

Abstract
DiGeorge syndrome (DGS) is a common genetic disease characterized by pharyngeal apparatus malformations and defects in cardiovascular, craniofacial and glandular development. TBX1 is the most likely candidate disease-causing gene and is located within a 22q11.2 chromosomal deletion that is associated with most cases of DGS. Here, we show that canonical Wnt—β-catenin signaling negatively regulates Tbx1 expression and that mesenchymal inactivation of β-catenin (Ctnnb1) in mice caused abnormalities within the DGS phenotypic spectrum, including great vessel malformations, hypoplastic pulmonary and aortic arch arteries, cardiac malformations, micrognathia, thymus hypoplasia and mislocalization of the parathyroid gland. In a heterozygous Fgf8 or Tbx1 genetic background, ectopic activation of Wnt—β-catenin signaling caused an increased incidence and severity of DGS-like phenotypes. Additionally, reducing the gene dosage of Fgf8 rescued pharyngeal arch artery defects caused by loss of Ctnnb1. These findings identify Wnt—β-catenin signaling as a crucial upstream regulator of a Tbx1—Fgf8 signaling pathway and suggest that factors that affect Wnt—β-catenin signaling could modify the incidence and severity of DGS.
Keywords: β-catenin, Tbx1, Fgf8, Pharyngeal arch, DiGeorge syndrome
INTRODUCTION
DiGeorge syndrome (DGS) is one of the most common genetic disorders with an incidence of 1 in 4000 live births. More than 90% of DGS cases are associated with hemizygous deletion of chromosome 22q11.2 (Lindsay, 2001; Scambler, 2000). Among the genes in this region, loss of the Tbx1 transcription factor is thought to be the major etiology of DGS phenotypes in humans (Baldini, 2003). The spectrum of DGS pathologies includes defects in pharyngeal arch artery formation and/or remodeling, cardiac outflow tract and ventricular and/or atrial septal defects, thymus and parathyroid aplasia/hypoplasia and craniofacial anomalies (Lindsay, 2001; Scambler, 2000). All of these phenotypes are caused by malformation of a transient embryonic structure called the pharyngeal apparatus (Wurdak et al., 2006). The pharyngeal apparatus comprises pharyngeal arches (PAs) and pharyngeal pouches. PAs are composed of ectoderm, endoderm, neural crest-derived mesenchyme and core mesoderm. Coordinated interaction among all of these cell types is necessary to form the tissues derived from the PAs.
In mice, heterozygosity of Tbx1 results in minor cardiovascular defects, whereas Tbx1-null mice display the most severe features characteristic of DGS (Guris et al., 2001; Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). Transgenic mice which have an additional human TBX1 gene and patients which have a mutation that stabilizes the TBX1 protein also develop DGS phenotypes (Liao et al., 2004; Torres-Juan et al., 2007; Zweier et al., 2007). This suggests that the amount of TBX1 protein is crucial for normal development and that either loss or gain of TBX1 can cause DGS phenotypes. Consistent with a role in PA development, Tbx1 is expressed in the pharyngeal arch endoderm, core mesoderm, anterior heart field and head mesenchyme, but is absent in neural crest-derived mesenchyme (Chapman et al., 1996; Torres-Juan et al., 2007; Vitelli et al., 2002a).
In PA development, Tbx1 regulates fibroblast growth factor (Fgf) signaling by regulating the expression of Fgf8 and fibroblast growth factor receptor 1 (Fgfr1) (Hu et al., 2004; Park et al., 2006). Tbx1 and Fgf8 compound heterozygotes result in more severe phenotypes than Tbx1 heterozygotes, indicating that these genes interact genetically (Vitelli et al., 2002b). Sonic hedgehog (Shh) promotes Tbx1 expression in the PA region through a Fox transcription factor-dependent mechanism (Garg et al., 2001; Yamagishi et al., 2003).
Wnt proteins are highly conserved, secreted, cysteine-rich glycoproteins that bind to frizzled (Fzd) receptors. In vertebrates, 19 Wnt and 10 Fzd genes have been identified. Activation of Wnt signaling results in increased cytosolic β-catenin (Ctnnb1). Translocation of β-catenin to the nucleus allows interactions with transcription factors in the T-cell factor/lymphocyte-enhancing factor (Tcf/Lef) family and regulates the transcription of numerous genes implicated in proliferation, differentiation and other cellular processes (Clevers, 2006). Limited genetic evidence suggests that Wnt—β-catenin signaling might be involved in pharyngeal apparatus development. Inactivation of Wnt1 and Wnt3, as well as conditional deletion of β-catenin in neural crest-derived cells, results in neural crest defects, including components of the first PA and cardiac outflow tract (Brault et al., 2001). A recent study indicates that inactivation of β-catenin in anterior heart field progenitors, using conditional targeting genes that are expressed early in development (e.g. Isl1-Cre, SM22-Cre and Mef2c-Cre), causes defects in the outflow tract and right ventricle by inhibiting the proliferation of Isl1-positive anterior heart field progenitor cells (Ai et al., 2007; Cohen et al., 2007; Kwon et al., 2007; Lin et al., 2007; Qyang et al., 2007). Also, constitutive activation of β-catenin signaling in anterior heart field progenitor cells causes enhanced progenitor cell proliferation and inhibition of differentiation (Ai et al., 2007; Cohen et al., 2007; Qyang et al., 2007). The involvement of Wnt—β-catenin signaling in pharyngeal apparatus development is suggested by the expression of β-catenin in PA mesenchyme.
Here, we show that canonical Wnt—β-catenin signaling is active in PA mesenchyme, where it functions to negatively regulate expression of Tbx1 and downstream signaling pathways, including Fgf signaling [Fgf8, Fgfr1 and Pea3 (Etv4 — Mouse Genome Informatics)] and Gcm2. Mesenchymal deletion of β-catenin disrupts PA artery remodeling and neural crest cell differentiation, leading to abnormalities in the great vessels. Other consequences of loss of β-catenin include craniofacial defects, thymic hypoplasia and detachment and mislocalization of the parathyroid gland. Complementary gain-of-function studies show opposite effects on Tbx1 expression and downstream signaling but surprisingly similar DGS-like phenotypes. These findings indicate that Wnt—β-catenin signaling is a crucial upstream factor that regulates the level of Tbx1 and downstream signaling molecules that are important for PA development.
MATERIALS AND METHODS
Mice
All mouse strains, including Ctnnb1F/F, Ctnnb1F(ΔEx3)/+, Fgf8lacZ/+, Tbx1−/+, Dermo1-Cre (Twist2-Cre — Mouse Genome Informatics), Wnt1-Cre and ROSA26 reporter (R26R), have been previously described (Brault et al., 2001; Danielian et al., 1998; Grieshammer et al., 2005; Harada et al., 1999; Jerome and Papaioannou, 2001; Soriano, 1999; Sosic et al., 2003). To inactivate Ctnnb1 in PA mesenchyme, Dermo1-Cre; Ctnnb1F/+ mice were mated with Ctnnb1F/F mice to generate mice with the genotype Ctnnb1F/F; Dermo1-Cre. These mice are referred to as Ctnnb1Dermo1-Cre. Control mice were of the genotype Ctnnb1F/F or Ctnnb1F/+; Dermo1-Cre. To inactivate Ctnnb1 in neural crest cells, Wnt1-Cre; Ctnnb1F/+ mice were mated with Ctnnb1F/F mice to generate mice with the genotype Ctnnb1F/F; Wnt1-Cre. These mice are referred to as Ctnnb1Wnt1-Cre. Control mice were of the genotype Ctnnb1F/F or Ctnnb1F/+; Wnt1-Cre. To ectopically activate Ctnnb1, Ctnnb1F(ΔEx3)/+ mice were mated with Dermo1-Cre mice to generate mice with a genotype of Ctnnb1F(ΔEx3)/+; Dermo1-Cre. These mice are referred to as Ctnnb1(ΔEX3)Dermo1-Cre. Control mice were of the genotype Ctnnb1F(ΔEx3)/+ or Dermo1-Cre. For detecting expression of Cre recombinase, Dermo1-Cre mice were crossed with R26R mice. All mice were maintained on 129SV/J-C57B6/J mixed genetic background.
β-galactosidase staining
Embryos were dissected in ice-cold PBS and fixed overnight in Mirsky's Fixative (National Diagnostics). Samples were washed three times in PBT (PBS, 0.1% Tween-20) and incubated in staining solution (2 mM MgCl2, 35 mM potassium ferrocyanide, 35 mM potassium ferricyanide, 1 mg/mg X-Gal in PBT) at 37°C until color reaction was apparent. Samples were washed in PBT, fixed in 10% formalin and imaged under a dissecting microscope. Samples were then soaked in 30% sucrose overnight, embedded and frozen in OCT solution for cryosectioning. Sections (12 μm) were co-stained with Nuclear Fast Red solution, washed with PBS and tap water, dehydrated in a series of ethanol and xylene, mounted and photographed with a Zeiss Axioplan 2 microscope. All staining patterns are representative of at least three samples.
