACs might be a potentially useful therapeutic strategy to inhibit ADPKD disease progression.
MATERIALS AND METHODS
Antibodies
HDAC5 (Phospho-Ser498 human) antibody was obtained from Signalway Antibody. The non-phospho-specific anti-HDAC5 antibody and anti-MIM were purchased from Cell Signaling. Anti-MEF2C antibody was from Santa Cruz Biotechnology, Inc. Anti-PC1 antibody was a generous gift from Patricia Wilson (Wisconsin Medical College). Specific bands detected by the anti-HDAC5, anti-MIM or anti-PC1 antibody were verified against null adult kidneys or cell lines (see Fig. S1 in the supplementary material).
Fluid flow experiments
Confluent mouse embryonic kidney (MEK) cells were cultured for 1-2 additional days in the absence of interferon-γ to induce optimal differentiation (Nauli et al., 2003). Fluid [Hanks' Balanced Salt Solution with 20.0 mM HEPES buffer (pH 7.4) and 1% bovine serum albumin was used for calcium imaging and cell culture media for the other experiments] flow was applied at 0.2 ml/minute using a peristaltic pump in 35 mm dishes or 6-well plates, with tubings connected through two 18-gauge needles at opposing sides of a well (see Fig. S2A in the supplementary material). Timelapse imaging was performed on an Axiovert 200M inverted microscope (Karl Zeiss) equipped with a 20×/0.8 NA Plan-Apochromat objective, a Pecon XL-3 environmental chamber (Erbach, Germany) and an AxioCam HSm r1.1 digital monochrome camera (Karl Zeiss). In drug treatments, PMA (Sigma-Aldrich) was used at 100 nM, GÖ6983 (Sigma-Aldrich) at 10 μM, ionomycin (Sigma-Aldrich) at 1 μM and GdCl3 (Sigma-Aldrich) at 20 μM.
Computation of the flow field was performed with COMSOL Multiphysics Finite Element Software and the result is presented in Fig. S2B in the supplementary material. The total inflow is characterized by the flow rate F=0.2 ml/minute, which is nearly evenly distributed over the rectangular cross-section with dimensions 2 cm × 0.5 cm, i.e. over the area A=1 cm2. The average velocity, v, is computed as v=F/A=0.2 cm/minute=30 μm/second, with a calculated fluid shear stress of 0.020 dyn/cm2. The standard deviation was estimated by integration of the calculation presented above, equaling 8 μm/second or 0.0053 dyn/cm2.
Microarray analysis
Biotinylated cRNA was prepared from 5 μg total RNA using the standard Affymetrix one-cycle target labeling protocol. Samples were assayed using Affymetrix GeneChip Mouse Genome 430 2.0 Arrays, comprising probe sets representing over 39,000 transcripts based on Unigene Build 107 from June 2002. Data was analyzed using the R statistical environment. Affymetrix CEL files were processed and normalized using RMA (Irizarry et al., 2003). The linear modeling package Limma (Azzam et al., 2004) was used to derive gene expression coefficients using genotype and presence or absence of fluid flow as factors, and to identify differentially expressed genes. The data was also examined and characterized with GCOS and Partek (St Louis, MO) ANOVA. The data are currently accessible in the ArrayExpress database at http://www.ebi.ac.uk/microarray-as/ae/, accession number E-TABM-411.
Chromatin immunoprecipitation (ChIP) and luciferase assay
Chromatin was prepared from Flag-Mef2c- or Flag-Hdac5-expressing cells as described in the manufacturer's instructions (Upstate), with a cross-linking time of 15 minutes at 25°C and sonication to an average length of 200-700 bp. ChIP was performed using anti-Flag M2 monoclonal antibody (Sigma). Samples were analyzed by PCR. The following primers for MIM were used: forward, 5′-CCAGCCAGGGTTGCCATAGCCAC-3′ and reverse, 5′-TGACTCTTGCAAACGGTTCAATTAGGTGC-3′, which flank a 1 kb region in the MIM promoter that harbor a potential MEF2 binding motif.
Luciferase assay was carried out 54 hours post-transfection using a commercial kit (Promega) and an Orin microplate luminometer. Each transfection included 200 ng of pRL-TK renilla luciferase as internal control.
RNA interference
The oligonucleotide sequences used for Mef2c RNA interference (Dharmacon) are as follows: 5′-GAGGAUCACCGGAACGAAU-3′; 5′-UAGUAUGUCUCCUGGUGUA-3′; 5′-GAUAAUGGAUGAGCGUAAC-3′; 5′-CCAGAUCUCCGCGUUCUUA-3′. The oligonucleotides were transfected by using the DharmaFECT siRNA transfection reagent (Dharmacon).
MEK cells transduced with Pkd1 siRNA lentivitus (VIRHD/P/siPKD13297) and control lentivirus (Battini et al., 2008) were cultured for four weeks in complete medium in the presence of puromycin. Cells were harvested and PC1 expression was analyzed by immunoblot analysis.
Mouse strains and treatment
All experiments involving higher vertebrates were approved by the Institutional Animal Care and Use Committee of the Stowers Institute for Medical Research. All mouse strains used in this study were of the C57BL/6 background. Embryonic stem (ES) cells (Baygenomics) carrying a trap in intron 1 of the MIM gene were injected into the inner cell mass of C57BL6 blastocysts. Chimeric mice with a high (65%) contribution to coat color from 129 were bred for germline transmission. Mef2cloxP/loxP mice, originally provided by J. Schwarz (Albany Medical Center), and Sglt2-Cre mice (CNRS-Orleans, France) were crossed to generate Mef2cloxP/+ Sglt2-Cre mice, which were then paired to generate Mef2loxP/loxP Sglt2-Cre mice. Pkd2+/− mice were originally provided by S. Somlo (Yale University) and Hdac5−/− mice by E. Olson (University of Texas Southwestern Medical Center). To obtain embryos of various genotypes, the mutant mice were paired and the pregnant females were sacrificed on 18.5 days postcoitus (dpc) to collect embryonic kidneys. For trichostatin A (TSA) treatment experiments, pregnant Pkd2+/− females paired with Pkd2+/− males were subcutaneously injected daily, from 10.5 dpc to 18.5 dpc with 0.5 μg TSA (Biomol) per gram mouse body weight or DMSO (solvent of TSA) in phosphate buffered saline (PBS). At the end of this treatment, females were sacrificed and embryonic kidneys were collected.
