

Abstract
A palladium-catalyzed carboamination reaction of γ-N-arylamino alkenes with vinyl bromides that affords N-aryl-2-allyl pyrrolidines is described. These reactions proceed with high diastereoselectivity for the formation of trans-2,3- and cis-2,5-disubstituted pyrrolidines. Conditions for a tandem N-arylation/carboamination sequence that leads to the formation of an N-aryl-2-allyl pyrrolidine or indoline via the coupling of a primary γ-amino alkene, an aryl bromide, and a vinyl bromide are also reported. The mechanism of the carboamination reactions and the origin of unexpected products that formally derive from rearrangement of the vinyl bromide are discussed.
Keywords: Heterocycles, Homogeneous Catalysis, Pyrrolidines, Palladium, Tandem Reactions, Vinyl Halides
Introduction
The pyrrolidine ring is a common motif found in a number of biologically active small molecules.[1] As a result, a great deal of effort has been devoted to the development of synthetic methods for the construction of this core.[2] Some of the most interesting and useful of these methods have utilized palladium catalysis to effect ring closure of a β- or γ-amino alkene or alkyne with concomitant carbon—carbon bond formation.[3]–[5] For example, Larock and Weinreb have described the synthesis of 2-vinyl pyrrolidines via the coupling and cyclization of β-N-tosylamino alkenes with vinyl halides.[3] Tamaru has also described a palladium-catalyzed synthesis of pyrrolidines, which employs a Wacker-type carbonylative cyclization to afford products bearing ester substituents.[4]
We have recently reported a new method for the stereoselective synthesis of N-aryl-2-benzyl pyrrolidines via the carboamination of γ-N-arylamino alkenes with aryl bromides in the presence of a palladium(0) catalyst and a base (eq 1).[6]–[9] These reactions proceed in good yields and are effective with a variety of aryl bromide coupling partners. High diastereoselectivity for the formation of trans-2,3- and cis-2,5-disubstituted pyrrolidines is observed for most substrate combinations (dr >20:1).
 |
(1) |
 |
(2) |
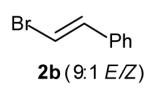
A significant extension of this methodology would involve the use of vinyl bromides as coupling partners to afford N-aryl-2-allyl pyrrolidines. However, initial attempts to couple 1 with β-bromostyrene using conditions identical to those employed in the analogous reactions of aryl bromides resulted in low yields of the desired pyrrolidine 3b due to competing N-vinylation of the substrate (eq 2). We reasoned that this problem could be circumvented by employing a palladium/phosphine catalyst system that would disfavor carbon—nitrogen bond-forming reductive elimination,[10] as the side product presumably results from relatively facile reductive elimination of an intermediate vinylpalladium amido complex.[11] Herein we report the use of a Pd2(dba)3/tri-2-furylphosphine catalyst system to effect the diastereoselective synthesis of N-aryl-2-allyl pyrrolidines via the palladium-catalyzed carboamination of γ-N-arylamino alkenes with vinyl bromides.
Results and Discussion
Catalyst optimization
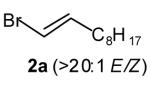
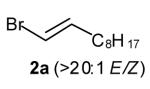
In our preliminary experiments we elected to optimize the palladium-catalyzed reaction of amine 1 with vinyl bromide 2a, which was easily prepared in isomerically pure form (eq 3, Table 1). Unfortunately, the Pd2(dba)3/dppb catalyst system, which provided high yields for the synthesis of 2-benzylpyrrolidines from γ-N-arylamino olefins and aryl bromides, afforded an inseparable mixture of the desired product 3a, regioisomer 4a, and unexpected isomer 5a (Table 1, entry 1). Curiously, N-vinylation was less problematic with this substrate combination than in the reaction of 1 with β-bromostyrene described above. After some optimization we found that 3a was obtained in 81 % isolated yield when the reaction was conducted with tri-2-furylphosphine as the ligand (Table 1, entry 4). These conditions provided good selectivity for the formation of 3a (versus 4a and 5a), and did not afford detectable amounts of N-vinylated side products.
Table 1.