RNA isolation, cDNA synthesis and quantitative RT-PCR analysis
E9.5 and E10.5 embryos were dissected by cutting from the first PA to the start of the forearm (the PA region excluding heart and outflow tract) and total RNA from this region was isolated using the RNeasy Kit (Qiagen) following the manufacturer's instructions. cDNA was synthesized using the SuperScript II First-Strand cDNA Synthesis Kit (Invitrogen). Quantitative RT-PCR was performed on an ABI 7500 Thermocycler using TaqMan probes for Tbx1 (ABI, Mm00448948_m1) and Fgf8 (ABI, Mm00438921_m1). Gapdh (ABI, Mm99999915_g1) was used to normalize samples. Amplification and analysis were performed according to the manufacturer's instructions. Results were graphed as relative expression compared with control, where control was scaled to 1. At least three independent dissections were used for each analysis.
India ink injection
Mouse embryos at various stages were dissected in PBS and injected with India ink by intra-cardiac perfusion using custom-made glass micropipettes. Samples were fixed in 10% formalin, washed in PBS, dehydrated in a series of graded methanol and cleared using BABB solution (1 benzyl alcohol to 2 benzyl benzoate). Samples were photographed on an Olympus SZX12 stereo microscope. All staining patterns are representative of at least three samples.
Wholemount immunohistochemistry
Embryos were isolated and fixed in 4% PFA overnight at 4°C. Samples were washed and dehydrated in a series of graded methanol and stored in 100% methanol at −20°C until used. Samples were rehydrated and treated with H2O2 overnight at 4°C. Samples were washed with PBT, blocked with blocking solution (2% skim milk, 0.1% Triton X-100 in PBS) and incubated with a primary antibody overnight at 4°C. Samples were washed with PBT five times and incubated with secondary antibody conjugated with HRP overnight at 4°C. After secondary antibody incubation, samples were washed five times with PBT and developed using HRP substrate (Vector Laboratories). Samples were photographed on an Olympus SZX12 stereo microscope.
Wholemount in situ hybridization
Embryos were dissected in diethylpyrocarbonate (DEPC)-treated PBS and fixed in 4% PFA. After washing, samples were dehydrated in methanol. Samples were rehydrated and washed with hybridization solution, and incubated overnight with digoxigenin-labeled RNA probes. After washing, samples were incubated with anti-digoxigenin antibody conjugated with alkaline phosphatase (Roche) and the color reaction was performed using alkaline phosphate substrate (Roche). Samples were photographed on an Olympus SZX12 stereo microscope.
Immunofluorescent staining
Sections (8 μm) from R26RDermo1-Cre embryos were incubated with anti-β-galactosidase antibody (Abcam, #ab9361, 1:500) and anti-neurofilament antibody (Developmental Studies Hybridoma Bank, 2H3, 1:500). Alexa 555-conjugated anti-mouse secondary antibody (Invitrogen, 1:200) and Alexa 488 anti-chicken secondary antibody (Invitrogen, 1:200) were used to visualize the staining pattern. Samples were photographed on an Olympus FV500 confocal microscope.
Skeletal preparation
E16.5 embryos were fixed for 24 hours in ethanol and 24 hours in acetone. Embryos were skinned and then stained with 0.3% Alcian Blue and 0.1% Alizarin Red for 24 hours, rinsed with H2O and incubated in 1% KOH/20% glycerol solution. Samples were cleared in a series of glycerol solutions (50%, 80% and 100%) for 1 week each and then stored in 100% glycerol. Samples were photographed on an Olympus SZX12 stereo microscope.
RESULTS
Canonical Wnt—β-catenin signaling is active in pharyngeal mesenchyme
The involvement of Wnt—β-catenin signaling in PA development is not well established. To investigate whether Wnt—β-catenin signaling is involved in PA development, we examined canonical Wnt—β-catenin signaling using the TOPGAL reporter mouse line (DasGupta and Fuchs, 1999). At E9.5, TOPGAL β-galactosidase activity was first found in core mesenchyme of the PAs and in pharyngeal mesoderm (Fig. 1A,A′). This finding was in agreement with other canonical Wnt—β-catenin reporters, such as BATGAL and Tcf/Lef-lacZ transgenic lines published by other groups (Cohen et al., 2007; Lin et al., 2007). Canonical Wnt—β-catenin signaling was also known to be activated in the cardiac outflow tract as early as E8.5 with continued activity through E10.5 (Cohen et al., 2007; Lin et al., 2007). These observations indicate that canonical Wnt—β-catenin signaling is continuously activated in PA mesenchyme during PA development.
Fig. 1.

Dermo1-Cre-mediated recombination during PA development. (A) TOPGAL β-galactosidase expression was observed in the core mesenchyme of pharyngeal arch (PA)1 and PA2 and in pharyngeal mesenchyme at E9.5. β-galactosidase expression was also detected in the endocardial cushion in cells undergoing epithelial (endocardial)-mesenchymal transition. (A′) Magnified view of A showing TOPGAL expression in PA core mesenchyme (arrows). (B) E9.5 R26RDermo1-Cre embryo showing trace β-galactosidase activity in pharyngeal mesenchyme (arrow). R26RDermo1-Cre was also active in the endocardial cushion (arrowhead). However, Dermo1-Cre was not active in neural crest-derived mesenchyme in the PAs at this stage. (C) E10.5 R26RDermo1-Cre embryos showing β-galactosidase activity in all PA mesenchymal cells including neural crest-derived mesenchyme. No Cre activity was found in the myocardium. (D-G) Co-immunohistochemistry showing β-galactosidase (green) and β-catenin expression (red) in PA mesenchyme at E10.5. (D,E) R26RDermo1-Cre embryos showing that β-galactosidase expression is restricted to PA mesenchyme and does not overlap with DAPI-stained (blue) nuclei in ectoderm and endoderm (arrow). (F) Ctnnb1+/+ embryos show staining for β-catenin in PA mesenchyme, ectoderm and endoderm. (G) In Ctnnb1Dermo1-Cre mice, β-catenin expression was decreased in PA mesenchyme. h, heart; hm, xxx? xxx; ov, otic vesicle; pa1, first pharyngeal arch; pa2, second pharyngeal arch; pm; pharyngeal mesenchyme. Scale bar: 400 μm in C; 200 μm in A′,B,D-G.
Inactivation of mesenchymal β-catenin results in DGS-like phenotypes
To investigate the function of canonical Wnt—β-catenin signaling in PA mesenchyme, we examined conditional knockout mice in which a floxed allele of Ctnnb1 was inactivated with Dermo1-Cre. The Dermo1-Cre allele expresses Cre recombinase in most mesenchymal regions. To examine Dermo1-Cre activity in pharyngeal mesenchyme, we crossed Dermo1-Cre mice with ROSA26 reporter (R26R) mice (Soriano, 1999) to generate Dermo1-Cre; R26R embryos (referred to as R26RDermo1-Cre). In R26RDermo1-Cre embryos, Cre recombinase activity using R26R-β-galactosidase staining was detected as early as E9.5 in PA core mesenchyme, head mesenchyme and pharyngeal mesenchyme (Fig. 1B). However, at this stage, Cre recombinase activity was absent from mesenchyme that originated from neural crest-derived cells. At E10.5, β-galactosidase activity was detected throughout pharyngeal mesenchyme regions, including mesenchyme that originated from neural crest (Fig. 1C-E). Dermo1-Cre was then used to inactivate a floxed allele of β-catenin by generating Dermo1-Cre; Ctnnb1F/F embryos (referred to as Ctnnb1Dermo1-Cre). Immunohistochemistry was performed to determine the extent to which Wnt—β-catenin was deleted in tissue expressing Cre recombinase. At E10.5, expression of β-catenin was markedly reduced in PA mesenchyme of Ctnnb1Dermo1-Cre embryos, although its expression was retained in pharyngeal arch epithelia (Fig. 1F,G).