RESULTS
Transcriptional profiling to identify potential targets of PC1-dependent mechanosensation
In order to identify pathways downstream of polycystins and the calcium signal in response to fluid shear stress, we established an in vitro system where fluid was applied using a peristaltic pump at a flow rate of 0.2 ml/minute, which correlates to an average flow rate of 30 μm/second, or calculated shear stress of 0.020 dyn/cm2, across polarized monolayers of kidney epithelial cells grown on collagen-coated coverslips in 35 mm culture dishes or 6-well plates (see Fig. S2A,B in the supplementary material). This rate is within the range of flow rates observed in renal tubules and in line with a previous work examining the effect of fluid flow on calcium channel activation in epithelial cells (Praetorius and Spring, 2001). To monitor calcium fluxes, we used GCaMP2, a genetically coded calcium sensor that produces high signal-to-noise at physiological calcium concentrations (Nakai et al., 2001; Tallini et al., 2006). In transfected cells expressing GCaMP2 at moderate (but not extremely high) levels, fluid flow using the setup described above induced calcium transients within 60-120 seconds after the start of the flow and, in several cells, imaged repeated calcium transients ~2 minutes apart were observed (see Fig. S3 in the supplementary material). The duration of the calcium peaks was in the range of 10-20 seconds, similar to that from single-cell measurements in a recent study using Fura-2 calcium sensor in Dolichos biflorus agglutinin (DBA)-positive mouse embryonic kidney (MEK) cells (Li et al., 2007).
Using this experimental setup, we performed an expression microarray analysis to identify genes whose expression levels were altered in response to fluid flow in a PC1-dependent manner. To this end, Pkd1+/+ and Pkd1−/− MEK cell lines were used, which were established previously from the collecting ducts (DBA-positive) of SV40 large T antigen-immortalized Pkd1+/+ and Pkd1−/− mouse embryos of the same genetic background (Li et al., 2005b; Nauli et al., 2003). These cells were grown confluent and differentiated in culture for 1-2 days, at which point cilia were clearly present in ~40% of the cells (see Fig. S2C in the supplementary material). Analysis of triplicate RNA samples from independent experiments using the Affymetrix mouse gene chip allowed us to identify genes that were differentially regulated in response to fluid flow between Pkd1+/+ cells and Pkd1−/− cells. Two different methods of analysis (ANOVA and Limma) were performed, which identified overlapping sets of genes encompassing a wide range of cellular functions, such as G-protein signaling, cytoskeleton regulation, cell cycle control, cell-matrix interactions, transcriptional regulation, etc. (see Fig. S4 and Table S1 in the supplementary material).
Fluid flow induces HDAC5 phosphorylation, nuclear export and MEF2C target genes
We were particularly intrigued by the presence of histone deacetylase 5 (Hdac5) and myocyte enhancer factor 2C (Mef2c) among the PC1-dependent, flow-induced genes (see Table S1 in the supplementary material) because these proteins are known to function in the same pathway in the control of cardiac hypertrophy in response to stress and calcium channel activation (McKinsey et al., 2002; Olson et al., 2006). MEF2 targets include not only structural proteins important for cardiac muscle differentiation, but also members of the MEF2 and class II HDAC families through positive-feedback loops (Haberland et al., 2007; Wang et al., 2001). Based on this knowledge, we reasoned that the presence of MEF2C and HDAC5 among the fluid flow-induced genes could be a reflection of activation of MEF2-based transcription downstream of the calcium rise elicited by polycystins.
A well-known post-translational mechanism for stress-induced MEF2C activation in myocardial cells involves phosphorylation and nuclear export of HDAC5, which represses MEF2-dependent transcripts in the resting state (McKinsey et al., 2000). Kinases activated by stress-induced Ca2+ increase, such as CaM kinase and protein kinase C or D, phosphorylate HDAC5 at two 14-3-3 binding sites, an event that leads to disruption of HDAC5-MEF2C interaction and translocation of HDAC5 from the nucleus to the cytosol (McKinsey et al., 2002). HDAC5 dissociation from MEF2C frees a binding site for the histone acetyl transferase p300/CBP, an activator of MEF2C. Mutually exclusive interaction of MEF2C with HDAC5 or p300/CBP generates a binary switch for activation of MEF2C target genes (McKinsey et al., 2001).
We determined whether fluid flow across monolayers of MEK cells led to endogenous HDAC5 phosphorylation using a phospho-specific antibody against the HDAC5 Ser489 site (equivalent of Ser498 of human HDAC5) (Bossuyt et al., 2008). Although the level of HDAC5 phosphorylation was low prior to fluid flow onset, an increase in phosphorylation was observed 30 minutes after the start of fluid flow and maintained for several hours thereafter (Fig. 1A,B). The increase in HDAC5 phosphorylation was accompanied by an increase in the level of total HDAC5 (see Fig. S5A,C in the supplementary material), consistent with the flow-induced gene expression of HDAC5 observed in the microarray analysis (see Table S1 in the supplementary material). Fluid flow-induced HDAC5 phosphorylation and increase in protein level were absent in Pkd1−/− MEK cells (Fig. 1A,B; see also Fig. S5B,C in the supplementary material) and blocked in wild-type MEK cells by Pkd1 siRNA (Battini et al., 2008) (Fig. 1C), which reduced PC1 expression by 85% (see Fig. S6 in the supplementary material). HDAC5 phosphorylation was also blocked by gadolinium (GdCl3), an inhibitor of stretch-activated cation channels previously shown to inhibit fluid flow-induced calcium response (Praetorius et al., 2003). By contrast, HDAC5 phosphorylation was induced by the calcium ionophore ionomycin in the absence of fluid flow (Fig. 1D). Treatment with PMA, an activator of protein kinase C (PKC), stimulated HDAC5 phosphorylation in the absence of fluid flow, whereas the PKC inhibitor GÖ6983 inhibited flow-induced HDAC5 phosphorylation (Fig. 1D). These data suggest that PC1, calcium channel activity and a PKC isoform are required for fluid flow-induced HDAC5 phosphorylation.
Fig. 1.

Fluid flow-induced HDAC5 phosphorylation in MEK cells. (A) Immunoblot analysis of HDAC5 phosphorylation using a phospho-specific antibody against serine 489 after Pkd1+/+ and Pkd1−/− MEK cells were stimulated with fluid flow at 0.2 ml/minute for up to 4 hours. (B) Quantification of HDAC5 phosphorylation in the above experiment, normalized first against actin and then against the t0 values. (C) Immunoblot analysis of HDAC5 phosphorylation in response to fluid flow for 4 hours in Pkd1 siRNA or control lentivirus-transduced MEK cells. (D)Effects of a PKC activator (PMA), a PKC inhibitor (GÖ6983), a calcium ionophore (ionomycin) and a calcium channel blocker (GdCl3) on HDAC5 phosphorylation with or without fluid flow in MEK cells.