Catalyst optimization.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | 3a/4a[b] | 3a/5a[b] | Isolated yield[c] | ||
| 1 | dppb | 20:1 | 3:1 | 60 | ||
| 2 | dppe[d] | 15:1 | 5:1 | |||
| 3 | PCy3[d] | 13:1 | 4:1 | |||
| 4 | P(2-furyl)3 | 21:1 | 19:1 | 81 | ||
Conditions: amine 1 (1.0 equiv), vinyl bromide 2a (1.1 equiv), NaOt-Bu (1.2 equiv), Pd2(dba)3 (1 mol%), Ligand (2 mol% of bidentate ligand or 4 mol% of monodentate ligand), toluene (0.25 M), 60 °C.
Ratios determined by GC analysis of crude reaction mixtures.
Isolated yields refer to the total yield of an inseparable mixture of products 3a, 4a, and 5a.
This reaction was conducted at 110 °C.
Scope and diastereoselectivity
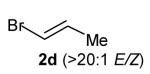
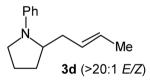
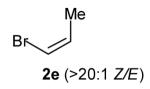
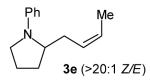
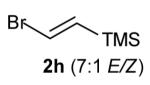
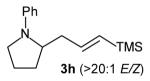
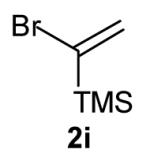
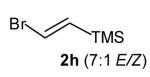
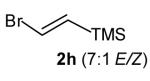
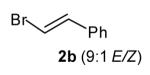
With optimized conditions in hand, we proceeded to examine the reaction of 1 with a variety of vinyl bromides. As shown in Table 2, N-aryl-2-allylpyrrolidines 3a–3h were obtained in good to excellent yields with 13:1 to >20:1 selectivity for 3 versus regioisomer 4. Isomers 5, which derive from formal rearrangement of the vinyl bromide, were observed in only two reactions, both of which employed E-substituted vinyl bromides (Table 2, entries 1 and 4). Notably, an analogous rearranged product was not observed in the reaction of the corresponding Z-substituted vinyl bromide 2e (Table 2, entry 5). Side products resulting from N-vinylation or Heck vinylation of the alkene were generally not observed in reactions catalyzed by Pd2(dba)3/tri-2-furylphosphine, even with the previously problematic β-bromostyrene (2b). All products were obtained as a single olefin stereoisomer,[12] and reactions of Z-substituted vinyl bromides afforded the desired pyrrolidine products with retention of the vinyl bromide geometry (Table 2, entries 3 and 5). However, although the reaction of 1 with (2-bromovinyl)trimethylsilane (2h) produced a single pyrrolidine product (Table 2, entry 8), the analogous reaction of 1 with the isomeric (1-bromovinyl)trimethylsilane (2i) afforded an inseparable 7:4 mixture of pyrrolidines 3i and 3h (Table 2, entry 9).
Table 2.
Palladium-catalyzed synthesis of N-phenyl-2-allylpyrroldines.[a]
| Entry | Vinyl bromide | Product (E/Z ratio)[b] | 3/4[b] | Yield [%][b] |
|---|---|---|---|---|
| 1 |

|
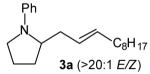
|
14:1 | 81[c] |
| 2 |
|
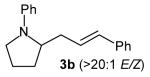
|
17:1 | 85 |
| 3 |

|

|
17:1 | 89 |
| 4 |
|
|
13:1 | 78[d] |
| 5 |
|
|
>20:1 | 88 |
| 6 |
|
|
19:1 | 77 |
| 7 |
|

|
>20:1 | 77 |
| 8 |
|
|
>20:1 | 87 |
| 9 |
|

|
>20:1 | 88 |
Conditions: amine 1 (1.0 equiv), vinyl bromide 2 (1.1–2.0 equiv), NaO t-Bu (1.2 equiv), Pd2(dba)3 (1 mol%), P(2-furyl)3 (4 mol %), toluene (0.25 M), 60–110 °C.
Product ratios refer to the isolated material, as judged by 1H NMR analysis. Product ratios and yields refer to the average of two or more experiments.
