

SUMMARY
Despite similarities between tumor initiating cells with stem-like properties (TICs) and normal neural stem cells, we hypothesized that there may be differences in their differentiation potentials. We now demonstrate that both bone morphogenetic protein (BMP)-mediated and ciliary neurotrophic factor (CNTF)-mediated Jak/STAT-dependent astroglial differentiation is impaired due to EZH2-dependent epigenetic silencing of BMP receptor 1B (BMPR1B) in a subset of glioblastoma TICs. Forced expression of BMPR1B either by transgene expression or demethylation of the promoter restores their differentiation capabilities and induces loss of their tumorigenicity. We propose that deregulation of the BMP developmental pathway in a subset of glioblastoma TICs contributes to their tumorigenicity both by desensitizing TICs to normal differentiation cues, and by converting otherwise cytostatic signals to pro-proliferative signals.
SIGNIFICANCE
Elucidation of the differentiation pathways operative and/or aberrant in both normal stem cells and TICs will be critical to fully understand the pathogenesis of primary human tumors and may help lead to better therapies. To this end, we utilized several TICs isolated from primary glioblastomas and compared them to normal human NSCs and mouse NSCs from various developmental stages. We demonstrate a major differentiation block in a subset of glioblastoma TICs is caused by the Polycomb repressor complex (PRC) mediated epigenetic silencing of the BMPR1B promoter, analogous to early embryonic NSCs. We provide here an example of a temporally deregulated and aberrantly fixed developmental block to differentiation contributing to the pathogenesis of a subset of human GBMs.
INTRODUCTION
A growing body of literature indicates that only a small subpopulation of tumor cells are clonogenic and are capable of repopulating a tumor mass. These clonogenic cells have been found in some tumor types to be highly undifferentiated with the capability of both self-renewal and at least partial differentiation, similar to tissue-specific normal stem cells, leading to their designation as tumor stem cells (TSCs) or tumor initiating cells with stem-like properties (TICs)(Bonnet and Dick, 1997; Clarke et al., 2006; Jordan et al., 2006; Reya et al., 2001). For example, several groups have demonstrated the presence of TICs in various types of primary brain tumors including glioblastomas (GBMs), ependymomas, and medulloblastomas. These TICs have functional similarities to normal NSCs including their clonogenicity and capability for multi-lineage differentiation(Hemmati et al., 2003; Lee et al., 2006a; Singh et al., 2004; Taylor et al., 2005). Indeed, it has recently been reported that BMP acts as a key inhibitory regulator of TICs from GBMs, similar to adult NSCs(Piccirillo et al., 2006). Unlike normal stem cells, however, TICs harbor the genetic aberrations characteristics of their representative tumors (i.e. activation/amplification of oncogenes and loss of tumor suppressor genes) endowing them with a tumorigenic phenotype. A number of pathways that are commonly deregulated in gliomagenesis, such as the NOTCH, epidermal growth factor receptor (EGFR), and the mammalian target of rapamycin (mTOR) pathways, are also involved in astrocytic differentiation in normal NSCs(Hu et al., 2005; Purow et al., 2005; Sabatini, 2006; Viti et al., 2003), suggesting that differentiation pathways of TICs may not be operated entirely similar to those of normal NSCs. Nevertheless, exactly where along the developmental pathway of tissue-specific stem cell maturation and differentiation TICs arise and which, if any, of the intrinsic stem cell signaling pathways are perturbed in TICs remains largely unknown.
The differentiation pathways of normal NSCs are complex and differentially regulated by many cytokines. BMP-mediated PS-SMAD1/5/8 pathways and CNTF-mediated Jak/STAT pathways are the most widely studied cytokine-mediated pathways in the differentiation of NSCs.