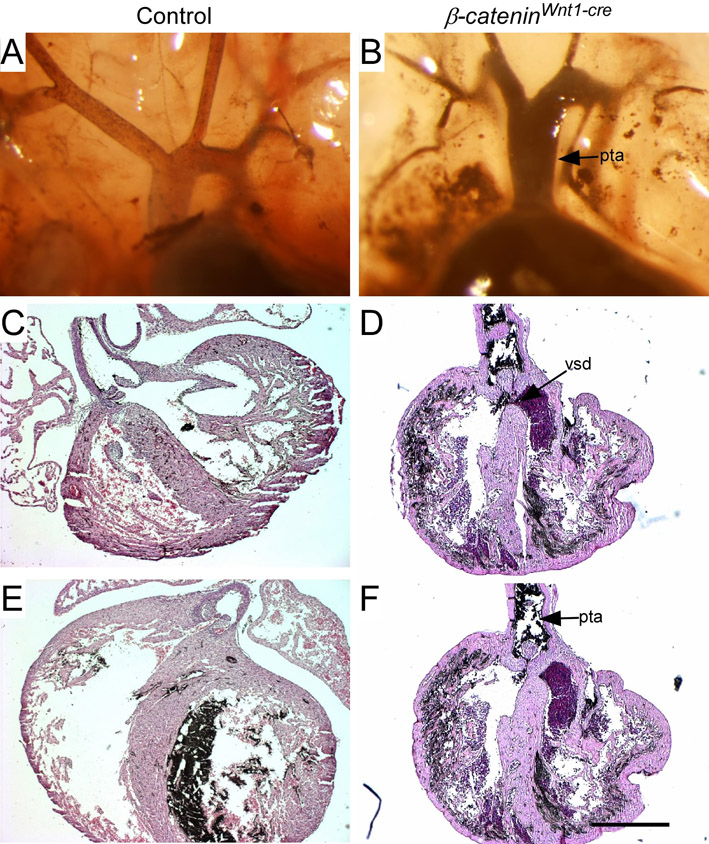
Beginning at E12.5, edema in the head and back, and hemorrhage around PA structures, was evident in Ctnnb1Dermo1-Cre embryos (see Fig. S1A,B in the supplementary material). This phenotype persisted through E17.5 (see Fig. S1C-F in the supplementary material; data not shown). Control embryos did not show any edema or hemorrhage at these stages of development. Local edema and hemorrhage indicated that Ctnnb1Dermo1-Cre embryos developed cardiovascular problems. Therefore, we analyzed the cardiovascular system of Ctnnb1Dermo1-Cre embryos and compared them with control embryos (Table 1). To examine great vessel morphology, India ink was injected intracardially and visualized at E16.5. In control embryos, the innominate and left common carotid artery normally branch off the aortic arch in close proximity (Fig. 2A; see Fig. S2A in the supplementary material). In Ctnnb1Dermo1-Cre embryos, the distance widens (arrow in Fig. 2B; see also Fig. S2B,C in the supplementary material) and the right subclavian artery originates aberrantly from the descending aorta (arrowhead in Fig. 2B; see also Fig. S2B,C in the supplementary material). Aortic arch hypoplasia was also evident in Ctnnb1Dermo1-Cre embryos (see Fig. S2B, dashed arrow, in the supplementary material). The pulmonary artery branches were hypoplastic in Ctnnb1Dermo1-Cre embryos compared with control embryos (Fig. 2C,D; see also Fig. S2C in the supplementary material). The heart of control embryos showed normal septation of the ventricle and the atrium and alignment of the aortic arch and pulmonary trunk (Fig. 2E,G). However, Ctnnb1Dermo1-Cre embryos contained ventricular and atrial septal defects with 100% penetrance (Fig. 2F; Table 1). Ctnnb1Dermo1-Cre embryos also showed other cardiac defects, including overriding aorta (53%), double-outlet right ventricle (27%) and persistent truncus arteriosus (6.7%; Fig. 2E-H; Table1; data not shown). In addition to these cardiovascular defects, 100% of Ctnnb1Dermo1-Cre embryos showed a small mandible (micrognathia; arrowhead in Fig. 2I,J,O,P; Table 1), low-set small external ears (arrow in Fig. 2I,J; Table 1), cleft palate (Table 1; data not shown), a detached and hypoplastic thymus (Fig. 2K,L), mislocalized parathyroid glands (next to the thymus; Fig. 2M,N), poorly calcified craniofacial bones and a missing tympanic ring (arrow in Fig. 2O,P). This repertoire of phenotypes (Table 1) is within the spectrum of those of DGS.
Table1.
DiGeorge syndrome-like phenotypes in Ctnnb1Dermo1-Cre embryos

Fig. 2.

Mesenchymal deletion of β-catenin causes DGS-like phenotypes. (A-D) Vascular structure of the aortic arch branches in E16.5 embryos visualized with India ink injection into the left ventricle. (A,C) Control embryo showing a normal branching pattern. (B,D) Ctnnb1Dermo1-Cre mutants showing increased separation between the innominate and left common carotid artery (arrow), abnormal origin of the left subclavian artery (arrowhead), and pulmonary artery hypoplasia. (E-H) Hematoxylin and Eosin (H&E)-stained sections of E16.5 control (E,G) and Ctnnb1Dermo1-Cre (F,H) embryos. Ctnnb1Dermo1-Cre embryos showed ventricular and atrial septal defects (F) and overriding aorta (H) anomalies. (I-L) Micrognathia (small mandible, arrowhead) and small outer ear (arrow) in E16.5 Ctnnb1Dermo1-Cre (J) compared with control (I) embryos. (K,L) Detached and decreased thymus size in Ctnnb1Dermo1-Cre embryos. (M,N) H&E-stained section of the neck region showing normal location of parathyroid gland (pth) next to the thyroid gland (thy) in control (M). Ctnnb1Dermo1-Cre embryos (N) showing the parathyroid gland located next to the thymus (th). (O,P) Skeleton preparation stained with Alizarine Red and Alcian Blue showing decreased calcification of cranial bones, missing tympanic ring (arrow) and shortened mandible (arrowhead) in Ctnnb1Dermo1-Cre (P) compared with control (O) embryos. aa, aortic arch; asd, atrial septal defect; e, ear; ia, innominate artery; lca, left common carotid artery; lpa, left pulmonary artery; lsa, left subclavian artery; m, mandible; oa, overriding aorta; pt, pulmonary trunk; pth, parathyroid gland; rca, right common carotid artery; rpa, right pulmonary artery; rsa, right subclavian artery; th, thymus; thy, thyroid gland; tr, tympanic ring; vsd, ventricular septal defect. Scale bars: 200 μm in E-H; 50 μm in M,N.
Mesenchymal Ctnnb1 is necessary for PA artery development
Many defects in the great vessels originate from defects in early PA artery formation and/or remodeling (Graham, 2003). To examine the origin of the great vessel anomalies in Ctnnb1Dermo1-Cre embryos, India ink injection was used to visualize the PA arteries in E10.5-E13.5 embryos. At E10.5, there was no difference in Ctnnb1Dermo1-Cre embryos compared with controls (Fig. 3A,B). At this time, a dorsal aorta joins the PA arteries dorsally. At E11.5, in control embryos, the dorsal aorta between the third and fourth PA arteries normally became hypoplastic and completely regressed by E13.5 (arrow in Fig. 3C,E). However, in Ctnnb1Dermo1-Cre embryos, the dorsal aorta between the third and fourth PA arteries remained dilated (at E11.5) and still patent (at E13.5) (arrow in Fig. 3D,F). These data indicate that Ctnnb1Dermo1-Cre embryos have defects in PA artery remodeling.
Fig. 3.

Mesenchymal β-catenin is required for normal PA artery remodeling. (A,B) At E10.5, both control (A) and Ctnnb1Dermo1-Cre (B) embryos showed similar aortic arch artery structures. (C,D) At E11.5, there is a normal atrophy of the dorsal aorta between PA artery 3 and 4 (C, arrow). Ctnnb1Dermo1-Cre mutant embryos (D) do not show regression of the PA artery 3 and 4 channel (arrow). (E,F) By E13.5, the dorsal aorta between PA artery 3 and 4 is normally lost (E, red dashed line, arrow in inset). Ctnnb1Dermo1-Cre mutant embryos (F) retained the dorsal aorta between PA artery 3 and 4 (arrow in inset). (G-J) In Ctnnb1Wnt1-Cre mutant embryos (H,J), the PA artery remodeling occurred normally with no apparent difference from controls (G,I) at E11.5 and E12.5, respectively. Arrows indicate the dorsal aorta spanning PA artery 3 to 4. 3, third pharyngeal arch; 4, fourth pharyngeal arch; 6, sixth pharyngeal arch; ia, innominate artery; lca, left common carotid artery; lva, left vertebral artery; rca, right common carotid artery.