This material contained ~5 % of 5a as an inseparable impurity.
This material contained ~10 % of 5d as an inseparable impurity.
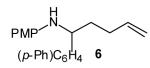
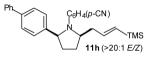
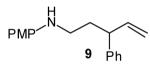
To probe the diastereoselectivity of these reactions, several γ-N-arylamino alkenes with substituents at the 1- or 3-position were prepared and subjected to our optimized reaction conditions. As shown in Table 3, reactions of amines 6—9 with various vinyl bromides afforded 2,3- and 2,5-disubstituted pyrrolidines in good yields with good to excellent levels of diastereoselectivity (dr 7:1 to >20:1). These reactions were effective with substrates bearing both electron-rich (Table 3, entries 1–2 and 4–8) and electron-poor (Table 3, entry 3) N-aryl substituents. The observed preference for the formation of trans-2,3- and cis-2,5-disubstituted pyrrolidines is analogous to that observed in the previously described reactions of related substrates with aryl bromides, and is complementary to the selectivity for the formation of cis-2,3-disubstituted N-tosyl pyrrolidines described by Larock and Weinreb.[3]
Table 3.
Stereoselective synthesis of disubstituted N-aryl-2-allylpyrroldines.[a]
| Entry | Amine | Vinyl bromide | Product (E/Z ratio)[b] | Regio[b] | Yield [%][b] |
|---|---|---|---|---|---|
| 1 |
|
|
|
>20:1 | 72 (dr 7:1) |
| 2 |
|
|
20:1 | 82 (dr >20:1) |
|
| 3 |
|
|
|
>20:1 | 71 (dr >20:1) |
| 4 |
|
|
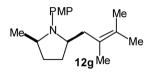
|
>20:1 | 84 (dr >20:1) |
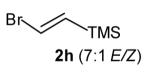
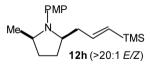
| 5 |
|
|
7:1 | 79 (dr >20:1) |
|
| 6 |
|
|
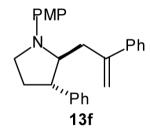
|
>20:1 | 55 (dr 10:1) |
| 7 |
|
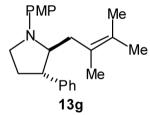
|
>20:1 | 64[c] (dr 10:1) |
|
| 8 |
|

|
3.6:1 | 61[c] (dr >20:1) |
Conditions: amine (1.0 equiv), vinyl bromide 2 (1.4–2.0 equiv), NaOt-Bu (1.2 equiv), Pd2(dba)3 (1 mol %), P(2-furyl)3 (4 mol%), toluene (0.25 M ), 110 °C.
Product ratios refer to the isolated material, as judged by 1H NMR analysis. Product ratios and yields refer to the average of two or more experiments.
Dppe (2 mol %) used as ligand in this reaction. PMP = p-methoxyphenyl.
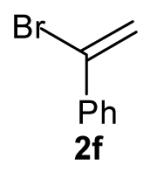
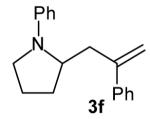
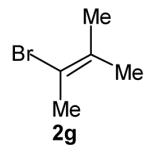
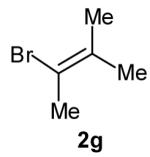
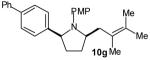
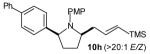
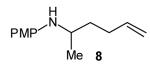
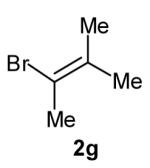
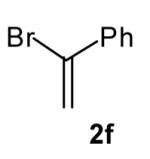
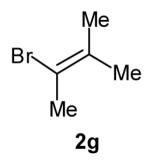
The size of the vinyl bromide had a notable effect on the stereoselectivity of the carboamination reactions. In reactions of 1-substituted amines 6–8 the highest selectivities (dr >20:1) were observed in cases where the vinyl bromide and/or the substituent on the amine were small (Table 3, entries 2–5). In contrast, the reaction of amine 6, which bears a 1-(p-biphenyl) substituent, with hindered vinyl bromide 2g provided 10g in good yield but with somewhat lower (dr 7:1) diastereoselectivity (Table 3, entry 1). The reactions of 3-substituted amine 9 provided the highest selectivity (dr >20:1) when the relatively small β-bromostyrene (2b) was employed (Table 3, entry 8). Use of α-bromostyrene (2f) or tetrasubstituted vinyl bromide 2g afforded products with lower (dr 10:1) diastereoselectivity (Table 3, entries 6–7).