BMPs mediate a wide variety of biological responses in NSCs ranging from proliferation to differentiation to apoptosis depending on the NSC developmental stage as well as the milieu of the local microenvironments(Lim et al., 2000; Mehler et al., 2000). The prototypic receptors for BMP 2/4 signaling in mammalian brains are the type II receptor, BMPR2, and type I receptors, BMPR1A and BMPR1B. One of best-characterized downstream events following ligand binding is the activation of SMAD1/5/8 by serine-phosphorylation. It has been reported that BMP2/4 acts as a neuroepithelial proliferation signal at very early stages of embryonic central nervous system development; an effect mediated principally by BMPR1A(Chen and Panchision, 2007; Panchision et al., 2001). Later in development, BMP2/4 induces neuronal and astrocytic differentiation of NSCs, an event coinciding with increased expression of BMPR1B (Hall and Miller, 2004; Mehler et al., 2000). The latter astroglial differentiation (E17–18 to murine neonates) occurs within the context of acquisition of STAT3 responsiveness to CNTF/LIF(Bonni et al., 1997; He et al., 2005).
CNTF signaling pathways are largely restricted to astroglial differentiation in NSCs. CNTF, a member of interleukin 6 superfamily including leukemia inhibitory factor (LIF), can transduce its signal through the gp130 receptor complex and through the Jak protein family, with subsequent activation of STAT3 by tyrosine phosphorylation (PY-STAT3). Early embryonic NSCs are minimally responsive to CNTF/LIF-mediated STAT3 activation. A significant increase in the amount of STAT3 activation, either by modulation of DNA demethylation, down-regulation of specific neurogenic transcription factors, and/or a feed-forward STAT3 activation signaling loop, seem to be required for the onset of astrogenesis(He et al., 2005; Molne et al., 2000; Sun et al., 2001; Takizawa et al., 2001).
In an effort to better understand the operative and aberrant differentiation pathways in glioblastoma TICs, we utilized several TIC-like cell lines isolated from primary glioblastomas, human fetal brain derived NSCs, and embryonic mouse NSCs isolated from various developmental stages. We demonstrate here that astroglial differentiation mediated by both the BMP and CNTF-mediated Jak/STAT pathway is impaired in TICs derived from a subset of GBMs due to the epigenetic silencing of BMPR1B. We provide an example of a temporally deregulated and aberrantly fixed normal stem cell developmental block to differentiation contributing to the pathogenesis of a human tumor.
RESULTS
Deregulated responses to BMP and CNTF pathways in 0308-TIC
We established various TICs from adult GBM patients. These cells proliferate in the presence of FGF2 and EGF in serum-free media and differentiate in the presence of serum or in the absence of FGF2 and EGF. Transplantation of these cells into brains of immunodeficient SCID mice generated tumors phenocopying the features of parental human GBM tumors(Lee et al., 2006a). The results of our extensive single nucleotide polymorphism (SNP) array-based genomic profiling and sequence analyses of genes implicated in the gliomagenesis demonstrate that these TICs have genomic changes characteristics of typical primary human GBMs (Figure S1 in the Supplemental Data available with this article online).
In order to evaluate cytokine-mediated differentiation of these TICs, we performed in vitro differentiation assays (Figures 1 and S2). Since human NSCs isolated from various developmental stages were not available, we utilized mouse NSCs from various developmental stages extending from E10 brain-derived neuroepithelial cells to adult subventricular zone (SVZ)-derived NSCs. Spheroid-forming cells cultured in serum-free media with FGF2 and EGF, the same conditions used for expansion of various TICs (NBE condition)(Lee et al., 2006a), were dissociated and plated in the poly-ornithine coated plates for adherent cultures. These cells were cultured in the presence of BMP2 or CNTF without FGF2 and EGF for 3 days and subsequently processed for immunocytochemical staining (Figure 1A). All the TICs continuously cultured in NBE condition retained their undifferentiated state as assessed by both their morphology and expression of Nestin and Sox2 (Figure 1A). Similar to other TICs, withdrawal of FGF2/EGF induced differentiation of all of our TICs lines as demonstrated by loss of Nestin and Sox2 (neural stem/progenitor markers) immuno-positivity with concurrent induction of GFAP (astroglial marker) or TuJ1 (neuronal marker) expression. Most of the TICs lines cultured in the presence of BMP2 or CNTF revealed further induction of differentiation as determined by increased numbers of GFAP positive cells (Figure 1A and 1B). BMP2-treated 0308 TICs, however, retained their expression of Nestin and Sox2 and there was no significant increase in the number of GFAP positive cells. In addition, GFAP positive cells in CNTF-treated 0308 TICs are similar to non-treated control, suggesting that 0308 TICs are relatively insensitive to normal CNTF-mediated glial differentiation. Immunostaining of CNTF-treated 0308 TICs for S100 β, another astroglial marker, revealed similar results (Figure S3).