Deletion of Ctnnb1 in mesenchyme causes cell non-autonomous defects in neural crest differentiation
Neural crest cells contribute significantly to craniofacial and cardiovascular development by migrating through and populating the PAs (Jiang et al., 2000). Deletion of several important molecules, such as Tgfβ type II receptor and smoothened, in the neural crest cell lineage, causes DGS-like phenotypes (Goddeeris et al., 2007; Wurdak et al., 2005). To examine neural crest-derived cell migration and development, control and Ctnnb1Dermo1-Cre embryos were stained for neurofilament to identify components of the peripheral nervous system (PNS). Compared with controls, Ctnnb1Dermo1-Cre embryos showed hypoplasia of nerve fibers in dorsal root ganglia (arrowheads in Fig. 4A,B). The roots of the vagus (X) and hypoglossal (XII) nerves were hypoplastic (arrow in Fig. 4B) and the glossopharyngeal (IX) nerve was missing entirely (dashed arrow in Fig. 4B). Failure to form a normal PNS indicated that Ctnnb1Dermo1-Cre embryos have neural crest cell defects. These defects could be due to intrinsic defects in neuronal cells or to cell-autonomous or cell-non-autonomous defects after neural crest cell migration. To determine whether early neural crest cells migrated normally, Ap2a in situ hybridization was used to identify all neural crest-derived cells at E10.5 (Mitchell et al., 1991). Despite abnormal development of the neural crest-derived PNS, there was no difference in Ap2a staining at E10.5, suggesting that neural crest cell migration was intact (Fig. 4C,D). Importantly, Dermo1-Cre recombinase activity in neural crest-derived mesenchyme occurs at relatively late stages of development (starting at E10.5) (Fig. 1C). To determine whether Dermo1-Cre activity was present in neuronal cells derived from neural crest cells, immunostaining for β-galactosidase and neurofilament was performed at E11.5 on R26RDermo1-Cre embryos. Neurofilament-positive neuronal cells were negative for β-galactosidase, indicating that the neuronal component of neural crest cells does not activate Dermo1-Cre expression (Fig. 4E-G). This is consistent with Dermo1-Cre activation in PA mesenchyme and neural crest-derived mesenchyme after neural crest cell migration. To determine whether Ctnnb1 functions earlier in neural crest cell development, Wnt1-Cre was used to inactivate Ctnnb1 in neural crest cells beginning at E8.5 of development. Consistent with previous reports, mice with the genotype Ctnnb1F/F; Wnt1-Cre (referred to as Ctnnb1Wnt1-Cre) showed neural crest differentiation defects (Brault et al., 2001) that were similar to those seen in Ctnnb1Dermo1-Cre embryos (Fig. 4A,B,H,I).
Fig. 4.

Neural crest cell defects in Ctnnb1Dermo1-Cre embryos. (A,B) Wholemount immunostaining for neurofilament showing defective peripheral neuronal development in Ctnnb1Dermo1-Cre embryos (B) compared with control (A) at E10.5. Arrowheads, dorsal root ganglia; black arrow, root of the vagus and hypoglossal nerve; white arrow, glossopharyngeal nerve. (C,D) Wholemount ISH for Ap2a showing no difference between control (C) and Ctnnb1Dermo1-Cre embryos (D) at E10.5. Arrows indicate neural crest cell migratory paths. (E-G) Immunofluorescence staining of an E11.5 embryo for neurofilament (E, red) and β-galactosidase (F, green) showing no overlap (G) between β-galactosidase-positive cells and neurofilament-positive cells in dorsal root ganglia. (H,I) Wholemount immunostaining for neurofilament showing defective peripheral neuron development in Ctnnb1Wnt1-Cre (I), compared with control (H), embryos. Arrowheads, dorsal root ganglia; arrow, root of the vagus and hypoglossal nerve. V, trigeminal nerve; VII and VIII, acousticofacial nerve; IX, glossopharyngeal nerve; X, vagus nerve; XII, hypoglossal nerve; h, heart; ov, otic vesicle; pa1, first pharyngeal arch; pa2, second pharyngeal arch. Scale bar: 100 μm in E-G.
As previously observed, Ctnnb1Wnt1-Cre embryos showed persistent truncus arteriosus accompanied by ventricular septal defects (see Fig. S3 in the supplementary material) (Kioussi et al., 2002). To determine whether Wnt—β-catenin signaling within the neural crest is required for normal PA artery development and remodeling, India ink was injected to visualize the dorsal aorta. This analysis indicated that Ctnnb1Wnt1-Cre embryos had normal PA artery formation and remodeling compared with controls at E11.5 and E12.5 (Fig. 3G-J). These results suggest that the PA artery and neuronal phenotypes result from defects in surrounding tissue and that loss of Ctnnb1 in neural crest-derived cells does not directly affect PA artery and neuronal development in Ctnnb1Dermo1-Cre embryos.
Cell death during PA artery remodeling is decreased in Ctnnb1Dermo1-Cre embryos
In PA artery remodeling, apoptotic cells are observed in mesenchyme surrounding regressed PA arteries (Yashiro et al., 2007). To identify if this is also the case in the dorsal aorta, E12 embryos were stained using the TUNEL assay. Apoptotic cell death was evident in surrounding mesenchyme proximal to the dorsal aorta between the third and fourth PA arteries in control embryos (see Fig. S4A in the supplementary material). However, no apoptotic cells were detected in Ctnnb1Dermo1-Cre embryos at this stage (see Fig. S4B in the supplementary material).
Isl1-positive anterior heart field progenitor cells are not involved in DGS-like phenotypes caused by mesenchymal deletion of β-catenin
Recent studies indicate that Wnt—β-catenin signaling plays an important role in anterior heart field formation and proliferation by modulating Isl1-positive cells and/or directly regulating Isl1 expression (Ai et al., 2007; Cohen et al., 2007; Kwon et al., 2007; Lin et al., 2007; Qyang et al., 2007). Isl1 in situ hybridization and real-time qRT-PCR showed that there was no change in expression at E10.5 (Fig. 5A,B; data not shown) and that the length of the outflow tract and right ventricle was not changed (data not shown). These data suggest that Isl1-positive cells are intact and that there is no anterior heart field defect in the Ctnnb1Dermo1-Cre line.
Fig. 5.

Upregulation of the Tbx1 signaling pathway in Ctnnb1Dermo1-Cre embryos. (A,B) Wholemount ISH of lsl1 at E9.5 showing no difference in Isl1 expression between control and Ctnnb1Dermo1-Cre embryos. (C,D) Wholemount ISH of Tbx1 at E9.5 showing increased expression in the pharyngeal arches (arrows) in Ctnnb1Dermo1-Cre (D) compared with control (C) embryos. (E,F) Quantitative RT-PCR analysis showing increased Tbx1 expression in Ctnnb1Dermo1-Cre embryos (white bar) compared with control (black bar) at E9.5 (E) and E10.5 (F). (G,H) Wholemount ISH of Gcm2 (arrows) at E10.5 showing increased expression in Ctnnb1Dermo1-Cre (H) compared with control (G) embryos. (I,J) Wholemount ISH of Fgf8 at E9.5 showing increased expression in pharyngeal arches (arrow) in Ctnnb1Dermo1-Cre (J) compared with control (I) embryos. (K,L) Wholemount ISH of Fgfr1 at E10.5 showing increased expression in pharyngeal arches (arrows) in Ctnnb1Dermo1-Cre (L) compared with control (K) embryos. (M,N) Wholemount ISH of Pea3 at E9.5 showing increased expression in pharyngeal arches (arrows) in Ctnnb1Dermo1-Cre (N) compared with control (M) embryos. *, P<0.04 in E; *, P<0.005 in F.
Impaired Tbx1 signaling in Ctnnb1Dermo1-Cre embryos
Tbx1 has been identified as the most probable disease-causing gene for DGS (Baldini, 2002; Baldini, 2003). Tbx1 is expressed in PA endoderm and mesoderm and gain-of-function, as well as loss-of-function, mouse mutants of Tbx1 recapitulate defects associated with DGS. To test whether Tbx1 is regulated by Wnt—β-catenin signaling, Tbx1 expression was examined in Ctnnb1Dermo1-Cre embryos. At E9.5, Ctnnb1Dermo1-Cre embryos showed increased Tbx1 expression in PA mesenchyme compared with controls (Fig. 5C,D). Consistent with the observed increase of Tbx1 expression, quantitative RT-PCR analysis of PA-region tissue showed a significant (P<0.04 at E9.5 and P<0.005 at E10.5) increase in Tbx1 mRNA in Ctnnb1Dermo1-Cre embryos at E9.5 and E10.5 (Fig. 5E,F). Furthermore, the expression of Gcm2, a Tbx1 target gene and early marker for the parathyroid gland, was also increased in Ctnnb1Dermo1-Cre embryos compared with controls (Fig. 5G,H). Recent studies identified Fgf8 as a genetic modifier of Tbx1 in PA development (Hu et al., 2004). Hypomorphic alleles or conditional knockout of the Fgf8 gene recapitulates DGS phenotypes, similar to what was observed in Tbx1 hypomorph or knockout mice. Consistent with an established link between Tbx1 and Fgf8, at E9.5, Fgf8 expression was increased in PAs in Ctnnb1Dermo1-Cre embryos (Fig. 5I,J). Additionally, the expression of Fgfr1, one of the receptors for Fgf8, was increased in the PAs in Ctnnb1Dermo1-Cre embryos (Fig. 5K,L), consistent with previous data (Park et al., 2006). Also consistent with increased Fgf8 signaling, Pea3, a target of Fgf8/Fgfr signaling, was increased in pharyngeal mesenchyme in Ctnnb1Dermo1-Cre embryos (Fig. 5M,N).