Tandem reactions
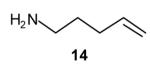
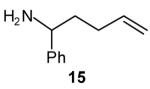
We have recently reported the development of palladium-catalyzed tandem N-arylation/carboamination reactions for the synthesis of N-aryl-2-benzyl pyrrolidines[13] and indolines[14] from appropriate primary amine precursors (eq 4, R = aryl). These reactions enable the modular assembly of heterocyclic compounds from very simple precursors and could be amenable to library synthesis. In these reactions, the primary amine substrate undergoes N-arylation in the presence of a monophosphine-supported palladium(0) catalyst.[15] After the first transformation is complete, an in situ ligand exchange process is effected via the addition of a bisphosphine to the reaction mixture, and then a second aryl bromide is added to provide the desired heterocycle in a one-pot process. An extension of this methodology to allow for the use of vinyl bromides as the second coupling partner would allow for the synthesis of an even broader array of compounds.
 |
(4) |
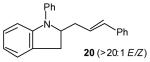
As shown in Table 4, the conditions reported previously[13],[14] for the synthesis of N-aryl-2-benzyl pyrrolidines and indolines via this tandem reaction were also effective for the synthesis of N-aryl-2-allyl pyrrolidines and indolines (eq 4, R = vinyl). Use of 2-(di-tert-butylphosphino)biphenyl as the ligand for the first step of the sequence was optimal with both the aliphatic and the aromatic amine substrates. In the second step, however, the ligand dppe provided the best results for the pyrrolidine forming reaction (Table 4, entries 1–2), whereas DPEphos was optimal in the reaction that afforded indoline products (Table 4, entries 3–4).
Table 4.
Synthesis of 2-allyl pyrrolidines and indolines via tandem reactions of primary amines.[a]
| Entry | Amine | Aryl bromide | Vinyl bromide | Product (E/Z ratio)[b] | Yield [%][b] |
|---|---|---|---|---|---|
| 1 |
|
|
|
|
65[c],[e],[f] |
| 2 |
|
|
|
|
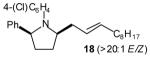
50[c],[e],[f] (>20:1 dr) |
| 3 |
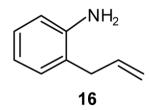
|

|
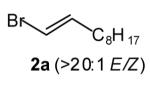
|
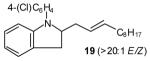
|
60[d],[f] |
| 4 |
|
|
|
69[d] |
Conditions: amine (1.0 equiv), aryl bromide (1.0 equiv), NaOt-Bu (2.4 equiv), Pd2(dba)3 (1 mol%), 2-di-t-butylphosphinobiphenyl(2 mol%), toluene (0.25 M), 60–80 °C, then dppe or DPEphos (2 mol%) and vinyl bromide (1.2 equiv), 105–110 °C.
Product ratios refer to the isolated material, as judged by 1H NMR analysis. Product ratios and yields refer to the average of two or more experiments.
This reaction was conducted with dppe as the second ligand.
This reaction was conducted with DPEphos as the second ligand.
This material contained ~3–10 % of an inseparable regioisomer analogous to 4a.
This material contained ~7–10 % of an inseparable impurity analogous to 5a.