To determine if HDAC5 was exported from the nucleus in response to fluid flow, FLAG-MEF2C and HDAC5-GFP constructs were co-transfected into Pkd1+/+ and Pkd1−/− MEK cells, and both tagged proteins correctly localized to the nucleus. Stimulation of MEK cells with fluid flow for 30 minutes led to translocation of HDAC5-GFP, but not FLAG-MEF2C, from the nucleus in Pkd1+/+ MEK cells, but not in Pkd1−/− MEK cells (Fig. 2A). Time-lapse movies showed that HDAC5-GFP nuclear export initiated several minutes after the start of the flow stimulation and stayed in the cytoplasm for the duration of the observation (Fig. 2B). By contrast, a HDAC5-GFP mutant lacking the 14-3-3 phosphorylation sites (HDAC5S250/489A) was not exported (Fig. 2C), consistent with a requirement for phosphorylation of these regulatory sites in fluid flow-induced nuclear export of HDAC5. HDAC5 nuclear export also failed in Pkd1−/− MEK cells and cells treated with GdCl3 (Fig. 2C) or the PKC inhibitor GÖ6983, whereas treatment with PMA stimulated HDAC5 nuclear exit even without fluid flow (Fig. 2D). These results suggest that the fluid flow-induced HDAC5 phosphorylation and nuclear export occurs in a PC1 and calcium-dependent manner and PKC is an upstream kinase required for these events.
Fig. 2.

Fluid flow-induced HDAC5 nuclear export in MEK cells. (A) Pkd1+/+ and Pkd1−/− MEK cells co-transfected with FLAG-MEF2C (red) and HDAC5-GFP (green) were stimulated with fluid flow at 0.2 ml/minute for 30 minutes (no-flow controls are also included). The cells were fixed, stained and imaged using confocal microscopy. (B) Time-lapse images of HDAC5-GFP translocation stimulated by fluid flow. Time (minutes) after flow initiation is indicated on each panel. The graph shows quantification of fluorescence ratio (nuclear/cytosolic) over time. (C) Quantification of HDAC5-GFP distribution in the cytosol or nucleus in Pkd1+/+ and Pkd1−/− cells with or without fluid flow stimulation, in the presence of GdCl3 and HDAC5S250/S489A mutations as indicated. Percentages of cells (over total) with HDAC5-GFP in the nucleus or cytosol or in both compartments are shown. (D) Quantification of HDAC5-GFP distribution in the cytosol or nucleus in cells treated with DMSO (solvent control), 100 nM PMA or 10 μM Gö 6983 in the absence or presence of fluid flow, as indicated. For both C and D, more than 600 cells were counted for each experimental condition. Shown are the average and s.e.m. from three independent experiments. Scale bars: 16 μm.
As HDAC5 phosphorylation and nuclear export are key steps in the activation of MEF2C-based transcription, we investigated a potential target of MEF2C, a gene called missing in metastasis (MIM). MIM was one of the fluid flow-induced genes in our microarray analysis, which was confirmed by northern blot analysis (see Fig. S7A in the supplementary material). MIM is a multifunctional regulator of actin cytoskeletal dynamics (Machesky and Johnston, 2007) and was previously implicated in sonic hedgehog (SHH) signaling (Callahan et al., 2004). Coincidentally, MIM was also among a list of potential targets of MEF2 in the heart, although its function in cardiac myocytes has not been characterized (van Oort et al., 2006). To determine if MEF2C indeed associates with the promoter of MIM, ChIP was carried out using an anti-FLAG antibody in MEK cells transfected with FLAG-MEF2C with or without fluid flow stimulation. FLAG-MEF2C co-immunoprecipitated with a MIM promoter fragment containing a putative MEF2C binding site both in the absence or presence of fluid flow stimulation (Fig. 3A). ChIP using an anti-FLAG antibody also showed binding of FLAG-HDAC5 to the same promoter fragment in FLAG-HDAC5-transfected MEK cells (Fig. 3A).
Fig. 3.

MIM is a transcriptional target of MEF2C and HDAC5. (A) Chromatin immunoprecipitation (ChIP) demonstrating that MEF2C and HDAC5 bind to the promoter region of MIM. FLAG-MEF2C or FLAG-HDAC5 were transfected into wild-type MEK cells, which were either resting (−flow) or stimulated with fluid flow for 0.5 hours (+flow), and an anti-FLAG antibody was used for ChIP. PCR was performed using primers in the MIM promoter region as explained in Materials and methods. Input is DNA template before ChIP. Ctrl is ChIP using MEK cells transfected with the empty FLAG vector. In two independent experiments, each with three independent cultures for flow and non-flow conditions, binding of FLAG-HDAC5 appeared to be reduced in flow-stimulated samples compared with that in the non-flow samples; however, this difference could not be quantified as ChIP is non-quantitative owing to the PCR amplification step. (B) MEF2C is important for normal MIM expression in MEK cells. RNAi knockdown of MEF2C in Pkd1+/+ MEK cells led to reduced MIM expression, quantified by qPCR. The results shown are averages of three experiments. Ctrl, cells transfected with control siRNA. Error bars indicate ± s.d. (C) Reconstitution of MIM reporter expression in Pkd1−/− MEK cells. Luciferase activity averaged from three experiments are shown for Pkd1−/− cells tranfected with either empty vector (Ctrl), MEF2C alone, GATA6 alone, or MEF2C and GATA6 together. **, P<0.05 compared with control. Error bars indicate s.d. (D) HDAC5 negatively regulates MIM expression in MEK cells. Immunoblot and quantification show that HDAC5 S250/489A-GFP overexpression reduced MIM expression in Pkd1+/+ MEK cells.
To determine if MEF2C is important for MIM expression, Mef2c siRNA was introduced into wild-type MEK cells. The Mef2c siRNA knocked down Mef2c expression, as determined by qPCR, to 38% of that in cells transfected with a control siRNA, and MIM expression was reduced to 40% of that in the control cells (Fig. 3B). To test if MEF2C can indeed activate the MIM promoter, a 2 kb promoter fragment of MIM was cloned upstream of the luciferase reporter gene. The luciferase reporter construct was introduced into Pkd1−/− MEK cells, which showed a drastically reduced level of endogenous MEF2C and MIM expression compared with those in Pkd1+/+ cells (see Fig. S7B,C in the supplementary material). Transfection of a Mef2c-expressing plasmid into the Pkd1−/− cells only minimally stimulated MIM reporter expression; however, MEF2 members have been shown to function together with cell-type specific GATA transcription factors, which recruit MEF2 proteins to promoters through direct interactions between these proteins (Morin et al., 2000). A candidate for this cofactor is GATA6, also a flow-induced gene that showed reduced expression in Pkd1−/− MEK cells (see Table S1 and Fig. S7B,C in the supplementary material). Several GATA sites are present within 500 bp of the MEF2 binding motif in the MIM promoter. Co-transfection of Pkd1−/− cells with both Mef2c and Gata6, but not either construct alone, stimulated MIM reporter expression by 3-fold (Fig. 3C). Moreover, overexpression of the non-phosphorylatable HDAC5 (HDAC5S250/489A-GFP) in Pkd1+/+ cells strongly repressed MIM expression (Fig. 3D). The above results suggest that MEF2C and HDAC5 directly regulate MIM expression in MEK cells.