Activation of Wnt—β-catenin signaling in mesenchyme suppresses Tbx1 expression and signaling
The results shown above indicate that Wnt—β-catenin signaling is necessary to inhibit Tbx1 and downstream signaling molecules. To determine if Wnt—β-catenin signaling is sufficient to inhibit Tbx1 expression in vivo, a Cre-activatable constitutively active allele of Ctnnb1 (Ctnnb1F(ΔEx3)/+) was mated with Dermo1-Cre mice to generate embryos with the genotype Ctnnb1F(ΔEx3)/+; Dermo1-Cre [referred to as Ctnnb1(ΔEX3)Dermo1-Cre]. At E10.5, embryos expressing Ctnnb1(ΔEX3)Dermo1-Cre showed decreased expression of Tbx1 (Fig. 6A,B). Consistent with the observed decreased Tbx1 expression, qRT-PCR analysis of PA-region tissue showed a significant (40%-50%, P<0.002) decrease in Tbx1 mRNA in Ctnnb1(ΔEX3)Dermo1-Cre embryos at E9.5 and E10.5 (Fig. 6C,D). The expression of Gcm2 was also decreased in Ctnnb1(ΔEX3)Dermo1-Cre embryos (Fig. 6E,F). Interestingly, at E9.5 and E10.5, we observed no changes in the expression of Fgf8 using in situ hybridization or qRT-PCR (data not shown). However, Fgf8 is normally expressed in the PA region at earlier stages of development and, by E9.5, Fgf8 expression normally declines to nearly undetectable levels (Ilagan et al., 2006). To examine the effects of increased Wnt—β-catenin signaling at earlier stages (before Dermo1-Cre becomes active), we injected pregnant female mice with LiCl to inhibit Gsk3β and to activate β-catenin in the embryos. To monitor Fgf8 expression, we used an allele of Fgf8 (Fgf8lacZ/+) in which the β-galactosidase gene replaced the Fgf8 coding sequence. The β-galactosidase reporter has the advantage of a relatively long half-life, allowing a cumulative assessment of changes in gene expression (Ilagan et al., 2006). Pregnant mice carrying Fgf8lacZ/+ embryos were injected intraperitoneally with 50 μl of 1 M NaCl or 1 M LiCl at E8.5. Embryos stained for β-galactosidase activity at E9.5 showed decreased Fgf8 expression in pharyngeal mesenchyme in LiCl-treated Fgf8lacZ/+ embryos compared with NaCl-treated embryos (Fig. 6G-H′).
Fig. 6.
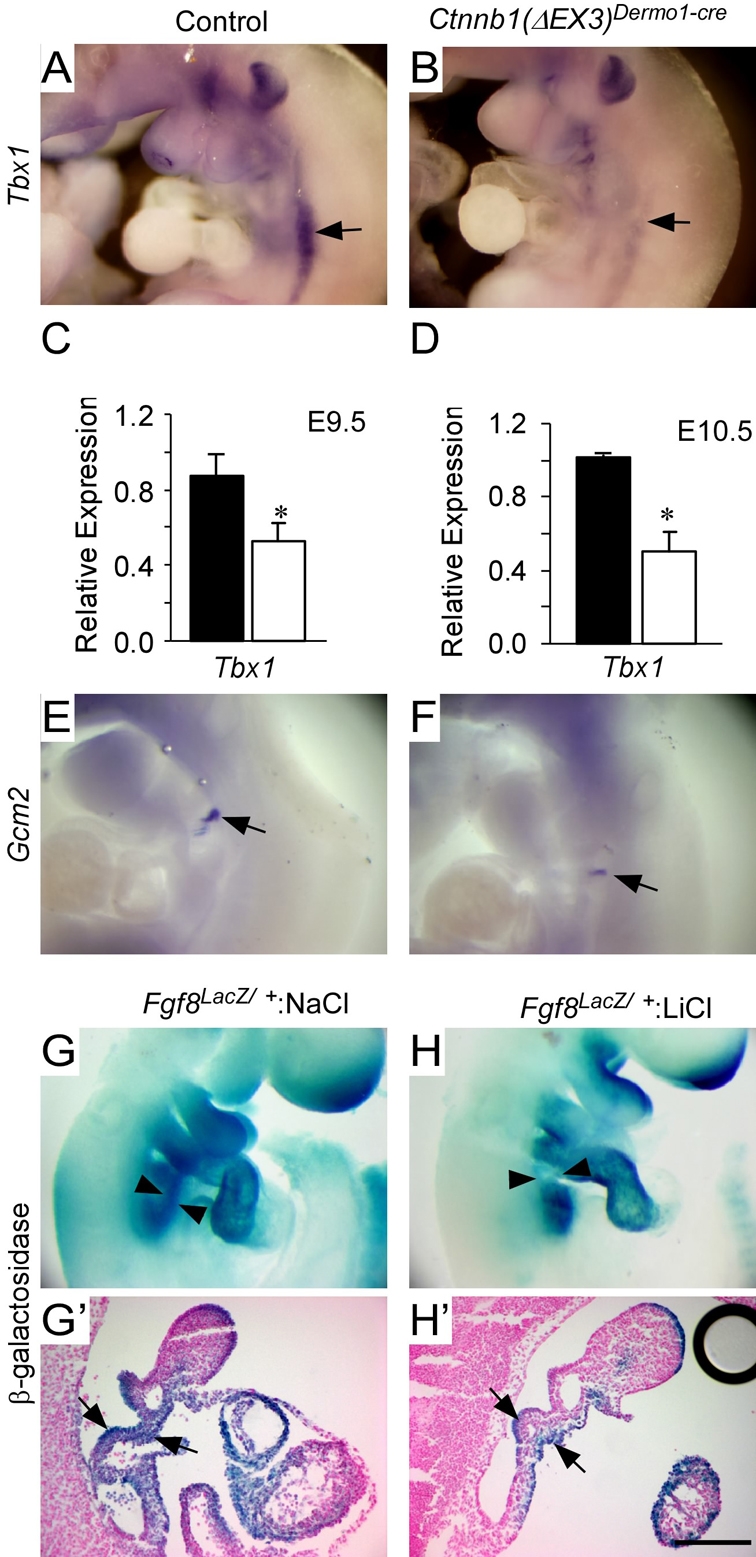
Decreased expression of the Tbx1 signaling pathway following activation of Wnt—β-catenin signaling. (A,B) Wholemount ISH of Tbx1 at E10.5 showing decreased expression in PAs (arrows) in Ctnnb1(ΔEX3)Dermo1-Cre (B) compared with control (A) embryos. (C,D) Quantitative RT-PCR analysis showing decreased Tbx1 expression in Ctnnb1(ΔEX3)Dermo1-Cre embryos (white bar) compared with control (black bar) at E9.5 (C) and E10.5 (D). (E,F) Wholemount ISH of Gcm2 at E10.5 showing decreased expression in PAs (arrows) in Ctnnb1(ΔEX3)Dermo1-Cre (F) compared with control (E) embryos. (G,H) Wholemount β-galactosidase staining of Fgf8lacZ/+ at E9.5 showing decreased expression in PAs (between arrowheads) in LiCl-treated Fgf8lacZ/+ embryos (H) compared with NaCl-treated Fgf8lacZ/+ embryos (G). (G′-H′) Sections of embryos from G and H showing decreased β-galactosidase activity in pharyngeal mesenchyme (between arrows) in LiCl-treated Fgf8lacZ/+ embryos (H′) compared with NaCl treated Fgf8lacZ/+ embryos (G′). *, P<0.02 in C; *, P<0.01 in D. Scale bar: 200 μm in G′,H′.