Figure 1. Immunohistochemical analysis of various TICs cultured in the presence of differentiation-inducing cytokines.

A) Representative microphotographs of 0308 and 0822 TICs cultured in the presence of various cytokines. Each column represents the same culture condition.,
(a–h) Immunohistochemistry of Nestin and Sox2 in 0308 TICs (a to d) and 0822 TICs (e to h).
(i–p) Immunohistochemistry of GFAP and TuJ1 in 0308 TICs (i to l) and 0822 TICs (m to p). DAPI staining (blue) was used to identify nuclei. White bars represent 10 micron.
B) Differentiation potentials of various TICs after exposure to cytokines. Error bars represent SD (performed in triplicate). * indicates p< 0.01.
Acutely isolated mouse NSCs from E11 brains differentiated predominantly along a neuronal lineage upon exposure to BMP2, while NSCs (passage 5) isolated from the SVZ of adult brains differentiated mainly along an astroglial lineage(Molne et al., 2000; Sun et al., 2001) (Figure S2). By contrast, CNTF treatment failed to induce astroglial differentiation in E11 NSCs compared to the efficient induction of astroglial differentiation in adult-derived NSCs (Figure S2), in agreement with previous reports(He et al., 2005; Molne et al., 2000). Thus, many TIC lines have normal cytokine-induced differentiation responses similar to adult-derived NSC whereas 0308-TICs behave more like E11 NSC cells (non-responsiveness to CNTF) although they do not demonstrate BMP-mediated neuronal differentiation. Taken together, these data imply that 0308-TICs, unlike other TICs, have defective/deregulated signal transduction responses to the BMP and CNTF pathways, despite the fact that they retain the ability to undergo terminal differentiation through the withdrawal of growth factors and/or the addition of serum(Lee et al., 2006a).
Downstream effectors of BMP and CNTF pathways in various TICs
In order to further understand the deregulated differentiation pathways in 0308 TICs, we evaluated downstream effectors of BMP and CNTF signaling in various TICs (Figure 2). Several groups have recently demonstrated that brain TSC-like cells are enriched in CD133 positive populations(Bao et al., 2006; Singh et al., 2004). We isolated CD133+ cells from 0308 and 0822 tumor cells by using fluorescence activated sorting (FACS) and confirmed that indeed these cells are enriched in tumorigenic potential in a SCID mouse transplantation model (data not shown). Similar to normal human NSCs, BMP2 induced strong activation of PS-SMAD1/5/8 in both 0308 and 0822 CD133+ cells. Similarly, CNTF induced strong activation of PY-STAT3 in 0822 CD133+ cells (Figure 2A). By contrast, CNTF induced only minimal activation of PY-STAT3 in 0308 CD133+ cells, consistent with the marginal CNTF-induced astroglial differentiation observed in 0308 cells (Figure 1 and 2B). We further examined the activation status of PS-SMAD1/5/8 and PY-STAT3 in additional TICs and normal NSCs isolated from both human and mouse brains (Figure 2 B and C). Normal human NSCs (passage 15) and other TICs elicited strong activation of PS-SMAD1/5/8 and PY-STAT3 upon BMP2 and CNTF challenge, respectively. In addition, adult mouse brain-derived NSCs respond to BMP and CNTF by showing a clear induction of PS-SMAD1/5/8 and PY-STAT3, respectively(Bonni et al., 1997; He et al., 2005). By contrast, mouse NSCs acutely isolated from earlier embryonic stages (E10 to E11) did not elicit CNTF-triggered PY-STAT3 activation, consistent with previous reports (He et al., 2005; Sun et al., 2001)(Figure 2C). In order to further confirm the activation status of the BMP and CNTF pathways in 0308 TICs, we performed real time RT-PCR analysis of genes known to be up-regulated through these pathways. Following exposure of 0308 TICs to BMP2, we found clear induction of genes downstream of the SMAD pathway including Id1, Id3, and Hes1, consistent with the observed PS-SMAD1/5/8 activation(Massague et al., 2005; Ying et al., 2003). By contrast, CNTF induced little activation of genes downstream of the Jak/STAT pathway including SOCS3 and JunB (data not shown) (Passegue et al., 2004). Taken together, these data suggest that the unique BMP response observed in 0308 TICs is unlikely attributed to suboptimal activation of PS-SMAD1/5/8, whereas, the lack of CNTF response is due, at least in part, to the failure to activate PY-STAT3.