Mef2c and MIM gene disruption in mouse leads to renal tubule dilation and cyst formation
To determine if MEF2C-based transcription is important for kidney epithelial organization and proliferation, we obtained a previously established Mef2c conditional knockout mouse line (Vong et al., 2005) because conventional Mef2c knockout resulted in early embryonic lethality (Lin et al., 1997). Kidney-specific disruption of Mef2c was accomplished using Sglt2 promoter-driven Cre (Rubera et al., 2004), which is expressed renal tubules and glomeruli (see Fig. S8 in the supplementary material). In young Mef2cloxp/loxP Sglt2-Cre mutant mice (up to 2-months-old), histological examination of the kidneys revealed no obvious abnormality (n=20); however, in mice 5-months-old or older, renal abnormalities were clearly observed in 9 out of 12 mice, including broadly distributed dilated tubules (Fig. 4B,D compared with 4A,C) and small bilateral cysts with flat lining cells (Fig. 4F compared with 4E). Marker staining showed that dilated tubules originated from distal tubules, proximal tubules and collecting ducts (Fig. 4H). Small glomerular cysts were also observed (Fig. 4G). Dilated tubules or cysts were associated with increased cell proliferation as indicated by Ki67 staining (Fig. 4I). These defects were not observed in kidneys of wild-type adult mice of the same age. This result indicates that MEF2C is important for the maintenance of normal tissue organization and repression of cell proliferation in differentiated kidney epithelia.
Fig. 4.

Kidney specific knock-out of Mef2C resulted in epithelial tubule dilations/cysts. (A-I) Kidneys from 5- to 6-month-old Mef2Cloxp/loxP Sglt2-Cre mice and their congenic wild-type controls were fixed in 4% formaldehyde, sectioned (5 μm) and stained with Hematoxylin and Eosin (A-G), tubule markers (H) or Ki67 (I). (A) A representative section of wild-type kidney. (B) A representative kidney section from a Mef2Cloxp/loxp Sglt2-Cre mouse. (C) Magnified region corresponding to the box in A. (D) Magnified region corresponding to the box in B, which reveals a broad distribution of dilated tubules (arrows). (E) A 40× magnified cortex region in wild-type mouse kidney, showing normal epithelial tubules. (F) A 40× magnified cortex region of a Mef2Cloxp/loxP Sglt2-Cre kidney, revealing dilated tubules and small cysts with flat lining cells (arrows). (G) Glomerular cysts (arrows) in a Mef2Cloxp/loxP Sglt2-Cre kidney. (H) Staining of wild-type and Mef2Cloxp/loxP Sglt2-Cre kidney sections against DAPI (blue) and various tubule markers (green): Na+/K+ ATPase a-1 (distal tubule), LTA (proximal tubule) and DBA (collecting duct). Arrows point to dilated tubules/cysts. (I) Representative Ki67 staining of wild-type and Mef2Cloxp/loxP Sglt2-Cre kidney sections, showing increased cell proliferation in the mutant kidney section. Scale bars: 1000 μm in A,B; 200 μm in C,D; 50 μm in E-G,I; 100 μm in H.
To assess the in vivo function of MIM, a MIM genetrap ES cell line was used to generate MIM-deficient mice (see Fig. S9A in the supplementary material). The genetrap was inserted at intron 1, truncating MIM after exon 1. X-gal staining of MIM+/− embryos showed that MIM is expressed abundantly in the embryonic heart, spinal cord, liver and the limb bud (data not shown). In E16.5 embryonic kidneys, MIM is expressed in branching collecting ducts, tubules and glomeruli (see Fig. S9B,C in the supplementary material). After birth, MIM is significantly expressed in the renal cortex and weakly expressed in the renal medula (see Fig. S9D,E in the supplementary material). MIM−/− animals appeared normal after birth, but by 5 months, kidneys in 50% of the mutant animals (n=40) exhibited multiple large cysts (Fig. 5B,C compared with 2A) with flat lining cells (Fig. 5D). As in Mef2c mutant kidneys, Ki67 staining showed drastically elevated proliferating cells (Fig. 5F compared with 5I). The above results suggest that MEF2C and its transcriptional targets, such as MIM, are required for preventing hyper-proliferation and cystogenesis in renal tubules.
Fig. 5.

MIM is required for normal renal epithelial organization. (A,B) Representative histology sections of wild-type (A) and MIM−/− (B) kidneys from 5-month-old mice. (C) A magnified image of the boxed region in B. (D) A magnified image of cyst lining cells (arrow) in the boxed region in C. (E,F) Representative Ki67 staining of wild-type (E) and MIM−/− (F) kidney sections, showing increased cell proliferation in the mutant kidney section. Scale bars: 1000 μm in A,B; 50 μm in C-F.
HDAC5 inhibition suppresses renal cyst formation in Pkd2−/− mouse embryos
A prediction based on a pathway where the calcium signal generated by polycytins deactivates HDAC5 is that loss-of-function mutations in HDAC5 should alleviate cyst formation in Pkd2−/−mice. We tested this possibility by crossing pairs of Pkd2+/− Hdac5+/− double mutant mice. Pkd2−/− embryos were known to die before or immediately after birth with many large renal cysts (Wu et al., 2000). Genotyping of 89 E18.5 embryos from 11 crosses showed that, for reasons unknown, no Pkd2−/− Hdac5+/+ embryos and only 1 Pkd2−/− Hdac5−/− embryo was observed from these crosses (expected and observed frequencies of all genotypes are listed in Table S2 in the supplementary material). However, kidneys of Pkd2−/− Hdac5+/− embryos (n=7) exhibited visibly reduced cyst formation compared with Pkd2−/− Hdac5+/+ kidneys from embryos of Pkd2+/− Hdac5+/+ parents of the same genetic background (Fig. 6C compared with 6D). Quantification of cyst area percentages further confirmed significantly reduced cyst formation in Pkd2−/− Hdac5+/− kidneys compared with that in Pkd2−/− Hdac5+/+ kidneys (Fig. 6H).