Enhancement of DGS pathology with activation of Wnt—β-catenin signaling
Because constitutive activation of Wnt—β-catenin signaling in mesenchyme decreased Tbx1 and downstream signaling cascades in PA mesenchyme, we hypothesized that constitutive activation of Wnt—β-catenin signaling might also enhance DGS phenotypes. Because Ctnnb1(ΔEX3)Dermo1-Cre embryos die between E11.5 and E12.5 (data not shown), we could not assess the consequences of activation of Wnt—β-catenin signaling at later developmental stages using this genetic system. However, one of the early DGS-like phenotypes is hypoplasia of the fourth PA artery. This phenotype occurs in embryos hypomorphic for both Tbx1 and Fgf8 (Lindsay et al., 2001; Vitelli et al., 2002b). To test whether increased Wnt—β-catenin signaling could enhance PA artery phenotypes on a sensitized genetic background (Fgf8lacZ/+ or Tbx1−/+), pregnant mice carrying Fgf8LacZ/+ and Tbx1−/+ embryos were treated with NaCl or LiCl from E8.5 to E9.5 and examined at E10.5 by dye injection into the heart. No abnormalities were seen in control embryos treated with NaCl or LiCl (Fig. 7A-B′; Table 2). NaCl-treated Fgf8lacZ/+ embryos also formed normal PA arteries, whereas 7 out of 22 (32%) LiCl-treated Fgf8LacZ/+ embryos showed hypoplastic fourth PA arteries (Fig. 7C-D′; Table 2). In the Tbx1−/+ genetic background, the incidence of fourth PA artery defects also became more severe in LiCl-treated embryos compared with NaCl-treated embryos, increasing from 78% in NaCl-treated to 100% in LiCl-treated Tbx1−/+ embryos (Fig. 7E-F′; Table 2). Increased severity of the phenotype was also evident as bilateral cases of fourth PA defects increased from 52% in NaCl-treated to 96% in LiCl-treated Tbx1−/+ embryos (Table 2). Additionally, the incidence of unilateral and bilateral aplasia of the fourth PA artery was also increased when Tbx1−/+ embryos were exposed to LiCl (Table 2). These data indicated that administration of LiCl enhances the severity of DGS phenotypes in Fgf8lacZ/+ and Tbx1−/+ genetic backgrounds, supporting the hypothesis that Wnt—β-catenin signaling is important for maintaining the proper level of Tbx1 signaling.
Fig. 7.
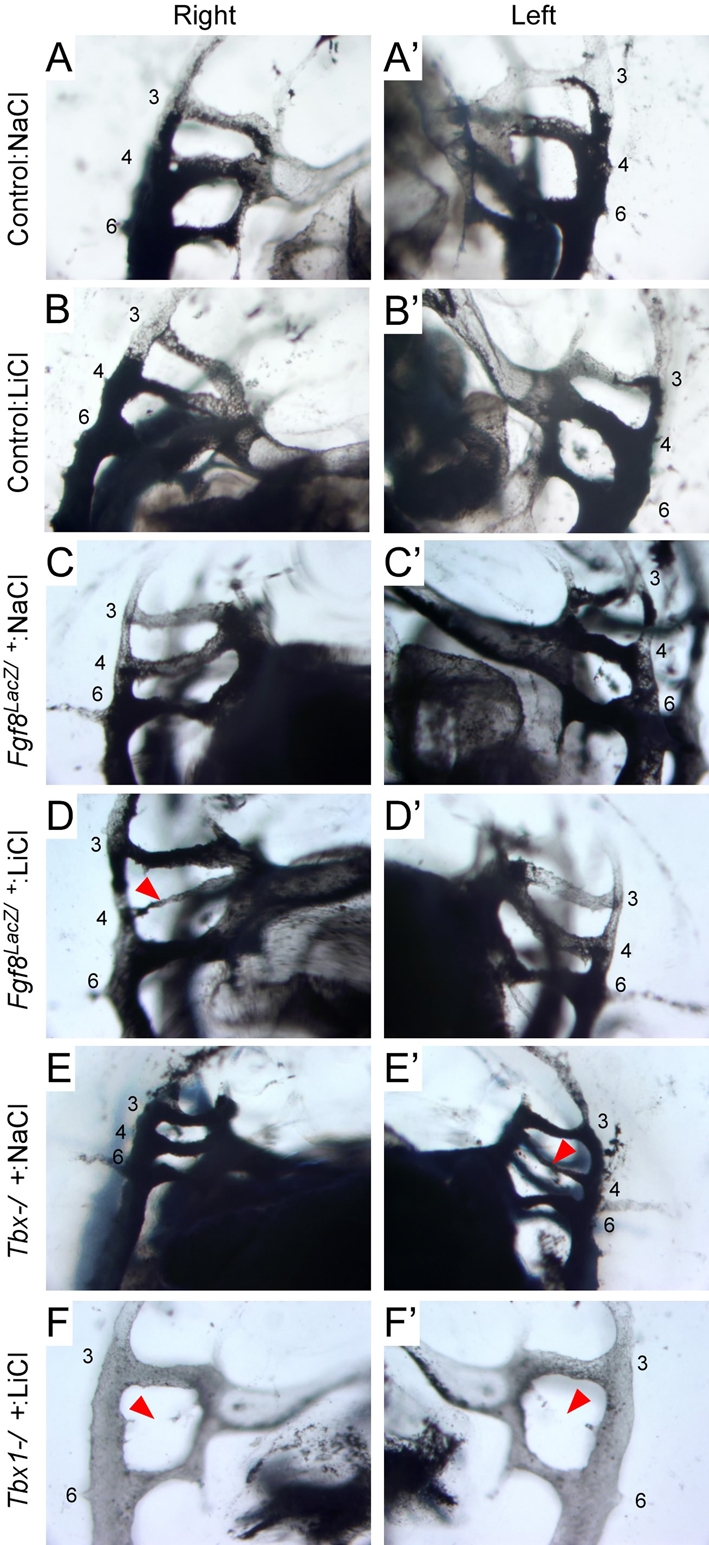
Activation of Wnt—β-catenin signaling enhances PA artery developmental anomalies. Pregnant mice were treated with NaCl or LiCl from E8.5 to E9.5. At E10.5, India ink was injected into the left ventricle to visualize PA artery morphology. (A-B′) Wild-type embryos treated with NaCl (A,A′) or LiCl (B,B′) show normal PA artery formation. (C-D′) Fgf8lacZ/+ embryos treated with LiCl (D,D′) showing unilateral fourth PA artery hypoplasia (arrowhead) compared with normal PA artery formation in NaCl-treated Fgf8lacZ/+ embryos (C,C′). (E-F′) Tbx1−/+ embryos treated with NaCl show unilateral fourth PA artery hypoplasia (arrowhead, E,E′). Tbx1−/+ embryos treated with LiCl show bilateral fourth PA artery aplasia (F,F′). Arrowhead indicates location of the fourth PA arteries. 3, third pharyngeal arch; 4, fourth pharyngeal arch; 6, sixth pharyngeal arch.
Table 2.
Incidence of fourth PA artery defects in Fgf8lacZ/+ and Tbx1−/+ embryos treated with NaCl or LiCl

Rescue of PA artery defects in Ctnnb1Dermo1-Cre; Fgf8lacZ/+ compound embryos
If gene dosage of Tbx1 and/or Fgf8 is crucial for DGS-like phenotypes, we hypothesize that in Ctnnb1Dermo1-Cre embryos, where both Tbx1 and Fgf8 expression is increased, reducing the gene dosage of Tbx1 or Fgf8 should reduce the severity of the DGS-like phenotype. To test this hypothesis, Ctnnb1Dermo1-Cre; Fgf8lacZ/+ compound embryos were injected with India ink at E12.5 and PA arteries were visualized (Fig. 8). Comparing Ctnnb1Dermo1-Cre, which shows a failure of regression of the channel between the third and fourth PA arteries (100% penetrance), Ctnnb1Dermo1-Cre;Fgf8lacZ/+ embryos showed normalization of regression of this aortic arch channel (8 of 10 channels regressed). Pea3 expression was also restored to normal in Ctnnb1Dermo1-Cre; Fgf8lacZ/+ embryos (see Fig. S5 in the supplementary material). Thus, reducing the gene dosage of Fgf8 can rescue the PA artery defect in Ctnnb1Dermo1-Cre embryos, and the Tbx1-Fgf8 signaling axis is at least in part responsible for the PA artery defects seen in Ctnnb1Dermo1-Cre embryos.
Fig. 8.

Reduced gene dosage of Fgf8 rescues PA artery defects in Ctnnb1Dermo1-Cre embryos. Embryos were injected with India ink at E12.5. (A) Control embryo showing normal regression of the dorsal aorta between the third and fourth PA arteries (arrow). (B) Ctnnb1Dermo1-Cre embryo showing a sustained dorsal aorta between the third and fourth PA arteries (arrow). (C) Ctnnb1Dermo1-Cre; Fgf8lacZ/+ compound embryo showing restored regression of the dorsal aorta between the third and fourth PA arteries (arrow). Boxed regions are magnified 2× and diagramed below each panel.