Figure 2. Evaluation of representative activators of Jak/STAT and BMP pathways in various TICs.

A) Western blot analysis of representative activators of Jak/STAT and BMP pathways in CD133+ TICs. Cells were stimulated with indicated cytokines (50 ng/ml) for 20 minutes. PY-STAT3 indicates detection of tyrosine 705-phosphorylated form of STAT3. STAT3 indicates detection of all STAT3 proteins regardless of phosphorylation status. PS-SMAD1/5/8 and SMAD1 indicate serine phosphorylated SMAD and total SMAD levels, respectively.
B and C) Western blot analysis of PY-STAT3 and PS-SMAD1/5/8 in various TICs and normal human NSCs (B) and mouse NSCs from different developmental stages (C).
D) Relative activation of PY-STAT3 and PS-SMAD1/5/8 in various TICs and normal NSCs upon cytokine exposure. Relative activation of PY-STAT3 in a given condition was measured by relative intensity of PY-STAT3 over that of STAT3 bands compared to non-treated control. Error bars represent SD.
BMP induces proliferation instead of differentiation of 0308 TICs
The inability of BMP2 to induce differentiation in 0308 TICs is reminiscent of its effects on early embryonic NSC where BMP acts as a transient mitogen(Chen and Panchision, 2007; Liu et al., 2004). Thus, we explored the mitogenic properties of BMP2 on various TICs. Proliferation kinetics determined both by cell number counting and BrdU incorporation assays (Figure 3A to C) demonstrated that BMP2 treatment of 0308 cells significantly increased BrdU incorporation and the total number of cells by day 4, similar to its effects on E11 NSC (Figure S4). By contrast, BMP2 caused no significant increase in proliferation or BrdU uptake in other TIC lines or in adult NSCs (Figure 3 A and C). In order to determine whether this mitogenic effect of BMP2 on 0308 TICs can be reflected by clonogenicity, we performed sphere-forming assays by using single-cell dissociated TICs (Figure S5). BMP-treated 0308 cells showed the enhanced clonogenic sphere formation albeit lower than FGF2/EGF-treated cells.
Figure 3. BMP induces proliferation instead of differentiation of 0308 TICs.

A) Proliferation kinetics of various TICs in the presence of BMP2. Cells were cultured with BMP2 (without FGF2 and EGF) for 6 days. Cell number (a) and BrdU positive cells (b) were counted and relative changes were calculated compared to non-treated cells. Error bars represent SD.
B) FACS analysis of BrdU incorporation in 0308 TICs. Percentages indicate BrdU-positive cells.
C) Proliferation kinetics of mouse NSCs from different developmental stages. Error bars represent SD.
D and E) Western blot analysis of BMPR1B and PY-STAT3 in serially cultured NSCs isolated from E11 brains and adult-SVZ derived mouse NSCs. DIV represents days in vitro. GAPDH was used to confirm equal loading of proteins.
F) Quantitative mRNA expression level of BMPR1B in various TICs and human NSCs determined by real time RT-PCR analysis. Error bars represent SD.