Fig. 6.

Effects of Hdac5 genetic or chemical inactivation on cyst formation in Pkd2−/− embryonic kidneys. (A-D) Representative embryonic kidney sections from E18.5 embryos of different genotypes. A, Pkd2+/+ Hdac5+/+; B, Pkd2+/+ Hdac5−/−; C, Pkd2−/− Hdac+/−; D, Pkd2−/− Hdac5+/+. (E-G) Representative histology sections of E18.5 Pkd2+/+ and Pkd2−/− embryonic kidneys from pregnant mothers injected with TSA or control solvent (DMSO) from 10.5 dpc to 18.5 dpc. E, Pkd2+/+ from TSA-treated mother; F, Pkd2−/− from TSA-treated mother; G, Pkd2−/− from DMSO-treated mother. (H) Quantification of the percentage of cystic areas over total kidney section areas of different genotypes or treatment, as indicated. The middle section of each kidney was quantified for all mice under each condition. Shown are mean and s.e.m. of all sections quantified for each condition. Asterisks, P<0.05 compared with the control to the left. (I-K) Magnified images of the box regions in E, F and G, respectively. Lining cells around a normal tubule (I), small cyst (J) and large cyst (K) are indicated by arrows. (L) Immunoblots showing reduced MIM and MEF2C expression in 18.5 dpc Pkd2−/− embryonic kidneys. (M) TSA stimulates MIM and MEF2C expression in Pkd2−/− embryonic kidneys. Scale bars: 200 μm in A-G; 50 μm in I-K.
To test further the possibility that reducing the activity of HDAC5 could suppress cyst formation in Pkd2−/− embryos, we injected trichostatin A (TSA), a chemical inhibitor against type I and II HDACs (Drummond et al., 2005), into pregnant Pkd2+/− female mice from 10.5 dpc after mating with Pkd2+/− males, and embryonic kidneys were analyzed at 18.5 dpc. In all Pkd2−/− embryos (n=7) from TSA-injected mothers, kidney cyst formation was drastically reduced compared with control DMSO-injected mothers (n=6; Fig. 6F compared with 6G,H). Even in the much smaller cysts that persisted in the Pkd2−/− kidneys from TSA-treated mothers, cyst lining cells exhibited a more normal cuboidal morphology as opposed to the flat morphology of Pkd2−/− cyst lining cells (Fig. 6I-K). Apart from the above effect on cystic kidneys, TSA injection did not affect the morphology of renal tubules in Pkd2+/+ embryonic kidneys (Fig. 6E,I). Immunoblot analysis showed that MIM and MEF2C expression levels were, respectively, 10-fold and 3-fold reduced in E18.5 Pkd2−/− embryonic kidneys compared with those in Pkd2+/+ embryonic kidneys (Fig. 6L). Pkd2−/− embryonic kidneys from TSA-treated mothers showed a 4-fold enhanced expression of MEF2C and a drastically increased expression of MIM compared with in Pkd2−/− kidneys from DMSO-treated mothers (Fig. 6M), suggesting that TSA stimulated the expression of MEF2C target genes.
DISCUSSION
The results of this study have revealed an unexpected role for HDAC5 and MEF2C as targets of polycystin mechanosensory function in renal epithelial cells. In this pathway, laminar fluid flow across the apical surface of epithelial cells induces phosphorylation and nuclear export of HDAC5 in a polycytin-1- and calcium-dependent manner, leading to activation of MEF2C-based transcription (summarized in Fig. 7). We found that conditional disruption of Mef2c or genetrap-based inactivation of MIM, one of the transcriptional targets of MEF2C, leads to renal tubule dilation and epithelial cysts in adult mice by 5 months of age, although the number and size of the cysts were not as severe as those in animals with conditional knockout or somatic inactivation of polycystins (Lantinga-van Leeuwen et al., 2007; Piontek et al., 2007; Wu et al., 2000). Interestingly, it also took 5 months for cysts to occur after inactivation of Pkd1 at postnatal day 14 (Piontek et al., 2007). These findings support the idea that there might be a developmental factor that contributes to the onset of cystogenesis in adult kidneys.
Fig. 7.
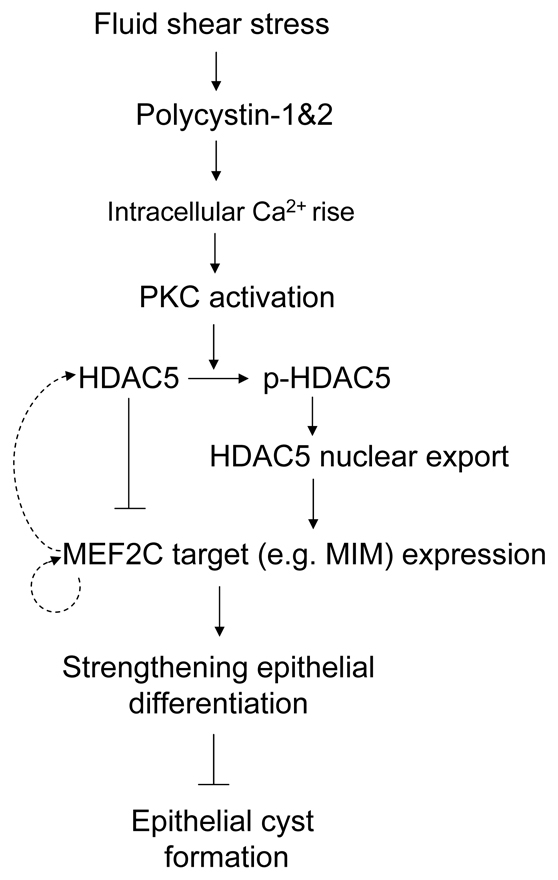
A schematic diagram depicting a pathway that connects polycystins and calcium signaling to HDAC5 and MEF2C-based transcription activation in the suppression of epithelial cyst formation. Fluid flow through lumens of renal tubules activates the polycystin 1 and 2 receptor-calcium channel complex. Downstream of the intracellular calcium rise, active PKC directly or indirectly causes phosphorylation of HDAC5 and its export from the nucleus, enabling activation of MEF2C-regulated transcripts, which probably encode proteins that maintain the normal differentiated state of renal epithelial cells. The observed increase of HDAC5 and MEF2C transcript levels in our microarray analysis might be explained by feedback loops, depicted with dotted lines.