Mechanism
The carboamination reactions of γ-N-arylamino alkenes 1 and 6–9 with vinyl bromides likely proceed through a mechanism similar to that described previously for the analogous transformation involving aryl bromides (Scheme 1).[6] The catalytic cycle presumably commences with oxidative addition of the vinyl bromide to palladium(0), which affords vinylpalladium bromide intermediate I. The reaction of I with the amine substrate and NaOt-Bu yields vinylpalladium amido intermediate II, which undergoes intramolecular amidopalladation to afford III. Carbon—carbon bond-forming reductive elimination from III provides the observed pyrrolidine 3 and regenerates the palladium(0) catalyst. The N-vinylated side product that is observed under the non-optimized reaction conditions presumably derives from carbon—nitrogen bond-forming reductive elimination of intermediate II.[11] This side reaction is effectively suppressed by the use of the relatively small tri-2-furylphosphine ligand. Interestingly, the fact that N-vinylation is more problematic than N-arylation with the Pd2(dba)3/dppb catalyst system suggests that vinyl sp2 carbon—nitrogen bond-forming reductive elimination is a more facile transformation than aryl sp2 carbon—nitrogen bond-forming reductive elimination in cases where the aryl and vinyl bromides have similar electronic properties (e.g., β-bromostyrene and 2-bromonaphthalene).[16]
Scheme 1.

Proposed mechanism of carboamination reaction.
As previously suggested,[6],[7] regioisomer 4 likely originates from β-hydride elimination of III to afford π-olefin complex IV (Scheme 2). Reinsertion of the olefin into the Pd—H bond with reversal of regiochemistry would provide V, which can undergo a second β-hydride elimination/reinsertion process to afford VI. Carbon—carbon bond-forming reductive elimination would generate regioisomer 4 with concomitant regeneration of the palladium(0) catalyst.
Scheme 2.

Proposed mechanism for formation of regioisomer 4.
The formation of isomer 5 as a side product in reactions involving E-substituted vinyl bromides 2a and 2d (Table 2, entries 1 and 4) and the formation of pyrrolidine 3h as a side product in the reaction of 1 with 2i (Table 2, entry 9) both appear to involve a formal rearrangement of the vinyl bromide coupling partner.[17],[18] One possible origin of these products involves an unusual β-hydride elimination reaction of vinylpalladium bromide intermediate I to afford π-alkynylpalladium hydride complex VII. Reinsertion of the alkyne into the Pd—H bond with reversal of regiochemistry would afford iso-I,[19] which could re-enter the catalytic cycle shown in Scheme 1 to afford 5 (Scheme 3). This mechanism has previously been invoked to explain the origin of similar rearranged products observed in cross-coupling reactions of (1-bromovinyl)trimethylsilane (2i).[17a—b,d] The fact that side products 5 were observed in reactions of amine 1 with E-substituted vinyl bromides 2a and 2d but not in reactions with Z-substituted vinyl bromide 2e (Table 2, entries 1, 4, and 5) also supports the mechanism shown in Scheme 3, as β-hydride elimination reactions of alkylpalladium complexes require a syn relationship between palladium and the involved β-hydrogen atom.[20] The relatively large amount of rearranged product 3h observed in the reaction of amine 1 with vinyl bromide 2i may be due to a β-silyl effect, which would stabilize the developing positive charge on the β-carbon atom in the transition state between I and VII.[21]
Scheme 3.

Proposed mechanism for formation of 5.
Conclusion
In conclusion, we have developed conditions for the synthesis of N-aryl-2-allyl pyrrolidines via the palladium-catalyzed carboamination of γ-N-arylamino alkenes with vinyl bromides. These reactions display broad scope with respect to vinyl halide structure and afford trans-2,3- and cis-2,5-disubstituted pyrrolidines with good to excellent levels of diastereoselectivity. Additionally, we have shown that tandem N-arylation/carboamination reactions of unsaturated primary amines are effective for the synthesis of N-aryl-2-allyl pyrrolidines and indolines. Efforts to apply this methodology to the total synthesis of structurally interesting and biologically active targets are currently underway.
Experimental Section
General remarks
All reactions were carried out under an argon atmosphere in flame-dried glassware. Tris(dibenzylidineacetone)dipalladium(0) and all phosphine ligands were purchased from Strem Chemical Co. and used without further purification. Toluene was purified using a GlassContour solvent purification system. Unless otherwise noted, yields refer to isolated yields of compounds estimated to be ≥95% pure as determined by 1H NMR and combustion analysis. The yields and product ratios reported in the experimental section and supporting information describe the result of a single experiment, whereas those reported in Tables 1–4 are averages of two or more experiments. Thus, the yields and ratios reported in the experimental section and supporting information may differ from those shown Tables 1–4.