DISCUSSION
Recent studies have identified essential roles for Wnt—β-catenin signaling in mesenchymal tissues such as bone and lung (Day et al., 2005; De Langhe et al., 2008; Hu et al., 2005; Yin et al., 2008). To examine developmental requirements for Wnt—β-catenin signaling in pharyngeal apparatus development, we have used the Dermo1-Cre allele to inactivate Ctnnb1 in the mesenchymal components of the PAs and surrounding mesenchyme. Phenotypes associated with mesenchymal loss of Ctnnb1 are within the spectrum of phenotypes seen in DGS. Because mutation or changes in gene copy number of the Tbx1 transcription factor are closely associated with the etiology of DGS, and mesenchymal Tbx1 is necessary and sufficient for pharyngeal arch development (Zhang et al., 2006), phenotypic similarities resulting from loss of mesenchymal Wnt—β-catenin signaling suggested possible genetic interactions between mesenchymal Wnt—β-catenin signaling and Tbx1. Here, we showed that loss of Wnt—β-catenin signaling in pharyngeal mesenchyme causes DGS-like phenotypes through a mechanism in which Wnt—β-catenin inhibits Tbx1 expression. Most cases of DGS are caused by deletion of chromosome 22q11.2, which includes the TBX1 gene. However, overexpression of human TBX1 in mice, or mutations that stabilize TBX1 in some DGS patients, also causes the same spectrum of DGS phenotypes (Torres-Juan et al., 2007; Zweier et al., 2007). This indicates that the level of expression of Tbx1 is crucial for PA formation. Changes in the level of TBX1 expression (either increase or decrease) might be responsible for PA malformations that are consistent with a diagnosis of DGS. Understanding the mechanisms that regulate the level of TBX1 could be important for understanding the severity of DGS phenotypes.
Shh signaling has been shown to activate Tbx1 expression in PA endoderm and mesoderm through regulation of the Foxa and Foxc transcription factors (Garg et al., 2001; Yamagishi et al., 2003). Recent data also indicate that retinoic acid signaling might function to inhibit Tbx1 expression in paraxial mesoderm or tongue muscle (Abe et al., 2008; Okano et al., 2008). Also, deletion of one copy of the Raldh2 gene in Crkl−/+; Tbx1−/+ compound heterozygous mice reduced thymic hypoplasia, consistent with negative regulation of Tbx1 or Crkl in thymic development (Guris et al., 2006). The observation that Wnt—β-catenin signaling negatively regulates Tbx1 expression suggests that the Tbx1 gene functions as an integrative node for multiple signaling pathways that are active in PA development.
PA mesenchyme comprises neural crest-derived mesenchyme and mesoderm-derived mesenchyme. Dermo1-Cre is first active in PA core mesenchyme and later becomes active in the remaining neural crest-derived mesenchyme. Defects in neural crest-derived tissues identified in Ctnnb1Dermo1-Cre embryos could result directly from loss of Ctnnb1 in neural crest-derived mesenchyme or indirectly from loss of Ctnnb1 in pharyngeal mesoderm. Comparison of phenotypes from Ctnnb1Dermo1-Cre and Ctnnb1Wnt1-Cre embryos showed normal PA artery remodeling in Ctnnb1Wnt1-Cre compared with Ctnnb1Dermo1-Cre embryos, suggesting that Wnt—β-catenin signaling in pharyngeal mesoderm-derived mesenchymal cells is important to the expression of DGS-like phenotypes. Also, deletion of Tbx1 causes neural crest defects even though Tbx1 is not expressed in neural crest cells (Vitelli et al., 2002a). These observations, in addition to defects in PNS development in Ctnnb1Dermo1-Cre embryos, demonstrate that mesenchymal cells function cell-non-autonomously through Tbx1 to regulate neural crest development.
Wnt—β-catenin signaling has been shown to promote Isl1-positive anterior heart field progenitor cell proliferation and inhibit their differentiation by directly regulating Isl1 transcription factor expression (Tzahor, 2007) Anterior heart field progenitor cells comprise the outflow tract and right ventricle in the heart (Laugwitz et al., 2008) As TOPGAL and BATGAL reporter expression indicated, Wnt—β-catenin signaling is first active in the pharyngeal mesenchymal region at E8.5 and remains active at later stages (Fig. 1) (Cohen et al., 2007; Lin et al., 2007). This suggests that Wnt—β-catenin signaling might be important to maintain pharyngeal mesenchyme, including mesenchyme in the anterior heart field. Reporter gene analysis to identify the expression of Dermo1-Cre indicates that Cre recombinase was first activated at E9.5 in the pharyngeal mesenchyme, including the anterior heart field. Therefore, the defects that result from inactivation of Ctnnb1 using the Dermo1-Cre driver might be due to disruption of anterior heart field progenitor cells. However, as Isl1-positive cells are not affected in Ctnnb1Dermo1-Cre embryos, the phenotype in Ctnnb1Dermo1-Cre embryos is probably independent of Wnt—β-catenin signaling in anterior heart field progenitor cells. We posit that Wnt—β-catenin signaling has dual functions in anterior heart field development. When the anterior heart field progenitor cells need to proliferate at early developmental stages, Wnt—β-catenin signaling promotes Isl1-positive cell proliferation and inhibits its differentiation. Later, Wnt—β-catenin signaling functions to inhibit Tbx1 expression and downstream signaling pathways to maintain proper levels of Tbx1 signaling. Sustained expression of Tbx1 causes defects in PA artery remodeling at E12.5.
The mechanism by which Wnt—β-catenin signaling regulates Tbx1 expression is not known. One possibility is that Tbx1 is regulated directly by Wnt—β-catenin signaling. However, no conserved Tcf/Lef binding sites are found in the regions flanking the Tbx1 gene (data not shown). Other possible mechanisms could include direct regulation of Tbx1 through interaction with another transcription factor or regulation of Tbx1 indirectly through modulation of other signaling pathways. However, Shh signaling was not changed in Ctnnb1Dermo1-Cre embryos (data not shown). Whether retinoic acid signaling, the only other known inhibitor of Tbx1, is involved remains to be explored.
DGS is a disease that has variable penetrance, ranging from life threatening cardiac defects to mild craniofacial defects. Because most cases of DGS are caused by haploinsufficiency of chromosome 22q11.2 and the severity of DGS is dependent on the level of TBX1 expression, mutations in other genes, which regulate TBX1 expression, are probable candidates for disease-modifying loci. Consistent with this, we identified that, in some genetic backgrounds such as Fgf8lacZ/+ and Tbx1−/+, treatment with LiCl results in an increased incidence of PA artery defects. Thus, potential teratogens that affect Wnt—β-catenin signaling could manifest as DGS in some genetic backgrounds.