During the preparation of this manuscript, a new paper was published demonstrating that fluid shear stress induced HDAC5 phosphorylation and nuclear export in endothelial cells, although the level of shear stress was ~1000-fold higher than that in renal tubules (Wang et al., 2009). Therefore, HDAC5 might be a common target of multiple mechanosensory pathways that respond to fluid flow or other mechanical stresses. Many details of this fluid flow sensing pathway in renal epithelial cells remain to be elucidated. For example, we do not yet know which isoform of PKC or other Ca2+-regulated kinase directly phosphorylates HDAC5. More important, there is not yet direct evidence supporting the notions that polycystins are the actual sensors of fluid shear stress and that the observed HDAC5 nuclear export was indeed induced by a calcium signal as a result of the channel activity of PC2. A related question is whether cilia are involved in the fluid flow-induced response observed in this study. Although ~40% of the MEK cells had visible cilia (see Fig. S2C in the supplementary material), the flow-induced HDAC5 nuclear export, which is dependent on calcium channel activation, was consistently observed in 70-80% of the cells. As a previous study involving physical bending of the cilium found that the stress-induced intra-cellular calcium increase can spread from cell to cell (Praetorius and Spring, 2001), a definitive experiment addressing the role of cilia in the observed response would probably require a complete inhibition of cilia formation or blocking the intercellular spreading of the calcium signal.
Cardiac hypertrophy in the adult heart is an adaptive mechanism of activation of an embryonic program to improve cardiac pump function in response to an increased workload or mechanical stress (Xu et al., 2006). The similarity between the cardiac hypertrophy pathway and the response to fluid flow in renal epithelial cells raises a question as to whether cystogenesis in ADPKD could in part be an outcome of a lack of the proper response to certain stress or an increase in workload on renal epithelial cells. The nephrons are frequently challenged with fluctuations in fluid shear stress and osmotic pressure, and these changes might be enhanced during certain developmental stages or under certain dietary, hormonal or pathological conditions. Renal hypertrophy is a well-known phenomenon that occurs after nephretomy or under other stress conditions (Hostetter, 1995). A recent study reported that treatment of PCK rats (a recessive PKD model) with 1-deamino-8-D-arginine vasopressin (dDAVP), an agonist of the arginine vasopressin V2 receptor, led to aggravated cystic disease, but the same treatment on wild-type rats resulted in renal hypertrophy (Wang et al., 2008). This observation and our finding that polycystins regulate a molecular pathway known to be involved in cardiac hypertrophy raise a question as to whether polycystins function at the crossroads between hypertrophy and hyperplasia in renal epithelial cells in response to stress or certain physiological stimuli.
MEF2C-based transcriptional activation leads to increased expression of contractile proteins and metabolic enzymes in cardiac tissues. If there is indeed a similar hypertrophic pathway in the kidney, MEF2C might cooperate with other tissue-specific transcription factors, such as GATA6, to control the expression of genes involved in strengthening the differentiated state of renal epithelial tubules in response to stress. Consistent with this, MIM, a founding member of the family of IMD-containing actin binding proteins, as well as another member of this family, BAIAP2L1 (see Table S1 in the supplementary material), were found to be a MEF2C target in epithelial cells. The IMD domain has the ability to induce membrane deformation and actin bundling in cultured cells (Machesky and Johnston, 2007). IMD proteins also possess a COOH-terminal WASP homology 2 (WH2) domain, which binds monomeric actin, and thus might be involved in actin polymerization. The actin cytoskeleton plays important roles in almost every aspect of epithelial cell and tissue organization (Leiser and Molitoris, 1993). Actin structures built through MIM and BAIAP2L1 might serve to strengthen epithelial cell polarity or cell-cell and cell-matrix interactions within epithelial tissues. Interestingly, several actin-interacting proteins (α-actinin, tropomysoin, troponin, mDia1, etc.) were found to bind polycystins and some were shown to modulate PC2 channel activity (Li et al., 2003a; Li et al., 2005a; Li et al., 2003b; Rundle et al., 2004). A recent study reported that polycystins regulate pressure sensing through stretch-activated channels in myocytes and this function requires an intact actin cytoskeleton and the actin-binding protein filamin (Sharif-Naeini et al., 2009). Therefore, it is also possible that the actin-regulatory activities of MIM and BAIAP2L1 are, in turn, involved in polycystin-mediated signaling, with the membrane-bending activity of IMD domains directly modulating the tension in the plasma membrane. The function of MIM might extend beyond cytoskeletal regulation; however, as in MIM−/− mouse kidneys, hyper-proliferation was evident in renal tubules. Also known as basal cell carcinoma-enriched gene 4 (BEG4), MIM was previously shown to be a target and modulator of sonic hedgehog (SHH) signaling in the skin (Callahan et al., 2004). A role in SHH signaling could underlie increased cell proliferation in MIM−/− kidneys.
Although MEF2C was the only MEF2 member whose expression level was found to increase in response to fluid flow in our microarray analysis, MEF2A, which functions in parallel with MEF2C in regulating cardiac hypertrophy (Xu et al., 2006), is also expressed in MEK cells (data not shown). The functional redundancy of MEF2C and MEF2A (Piontek et al., 2007) might account for the much milder cystic phenotype in Mef2c knockout kidneys than in polycystin knockout mice. Alternatively, this might be explained by the fact that Sglt2-Cre was not expressed in any renal tubules (see Fig. S8 in the supplementary material) and that MEF2C activation is likely to be one of several downstream pathways regulated by polycystins and important for the maintenance of epithelial architecture. Although the reduction in cyst severity in Pkd2−/− embryonic kidneys caused by Hdac5 heterozygosity is consistent with our proposed epistatic relationship between Pkd2 and Hdac5, this result does not rule out potential regulation by other members of the class II HDAC family. Furthermore, it is known that Class IIa HDACs, such as HDAC5, lack intrinsic enzymatic activity and require complex formation with HDAC3, a class I HDAC that is sensitive to TSA, for their transcriptional repression activity (Fischle et al., 2002). The restoration of the MIM expression level in TSA-treated Pkd2−/− embryonic kidneys is consistent with stimulation of MEF2C target gene expression through functional inhibition of HDAC5 or other class IIa HDACs. This might tip the balance toward upregulation of MEF2 target genes, leading to reinforcement of epithelial differentiation and suppression of cell proliferation. The anti-proliferative effect of HDAC inhibitors is in fact the basis for clinical studies of these molecules in anticancer therapies (Johnstone, 2002). Our findings raise the possibility that HDAC inhibitors could be explored as therapeutic agents for the treatment of ADPKD.