General procedure for palladium-catalyzed carboamination reactions with vinyl bromides
A flame-dried Schlenk tube was charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), tri-2-furylphosphine (4 mol %), and sodium tert-butoxide (1.2 equiv). The tube was purged with argon and toluene (4 mL/mmol amine substrate), the amine substrate (1.0 equiv), and the vinyl bromide (1.1–2.0 equiv) were added via syringe. The mixture was heated to 60–110 °C with stirring until the starting material had been consumed as judged by GC or 1H NMR analysis. The reaction mixture was cooled to room temperature, quenched with saturated aqueous ammonium chloride (2 mL), and diluted with ethyl acetate (10 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel.
(E)-1-Phenyl-2-[undec-2-enyl]pyrrolidine (3a)
Reaction of amine 1 (40 mg, 0.25 mmol) with vinyl bromide 2a (60 mg, 0.275 mmol, >20:1 E/Z) and sodium tert-butoxide (29 mg, 0.3 mmol) at 60 °C following the general procedure afforded the title compound (3a; 66 mg, 88 %, >20:1 E/Z) as a colorless oil. This material was obtained as a 14:1 mixture of inseparable regioisomers and contained ~6 % 5a, as judged by 1H NMR analysis; data are for the major isomer. 1H NMR (400 MHz, CDCl3) δ 7.28–7.22 (m, 2 H), 6.67 (t, J = 7.4 Hz, 1 H), 6.6 (d, J = 7.6 Hz, 2 H), 5.57–5.48 (m, 1 H), 5.47–5.38 (m, 1 H), 3.75–3.69 (m, 1 H), 3.47–3.40 (m, 1 H), 3.22–3.14 (m, 1 H), 2.48–2.41 (m, 1 H), 2.07–1.87 (m, 7 H), 1.44–1.25 (m, 12 H), 0.91 (t, J = 6.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 147.4, 133.4, 129.4, 126.9, 115.4, 112.0, 58.7, 48.5, 36.3, 33.0, 32.1, 29.9, 29.7, 29.5, 29.4, 23.5, 22.9, 14.3 (two aliphatic signals are incidentally equivalent); IR (film) 1598, 1505 cm−1. Anal calcd for C21H33N: C, 84.22; H, 11.11; N, 4.68. Found: C, 84.47; H, 11.29; N, 4.70.
General procedure for palladium-catalyzed tandem N-arylation/carboamination reactions
A flame-dried Schlenk tube was charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), 2-(di-tert-butylphosphino)biphenyl (2 mol %) and sodium tert-butoxide (2.4 equiv). The tube was purged with argon and toluene (1 mL), the amine substrate (1.0 equiv) and the aryl bromide (1.0 equiv) were added via syringe. The mixture was placed in a pre-heated oil bath at 60 °C with stirring until the starting materials had been consumed as judged by GC analysis. A solution of dppe (2 mol %) in toluene (1 mL) was added, and the temperature was increased to 110 °C. After 15 min of stirring at 110 °C the vinyl bromide (1.2–2.0 equiv) was added via syringe. Heating was continued until the intermediate arylamine had been consumed as judged by GC analysis. The reaction mixture was then cooled to room temperature, quenched with saturated aqueous ammonium chloride (2 mL) and diluted with ethyl acetate (10 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic extracts were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel.