In early embryonic NSCs, BMP signaling leads to both NSC proliferation and neuronal differentiation. This pleiotropic response may, in part, be due to the differential expression of the BMP receptor subtypes. Whereas BMPR1A is ubiquitously expressed in most NSCs, BMPR1B expression increases through the developmental maturation process of NSCs. Upon BMP signaling, BMPR1B expression is transcriptionally induced by BMPR1A-mediated PS-SMAD1/5/8 activation. Therefore, it has been proposed that early neural precursor cells express mainly BMPR1A whereas NSCs from later developmental stages to the adult stage express both BMPR1A and BMPR1B. Accordingly, BMPR1A-mediated signals may be mainly responsible for proliferation of early NSCs whereas BMPR1B-mediated signals likely play a role in NSC differentiation and survival. Thus, we examined the status of the BMPR subtypes in normal embryonic NSCs. Compared to adult SVZ-derived NSCs, acutely isolated E10-E11 neuroepithelial cells have very low level of BMPR1B expression, however, these cells showed progressive expression of BMPR1B following several days in culture (i.e. intrinsic cellular developmental clock; Figure 3D). Similarly to the 0308 TICs, neuroepithelial cells acutely isolated from E11 embryos demonstrated only marginal PY-STAT3 activation following CNTF challenge, however, after in vitro passage over several days the NSCs ultimately demonstrated significantly high CNTF-mediated PY-STAT3 activation comparable to that seen in adult NSCs (Figures 2C and 3E). Thus, the timing of BMPR1B expression in embryonic NSCs correlates with the induction of PY-STAT3 activation, events that occur immediately before the onset of increased potential for astroglial differentiation.
Given the similarity between 0308-TICs and early embryonic NSCs, we examined the expression of the BMPR subtypes in different TIC lines. Expression levels of BMPR2 and BMPR1A in various TICs were comparable as determined by real-time RT-PCR analysis and Western blot analysis (Figure S6). By contrast, BMPR1B expression was almost totally lacking in 0308-TICs compared to normal NSCs and other TICs (Figure 3F). Based on these data, we hypothesized that the lack of BMPR1B expression might be responsible for both the lack of CNTF–dependent PY-STAT3 activation and the aberrant BMP-mediated proliferation/differentiation responses observed in 0308 TICs.
Expression of BMPR1B in 0308 TICs abolishes BMP-induced proliferation and renders them responsive to CNTF
In order to test our hypothesis, we overexpressed a constitutively active BMPR1B mutant protein in 0308-TICs (Figure 4). This construct was generated by a point mutation in the glycine/serine domain of BMPR1B leading to constitutive phosphorylation of PS-SMAD1/5/8, albeit with low efficiency(Panchision et al., 2001). Polyclonal populations of BMPR1B-expressing TICs proliferated slower than the control transfected or parental 0308-TICs, however, still maintained expression of stem cell markers and clonogenicity in vitro, suggesting that BMPR1B expression itself is not sufficient to induce differentiation of the 0308 TIC under proliferation-conducive culture conditions (NBE condition) (data not shown). We then evaluated the proliferation kinetics of BMPR1B-0308 cells in the presence of BMP2. Unlike the parental and transfection control cells, BMP2 did not induce proliferation nor enhance clonal sphere formation in BMPR1B-overexpressing cells (Figures 4A and S5).
Figure 4. Overexpression of BMPR1B in 0308-TICs enhances astroglial differentiation.

A) Proliferation kinetics of BMPR1B-0308 cells cultured in BMP2 and CNTF (without FGF2 and EGF). Error bars represent SD.
B) Western blot analysis of representative activators of Jak/STAT and BMP pathways in 0308 and BMPR1B-0308 cells treated with indicated cytokines (BMP2: 50 ng/ml, CNTF: 50 ng/ml, LIF: 100 ng/ml). Exogenous BMPR1B expression was monitored by expression of HA tag which was fused with BMPR1B protein.
C) Increased astroglial differentiation in 0308-BMPR1B cells.
a. Representative immunohistochemical staining of BMPR1B-0308 cells cultured in the presence of BMP2 or CNTF (50 ng/ml each). Cytokines were added every other day (in the absence of FGF2 and EGF) for 5 days. White bar represents 10 micron.
b. Quantitation of Sox2 and GFAP-positive cells in (a). Error bars represent SD.