Supplementary Material
Acknowledgements
We are grateful to John Schwarz for kindly providing the Mef2CloxP/loxP mice, Eric Olson for the Hdac5−/− mice and Stefan Somlo for the Pkd2+/− mice, Patricia Wilson for the anti-PC1 antibody and Luca Gusella for Pkd1 and control siRNA lentivirus. We thank Michael Durnin for the generation of MIM genetrap mice, Blake Geppert for mouse maintenance and assistance in TSA injection experiments, Cathy Vogelweid for histology examination, Boris Rubinstein for help on calculating fluid flow rate, Mary Toth for assistance on manuscript preparation and Wei Bin Fang and Swarna Mehrotra for helpful discussion. This work was supported by the Stowers Institute for Medical Research (R.L.) and National Institutes of Health grant P50 DK057301 (D.P.W.). Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
S.X. and R.L. have a patent application(s) that is directed to (among other things) the use of histone deacetylase inhibitors to treat polycystic kidney disease and other related diseases.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.049437/-/DC1
References
- Azzam R., Chen S. L., Shou W., Mah A. S., Alexandru G., Nasmyth K., Annan R. S., Carr S. A., Deshaies R. J. (2004). Phosphorylation by cyclin B-Cdk underlies release of mitotic exit activator Cdc14 from the nucleolus. Science 305, 516-519 [DOI] [PubMed] [Google Scholar]
- Battini L., Macip S., Fedorova E., Dikman S., Somlo S., Montagna C., Gusella G. L. (2008). Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum. Mol. Genet. 17, 2819-2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari N. F., O'Connor A. K., Haycraft C. J., Yoder B. K. (2009). The primary cilium as a complex signaling center. Curr. Biol. 19, R526-R535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia A. K., Piontek K., Boletta A., Liu L., Qian F., Xu P. N., Germino F. J., Germino G. G. (2002). PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109, 157-168 [DOI] [PubMed] [Google Scholar]
- Boletta A., Germino G. G. (2003). Role of polycystins in renal tubulogenesis. Trends Cell Biol. 19, 484-492 [DOI] [PubMed] [Google Scholar]
- Bossuyt J., Helmstadter K., Wu X., Clements-Jewery H. (2008). Ca2+/calmodulin-dependent protein kinase II and protein kinase D overexpression reinforce the histone deacetylase 5 redistribution in heart failure. Circ. Res. 102, 695-702 [DOI] [PubMed] [Google Scholar]
- Callahan C. A., Ofstad T., Horng L., Wang J. K., Zhen H. H., Coulombe P. A., Oro A. E. (2004). MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 18, 2724-2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport J. R., Watts A. J., Roper V. C., Croyle M. J., van Groen T., Wyss J. M., Nagy T. R., Kesterson R. A., Yoder B. K. (2007). Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol. 17, 1586-1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond D., Noble C., Kirpotin D., Guo Z., Scott G., Benz C. (2005). Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 45, 495-528 [DOI] [PubMed] [Google Scholar]
- Eley L., Yates L. M., Goodship J. A. (2005). Cilia and disease. Curr. Opin. Genet. Dev. 15, 308-314 [DOI] [PubMed] [Google Scholar]
- Fischle W., Dequiedt F., Hendzel M. J., Guenther M. G., Lazar M. A., Voelter W., Verdin E. (2002). Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell. 9, 45-57 [DOI] [PubMed] [Google Scholar]
- Gattone V. H., Chen N. X., Sinders R. M., Seifert M. F., Duan D., Martin D., Henley C., Moe S. M. (2009). Calcimimetic inhibits late-stage cyst growth in ADPKD. J. Am. Soc. Nephrol. 20, 1527-1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham J. J. (2003). Lillian Jean Kaplan International Prize for advancement in the understanding of polycystic kidney disease. Understanding polycystic kidney disease: a systems biology approach. Kidney Int. 64, 1157-1162 [DOI] [PubMed] [Google Scholar]
- Grimm D. H., Praetorius H. A., Cai Y. (2002). A role for polycystin-2 in the cilium mechanostimulation pathway. J. Am. Soc. Nephrol. 13, F541-F552 [Google Scholar]
- Haberland M., Arnold M. A., McAnally J., Phan D., Kim Y., Olson E. N. (2007). Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell. Biol. 27, 518-525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetter T. H. (1995). Progression of renal disease and renal hypertrophy. Annu. Rev. Physiol. 57, 263-278 [DOI] [PubMed] [Google Scholar]
- Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249-264 [DOI] [PubMed] [Google Scholar]
- Johnstone R. W. (2002). Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287-299 [DOI] [PubMed] [Google Scholar]
- Köttgen M., Buchholz B., Garcia-Gonzalez M. A., Kotsis F., Fu X., Doerken M., Boehlke C., Steffl D., Tauber R., Wegierski T., et al. (2008). TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 182, 437-447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantinga-van Leeuwen I. S., Leonhard W. N., van der Wal A., Breuning M. H., de Heer E., Peters D. J. (2007). Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 16, 3188-3196 [DOI] [PubMed] [Google Scholar]
- Leiser J., Molitoris B. A. (1993). Disease processes in epithelia: the role of the actin cytoskeleton and altered surface membrane polarity Biochim. Biophys. Acta 1225, 1-13 [DOI] [PubMed] [Google Scholar]
- Li G., Vega R., Nelms K., Gekakis N., Goodnow C., McNamara P., Wu H., Hong N. A., Glynne R. (2007). A role for Alström syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 3, e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Dai Y., Guo L., Liu Y., Hao C., Wu G., Basora N., Michalak M., Chen X. Z. (2003a). Polycystin-2 associates with tropomyosin-1, an actin microfilament component. J. Mol. Biol. 325, 949-962 [DOI] [PubMed] [Google Scholar]
- Li Q., Shen P. Y., Wu G., Chen X. Z. (2003b). Polycystin-2 interacts with troponin I, an angiogenesis inhibitor. Biochemistry 42, 450-457 [DOI] [PubMed] [Google Scholar]
- Li Q., Montalbetti N., Shen P. Y., Dai X. Q., Cheeseman C. I., Karpinski E., Wu G., Cantiello H. F., Chen X. Z. (2005a). Alpha-actinin associates with polycystin-2 and regulates its channel activity. Hum. Mol. Genet. 14, 1587-1603 [DOI] [PubMed] [Google Scholar]
- Li X., Luo Y., Starremans P. G., McNamara C. A., Pei Y., Zhou J. (2005b). Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat. Cell Biol. 7, 1202-1212 [DOI] [PubMed] [Google Scholar]