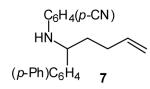
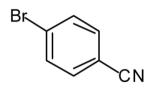
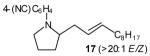
(E)-4-{2-[Undec-2-enyl]pyrrolidin-1-yl}benzonitrile (17)
Reaction of amine 14 (21 mg, 0.25 mmol) with 4-bromobenzonitrile (46 mg, 0.25 mmol), sodium tert-butoxide (58 mg, 0.6 mmol) and vinyl bromide 2a (110 mg, 0.5 mmol, >20:1 E/Z) following the general procedure afforded the title compound (17; 55 mg, 68 %, >20:1 E/Z) as a pale yellow oil. This material was obtained as a 30:1 mixture of inseparable regioisomers and contained ~7 % 4-[2-(2-methylenedecyl)pyrrolidin-1-yl]benzonitrile as judged by 1H NMR analysis; data are for the major isomer. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 9.2 Hz, 2 H), 6.52 (d, J = 8.4 Hz, 2 H), 5.56–5.47 (m, 1 H), 5.40–5.31 (m, 1 H), 3.80–3.73 (m, 1 H), 3.45–3.39 (m, 1 H), 3.25–3.17 (m, 1 H), 2.39–2.31 (m, 1 H), 2.12–1.91 (m, 7 H), 1.40–1.22 (m, 12 H), 0.88 (t, J = 7.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3) δ 149.6, 134.3, 133.7, 125.9, 121.2, 112.0, 96.8, 58.7, 48.3, 35.6, 32.9, 32.1, 29.7, 29.7, 29.6, 29.5, 29.4, 23.2, 22.9, 14.3; IR (film) 2213, 1607, 1520 cm−1. Anal calcd for C22H32N2: C, 81.43; H, 9.94; N, 8.63. Found: C, 81.51; H, 10.00; N, 8.72.
Supplementary Material
Acknowledgements
The authors thank the University of Michigan and the National Institutes of Health—National Institute of General Medical Science (GM-071650) for financial support of this work. JPW thanks the Camille and Henry Dreyfus Foundation for a new faculty award and Research Corporation for an innovation award. JEN acknowledges the University of Michigan for a Regents Fellowship and Pfizer for a Graduate Research Fellowship. Additional unrestricted support was provided by Amgen, Eli Lilly, and 3M.
Footnotes
Supporting Information for this article is available on the WWW under http://asc.wiley-vch.de
References and Notes
- [1] (a).Massiot G, Delaude C. Alkaloids. 1986;27:269–322. [Google Scholar]; (b) O’Hagan D. Nat. Prod. Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; (c) Lewis JR. Nat. Prod. Rep. 2001;18:95–128. doi: 10.1039/a909077k. [DOI] [PubMed] [Google Scholar]
- [2] (a).Pichon M, Figadere B. Tetrahedron: Asymmetry. 1996;7:927–964. [Google Scholar]; (b) Mitchinson A, Nadin A. J. Chem. Soc., Perkin Trans. 1. 2000:2862–2892. [Google Scholar]
- [3] (a).Harris GD, Jr., Herr RJ, Weinreb SM. J. Org. Chem. 1992;57:2528–2530. [Google Scholar]; (b) Larock RC, Yang H, Weinreb SM, Herr RJ. J. Org. Chem. 1994;59:4172–4178. [Google Scholar]
- [4] (a).Tamaru Y, Kimura M. Synlett. 1997:749–757. [Google Scholar]; (b) Tamaru Y, Hojo M, Higashimura H, Yoshida Z. -i. J. Am. Chem. Soc. 1988;110:3994–4002. [Google Scholar]; (c) Tamaru Y, Hojo M, Yoshida Z. —i. J. Org. Chem. 1988;53:5731–5741. [Google Scholar]; (d) Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J. Org. Chem. 1997;62:2113–2122. doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]
- [5].Jacobi PA, Liu H. Org. Lett. 1999;1:341–344. doi: 10.1021/ol990684j. [DOI] [PubMed] [Google Scholar]
- [6] (a).Ney JE, Wolfe JP. Angew. Chem. 2004;116:3689–3692. [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; (b) Ney JE, Wolfe JP. J. Am. Chem. Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For a related synthesis of N-acyl- and N-boc-2-benzyl pyrrolidines, see: Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447–6459.
- [8] (a).For a related synthesis of tetrahydrofurans, see: Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838. Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2005;70:3099–3107. doi: 10.1021/jo050022+.
- [9].For a recent report of an intramolecular carboamination reaction of N-(arylsulfonyl)-2-allylanilines in the presence of excess Cu(OAc)2, see: Sherman ES, Chemler SR, Tan TB, Gerlits O. Org. Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+.