Supplementary Material
Acknowledgements
We thank C. Smith for technical assistance, Dr V. E. Papaioannou for providing Tbx1−/+ mice, Dr G. M. Martin for providing Fgf8lacZ/+ mice, Dr J. Gordon for providing the Tbx1 probe, Dr N. R. Manley for providing the Gcm2 probe and Dr S. M. Evans for providing the Isl1 probe. This work was supported by NIH grant HD049808 and a gift from Edward and Linda Ornitz. Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.045534/-/DC1
References
- Abe M., Maeda T., Wakisaka S. (2008). Retinoic acid affects craniofacial patterning by changing Fgf8 expression in the pharyngeal ectoderm. Dev. Growth Differ. 50, 717-729 [DOI] [PubMed] [Google Scholar]
- Ai D., Fu X., Wang J., Lu M. F., Chen L., Baldini A., Klein W. H., Martin J. F. (2007). Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 104, 9319-9324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini A. (2002). DiGeorge syndrome: the use of model organisms to dissect complex genetics. Hum. Mol. Genet. 11, 2363-2369 [DOI] [PubMed] [Google Scholar]
- Baldini A. (2003). DiGeorge's syndrome: a gene at last. Lancet 362, 1342-1343 [DOI] [PubMed] [Google Scholar]
- Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., Sommer L., Boussadia O., Kemler R. (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253-1264 [DOI] [PubMed] [Google Scholar]
- Chapman D. L., Garvey N., Hancock S., Alexiou M., Agulnik S. I., Gibson-Brown J. J., Cebra-Thomas J., Bollag R. J., Silver L. M., Papaioannou V. E. (1996). Expression of the T-box family genes, Tbx1-Tbx5, during early mouse development. Dev. Dyn. 206, 379-390 [DOI] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469-480 [DOI] [PubMed] [Google Scholar]
- Cohen E. D., Wang Z., Lepore J. J., Lu M. M., Taketo M. M., Epstein D. J., Morrisey E. E. (2007). Wnt/beta-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 117, 1794-1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., McMahon A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323-1326 [DOI] [PubMed] [Google Scholar]
- DasGupta R., Fuchs E. (1999). Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 126, 4557-4568 [DOI] [PubMed] [Google Scholar]
- Day T. F., Guo X., Garrett-Beal L., Yang Y. (2005). Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8, 739-750 [DOI] [PubMed] [Google Scholar]
- De Langhe S. P., Carraro G., Tefft D., Li C., Xu X., Chai Y., Minoo P., Hajihosseini M. K., Drouin J., Kaartinen V., et al. (2008). Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS ONE 3, e1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V., Yamagishi C., Hu T., Kathiriya I. S., Yamagishi H., Srivastava D. (2001). Tbx1, a DiGeorge syndrome candidate gene, is regulated by sonic hedgehog during pharyngeal arch development. Dev. Biol. 235, 62-73 [DOI] [PubMed] [Google Scholar]
- Goddeeris M. M., Schwartz R., Klingensmith J., Meyers E. N. (2007). Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 134, 1593-1604 [DOI] [PubMed] [Google Scholar]
- Graham A. (2003). Development of the pharyngeal arches. Am. J. Med. Genet. 119A, 251-256 [DOI] [PubMed] [Google Scholar]
- Grieshammer U., Cebrian C., Ilagan R., Meyers E., Herzlinger D., Martin G. R. (2005). FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847-3857 [DOI] [PubMed] [Google Scholar]
- Guris D. L., Fantes J., Tara D., Druker B. J., Imamoto A. (2001). Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 27, 293-298 [DOI] [PubMed] [Google Scholar]
- Guris D. L., Duester G., Papaioannou V. E., Imamoto A. (2006). Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev. Cell 10, 81-92 [DOI] [PubMed] [Google Scholar]
- Harada N., Tamai Y., Ishikawa T., Sauer B., Takaku K., Oshima M., Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931-5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H., Hilton M. J., Tu X., Yu K., Ornitz D. M., Long F. (2005). Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development 132, 49-60 [DOI] [PubMed] [Google Scholar]
- Hu T., Yamagishi H., Maeda J., McAnally J., Yamagishi C., Srivastava D. (2004). Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development 131, 5491-5502 [DOI] [PubMed] [Google Scholar]
- Ilagan R., Abu-Issa R., Brown D., Yang Y. P., Jiao K., Schwartz R. J., Klingensmith J., Meyers E. N. (2006). Fgf8 is required for anterior heart field development. Development 133, 2435-2445 [DOI] [PubMed] [Google Scholar]
- Jerome L. A., Papaioannou V. E. (2001). DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 27, 286-291 [DOI] [PubMed] [Google Scholar]
- Jiang X., Rowitch D. H., Soriano P., McMahon A. P., Sucov H. M. (2000). Fate of the mammalian cardiac neural crest. Development 127, 1607-1616 [DOI] [PubMed] [Google Scholar]
- Kioussi C., Briata P., Baek S. H., Rose D. W., Hamblet N. S., Herman T., Ohgi K. A., Lin C., Gleiberman A., Wang J., et al. (2002). Identification of a Wnt/Dvl/beta-catenin -> Pitx2 pathway mediating cell-type-specific proliferation during development. Cell 111, 673-685 [DOI] [PubMed] [Google Scholar]
- Kwon C., Arnold J., Hsiao E. C., Taketo M. M., Conklin B. R., Srivastava D. (2007). Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc. Natl. Acad. Sci. USA 104, 10894-10899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugwitz K. L., Moretti A., Caron L., Nakano A., Chien K. R. (2008). Islet1 cardiovascular progenitors: a single source for heart lineages? Development 135, 193-205 [DOI] [PubMed] [Google Scholar]
- Liao J., Kochilas L., Nowotschin S., Arnold J. S., Aggarwal V. S., Epstein J. A., Brown M. C., Adams J., Morrow B. E. (2004). Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum. Mol. Genet. 13, 1577-1585 [DOI] [PubMed] [Google Scholar]
- Lin L., Cui L., Zhou W., Dufort D., Zhang X., Cai C. L., Bu L., Yang L., Martin J., Kemler R., et al. (2007). Beta-catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc. Natl. Acad. Sci. USA 104, 9313-9318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay E. A. (2001). Chromosomal microdeletions: dissecting del22q11 syndrome. Nat. Rev. Genet. 2, 858-868 [DOI] [PubMed] [Google Scholar]
- Lindsay E. A., Vitelli F., Su H., Morishima M., Huynh T., Pramparo T., Jurecic V., Ogunrinu G., Sutherland H. F., Scambler P. J., et al. (2001). Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410, 97-101 [DOI] [PubMed] [Google Scholar]
- Merscher S., Funke B., Epstein J. A., Heyer J., Puech A., Lu M. M., Xavier R. J., Demay M. B., Russell R. G., Factor S., et al. (2001). TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104, 619-629 [DOI] [PubMed] [Google Scholar]
- Mitchell P. J., Timmons P. M., Hebert J. M., Rigby P. W., Tjian R. (1991). Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 5, 105-119 [DOI] [PubMed] [Google Scholar]
- Okano J., Sakai Y., Shiota K. (2008). Retinoic acid down-regulates Tbx1 expression and induces abnormal differentiation of tongue muscles in fetal mice. Dev. Dyn. 237, 3059-3070 [DOI] [PubMed] [Google Scholar]
- Park E. J., Ogden L. A., Talbot A., Evans S., Cai C. L., Black B. L., Frank D. U., Moon A. M. (2006). Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 133, 2419-2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qyang Y., Martin-Puig S., Chiravuri M., Chen S., Xu H., Bu L., Jiang X., Lin L., Granger A., Moretti A., et al. (2007). The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/beta-catenin pathway. Cell Stem Cell 1, 165-179 [DOI] [PubMed] [Google Scholar]
- Scambler P. J. (2000). The 22q11 deletion syndromes. Hum. Mol. Genet. 9, 2421-2426 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- Sosic D., Richardson J. A., Yu K., Ornitz D. M., Olson E. N. (2003). Twist regulates cytokine gene expression through a negative feedback loop that represses NF-kappaB activity. Cell 112, 169-180 [DOI] [PubMed] [Google Scholar]
- Torres-Juan L., Rosell J., Morla M., Vidal-Pou C., Garcia-Algas F., de la Fuente M. A., Juan M., Tubau A., Bachiller D., Bernues M., et al. (2007). Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: a new susceptibility factor for mental retardation. Eur. J. Hum. Genet. 15, 658-663 [DOI] [PubMed] [Google Scholar]
- Tzahor E. (2007). Wnt/beta-catenin signaling and cardiogenesis: timing does matter. Dev. Cell. 13, 10-13 [DOI] [PubMed] [Google Scholar]
- Vitelli F., Morishima M., Taddei I., Lindsay E. A., Baldini A. (2002a). Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 11, 915-922 [DOI] [PubMed] [Google Scholar]
- Vitelli F., Taddei I., Morishima M., Meyers E. N., Lindsay E. A., Baldini A. (2002b). A genetic link between Tbx1 and fibroblast growth factor signaling. Development 129, 4605-4611 [DOI] [PubMed] [Google Scholar]
- Wurdak H., Ittner L. M., Lang K. S., Leveen P., Suter U., Fischer J. A., Karlsson S., Born W., Sommer L. (2005). Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 19, 530-535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak H., Ittner L. M., Sommer L. (2006). DiGeorge syndrome and pharyngeal apparatus development. BioEssays 28, 1078-1086 [DOI] [PubMed] [Google Scholar]
- Yamagishi H., Maeda J., Hu T., McAnally J., Conway S. J., Kume T., Meyers E. N., Yamagishi C., Srivastava D. (2003). Tbx1 is regulated by tissue-specific forkhead proteins through a common Sonic hedgehog-responsive enhancer. Genes Dev. 17, 269-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K., Shiratori H., Hamada H. (2007). Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 450, 285-288 [DOI] [PubMed] [Google Scholar]
- Yin Y., White A. C., Huh S. H., Hilton M. J., Kanazawa H., Long F., Ornitz D. M. (2008). An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev. Biol. 319, 426-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Huynh T., Baldini A. (2006). Mesodermal expression of Tbx1 is necessary and sufficient for pharyngeal arch and cardiac outflow tract development. Development 133, 3587-3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier C., Sticht H., Aydin-Yaylagul I., Campbell C. E., Rauch A. (2007). Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am. J. Hum. Genet. 80, 510-517 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}