- Lin Q., Schwarz J., Bucana C., Olson E. N. (1997). Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276, 1404-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L. M., Johnston S. A. (2007). MIM: a multifunctional scaffold protein. J. Mol. Med. 85, 569-576 [DOI] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Olson E. N. (2001). Control of muscle development by dueling HATs and HDACs Curr. Opin. Genet. Dev. 11, 497-504 [DOI] [PubMed] [Google Scholar]
- McKinsey T. A., Zhang C. L., Olson E. N. (2002). MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 27, 40-47 [DOI] [PubMed] [Google Scholar]
- Mochizuki T., Wu G., Hayashi T., Xenophontos S. L., Veldhuisen B., Saris J. J., Reynolds D. M., Cai Y., Gabow P. A., Pierides A., et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339-1342 [DOI] [PubMed] [Google Scholar]
- Morin S., Charron F., Robitaille L., Nemer M. (2000). GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 19, 2046-2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao S., Yamaguchi T., Kusaka M., Maser R. L., Takahashi H., Cowley B. D., Grantham J. J. (2003). Renal activation of extracellular signal-regulated kinase in rats with autosomal-dominant polycystic kidney disease. Kidney Int. 63, 427-437 [DOI] [PubMed] [Google Scholar]
- Nagao S., Nishii K., Yoshihara D., Kurahashi H., Nagaoka K., Yamashita T., Takahashi H., Yamaguchi T., Calvet J. P., Wallace D. P. (2008). Calcium channel inhibition accelerates polycystic kidney disease progression in the Cy/+ rat. Kidney Int. 73, 269 [DOI] [PubMed] [Google Scholar]
- Nakai J., Ohkura M., Imoto K. (2001). A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat. Biotechnol. 19, 137-141 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., Elia A. E., Lu W., Brown E. M., Quinn S. J., et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129-137 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Rossetti S., Kolb R. J., Alenghat F. J., Consugar M. B., Harris P. C., Ingber D. E., Loghman-Adham M., Zhou J. (2006). Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol. 17, 1015-1025 [DOI] [PubMed] [Google Scholar]
- Olson E. N., Backs J., McKinsey T. A. (2006). Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation. Novartis Found. Symp. 274, 3-12; discussion 13-19, 152-155, 272-156 [PubMed] [Google Scholar]
- Patel V., Li L., Cobo-Stark P., Shao X., Somlo S., Lin F., Igarashi P. (2008). Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum. Mol. Genet. 17, 1578-1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontek K., Menezes L. F., Garcia-Gonzalez M. A., Huso D. L., Germino G. G. (2007). A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat. Med. 13, 1490-1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius H. A., Spring K. R. (2001). Bending the MDCK cell primary cilium increases intracellular calcium. J. Membr. Biol. 184, 71-79 [DOI] [PubMed] [Google Scholar]
- Praetorius H. A., Frokiaer J., Nielsen S., Spring K. R. (2003). Bending the primary cilium opens Ca2+-sensitive intermediate-conductance K+ channels in MDCK cells. J. Membr. Biol. 191, 193-200 [DOI] [PubMed] [Google Scholar]
- Rossetti S., Strmecki L., Gamble V., Burton S., Sneddon V., Peral B., Roy S., Bakkaloglu A., Komel R., Winearls C. G., et al. (2001). Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am. J. Hum. Genet. 68, 46-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubera I., Poujeol C., Bertin G., Hasseine L., Counillon L., Poujeol P., Tauc M. (2004). Specific Cre/Lox recombination in the mouse proximal tubule. J. Am. Soc. Nephrol. 15, 2050-2056 [DOI] [PubMed] [Google Scholar]
- Rundle D. R., Gorbsky G., Tsiokas L. (2004). PKD2 interacts and co-localizes with mDia1 to mitotic spindles of dividing cells: role of mDia1 IN PKD2 localization to mitotic spindles. J. Biol. Chem. 279, 29728-29739 [DOI] [PubMed] [Google Scholar]
- Sharif-Naeini R., Folgering J. H., Bichet D., Duprat F., Lauritzen I., Arhatte M., Jodar M., Dedman A., Chatelain F. C., Schulte U., et al. (2009). Polycystin-1 and -2 dosage regulates pressure sensing. Cell 139, 587-596 [DOI] [PubMed] [Google Scholar]
- Tallini Y. N., Ohkura M., Choi B. R., Ji G., Imoto K., Doran R., Lee J., Plan P., Wilson J., Xin H. B., et al. (2006). Imaging cellular signals in the heart in vivo: cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. USA 103, 4753-4758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort R. J., van Rooij E., Bourajjaj M., Schimmel J., Jansen M. A., van der Nagel R., Doevendans P. A., Schneider M. D., van Echteld C. J., De Windt L. J. (2006). MEF2 activates a genetic program promoting chamber dilation and contractile dysfunction in calcineurin-induced heart failure. Circulation 114, 298-308 [DOI] [PubMed] [Google Scholar]
- Vong L. H., Ragusa M. J., Schwarz J. J. (2005). Generation of conditional Mef2cloxP/loxP mice for temporal- and tissue-specific analyses. Genesis 43, 43-48 [DOI] [PubMed] [Google Scholar]
- Wang D. Z., Valdez M. R., McAnally J., Richardson J., Olson E. N. (2001). The Mef2c gene is a direct transcriptional target of myogenic bHLH and MEF2 proteins during skeletal muscle development. Development 128, 4623-4633 [DOI] [PubMed] [Google Scholar]
- Wang W., Ha C. H., Jhun B. S., Wong C., Jain M. K., Jin Z. G. (2009). Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Wu Y., Ward C. J., Harris P. C., Torres V. E. (2008). Vasopressin directly regulates cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 19, 102-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watnick T., Germino G. G. (1999). Molecular basis of autosomal dominant polycystic kidney disease. Semin. Nephrol. 19, 327-343 [PubMed] [Google Scholar]
- Wilson P., Goilav B. (2007). Cystic disease of the kidney. Annu. Rev. Pathol. 2, 341-368 [DOI] [PubMed] [Google Scholar]
- Wu G., Markowitz G. S., Li L., D'Agati V. D., Factor S. M., Geng L., Tibara S., Tuchman J., Cai Y., Park J. H., et al. (2000). Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat. Genet. 24, 75-78 [DOI] [PubMed] [Google Scholar]
- Xu J., Gong N. L., Bodi I., Aronow B. J., Backx P. H., Molkentin J. D. (2006). Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J. Biol. Chem. 281, 9152-9162 [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Nagao S., Wallace D. P., Belibi F. A., Cowley B. D., Pelling J. C., Grantham J. J. (2003). Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 63, 1983-1994 [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Hempson S. J., Reif G. A., Hedge A. M., Wallace D. P. (2006). Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 17, 178-187 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.