- [10] (a).See reference 6b. For a discussion of ligand effects on the rate of carbon—nitrogen bond-forming reductive elimination, see: Hamann BC, Hartwig JF. J. Am. Chem. Soc. 1998;120:3694–3703. For a discussion of ligand effects on the rate of carbon—carbon bond-forming reductive elimination, see: Gillie A, Stille JK. J. Am. Chem. Soc. 1980;102:4933–4941.
- [11] (a).For recent reports of palladium-catalyzed N-vinylation reactions, see: Dehli JR, Legros J, Bolm C. Chem. Commun. 2005:973–986. doi: 10.1039/b415954c. and references cited therein. Barluenga J, Fernandez MA, Aznar F, Valdes C. Chem. Eur. J. 2004;10:494–507. doi: 10.1002/chem.200305406.
- [12].Pyrrolidines 3b and 3h were obtained with >20:1 E/Z ratios even though they were derived from geometrically impure vinyl bromides 2b and 2h. These vinyl bromides were present in excess, and (E)-vinyl bromides have been shown to undergo oxidative addition faster than the corresponding (Z)-vinyl bromides. See: Jutand A, Negri S. Organometallics. 2003;22:4229–4237.
- [13].Yang Q, Ney JE, Wolfe JP. Org Lett. 2005;7:2575–2578. doi: 10.1021/ol050647u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906–13907. doi: 10.1021/ja0460920. [DOI] [PubMed] [Google Scholar]
- [15] (a).For recent reviews on palladium-catalyzed N-arylation reactions, see: Muci AR, Buchwald SL. 2002;219:131–209. Hartwig JF. In: Modern Arene Chemistry. Astruc D, editor. Wiley-VCH; Weinheim, Germany: 2002. pp. 107–168. Schlummer B, Scholz U. Adv. Synth. Catal. 2004;346:1599–1626.
- [16].Previous competition experiments have shown that N-vinylation occurs preferentially to N-arylation when amines are treated with 1:1 mixtures of aryl and vinyl halides under catalytic amination conditions. However, the rate of oxidative addition of vinyl halides is faster than that of aryl halides. Thus, the competition experiments reported do not give a direct measure of the relative rates of reductive elimination. For details of these previous experiments, and references concerning the relative rates of oxidative addition of aryl and vinyl halides, see reference 11b.
- [17] (a).For reports of rearranged products in palladium-catalyzed coupling reactions of (1-bromovinyl)trimethylsilane (2i), see: Cazes B, Colovray V, Gore J. Tetrahedron Lett. 1988;29:627–630. Carpita A, Rossi R, Scamuzzi B. Tetrahedron Lett. 1989;30:2699–2702. Ennis DS, Gilchrist TL. Tetrahedron. 1990;46:2623–2632. Sasmal PK, Maier ME. Org. Lett. 2002;4:1271–1274. doi: 10.1021/ol025570d.
- [18].To the best of our knowledge, the formation of a side product analogous to 5 in a palladium-catalyzed coupling reaction of an E-substituted 1-bromo-1-alkene has been reported only once. See: Flamme EM, Roush WR. Org. Lett. 2005;7:1411–1414. doi: 10.1021/ol050250q.
- [19] (a).This reinsertion step fundamentally involves the hydropalladation of an alkyne. For examples of other reactions involving alkyne hydropalladation, see: Grushin VV. Chem. Rev. 1996;96:2011–2034. doi: 10.1021/cr950272y. and references cited therein. Trost BM. Acc. Chem. Res. 1990;23:34–42. Ji J, Wang Z, Lu X. Organometallics. 1996;15:2821–2828. Kadota I, Lutete LM, Shibuya A, Yamamoto Y. Tetrahedron Lett. 2001;42:6207–6210.
- [20].Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- [21] (a).β-hydride elimination reactions have been shown to involve the buildup of positive charge at the β-carbon atom in the transition state. See: Mueller JA, Goller CP, Sigman MS. J. Am. Chem. Soc. 2004;126:9724–9734. doi: 10.1021/ja047794s. Mueller JA, Sigman MS. J. Am. Chem. Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.