D) Transcriptional activity assay of synthetic STAT3-responsive promoter (a) and genomic GFAP promoter (b) in 0308 cells and human NSCs. Cells were co-transfected with a plasmid containing a STAT3-responsive or genomic GFAP promoter driving luciferase expression and a plasmid containing a CMV promoter driving beta-galactosidase expression. Y axis represents relative activity of luciferase versus beta-galactosidase activity. Error bars represent SD.
E) Quantitation of GFAP-positive cells after modulation of STAT3 activity in 0308 and 0308-BMPR1B cells. Cells were transiently transfected with either empty vector control, STAT3D, or STAT3C vectors and processed for GFAP staining 4 days after transfection. % of GFAP positive cells among transfected cells are shown. Data shown were from a representative experiment and numbers in parenthesis represent SD.
F) Western blot analysis of PY-STAT3 and PS-SMAD1/5/8 in tetracycline inducible U251-BMPR1B cells. Cells were serum-starved overnight prior to the addition of various cytokines (B, BMP2; T, TGF-β; C, CNTF; L, LIF). Actively proliferating U251 or U251-BMPR1B cells in serum containing media reveals PY-STAT3 (right).
Next, we examined main effectors of BMP2 and CNTF-mediated signal transduction pathways in these cells. The basal levels of PS-SMAD1/5/8 in BMPR1B-overexpressing 0308 cells were similar or slightly higher than the parental cells, however, BMPR1B-overexpressing cells significantly upregulated phosphorylation of STAT3 following exposure to CNTF/LIF (Figures 4B and S7). We then evaluated the effects of BMPR1B over-expression on cytokine-mediated differentiation of 0308 TICs (Figure 4C). After 5 days of culture in the presence of BMP2, BMPR1B over-expressing cells demonstrated a more differentiated morphology than BMP-treated 0308 TICs (Figure 4C). Furthermore, BMP2 induced a dramatic down-regulation in Sox2 and a significant upregulation in GFAP expression in BMPR1B-overexpressing 0308 TICs, similar to the response of adult-derived normal NSCs (Figure 4C). Finally, CNTF treatment of 0308-BMPR1B cells significantly increased the number of GFAP/S100β positive cells consistent with the robust induction of PY-STAT3 in the BMPR1B-treansfected 0308 cells (Figure 4 B to C and Figure S3).
Increased astroglial differentiation potential by BMPR1B overexpression is mediated by PY-STAT3 activation
In order to confirm PY-STAT3 activity following exposure of 0308-BMPR1B cells to CNTF, we performed transcription reporter assays using a synthetic STAT3 responsive promoter (Figure 4D). Indeed, transcriptional activity of PY-STAT3 was significantly increased in 0308-BMPR1B cells following CNTF exposure compared to the parental 0308 cells. Additionally, CNTF treatment activated transcription from a genomic GFAP promoter in 0308-BMPR1B cells, consistent with the known temporal association between phosphorylation and activation of PY-STAT3 and astrocytic differentiation (Figure 4D). It has been reported that CNTF-triggered STAT3 phosphorylation is critical for astrocytic differentiation of normal NSCs(He et al., 2005). To evaluate the importance of STAT3 activity on astrocytic differentiation of 0308 cells, we modulated STAT3 activity by over-expressing either a dominant negative form of STAT3 (STAT3D) or a constitutively active form of STAT3 (STAT3C)(Bromberg et al., 1999). Inhibition of STAT3 activity in BMPR1B-0308 cells decreased the number of GFAP positive cells after CNTF treatment (Figure 4E), suggesting that STAT3 activity is necessary for accelerated astrocytic differentiation. Over-expression of STAT3C alone in 0308 cells, however, did not increase the number of GFAP-positive cells, indicating that additional factors appear to be required for astroglial differentiation of 0308 cells (Viti et al., 2003).
To explore whether overexpression of BMPR1B can cause a general activation of the Jak/STAT pathways leading to glial differentiation or an anti-proliferative effect in any glioma cell line, we overexpressed BMPR1B in a standard U251 glioma non-stem cell line. Like most glial tumor cell lines, U251 cells have high basal PY-STAT3 activity when cultured in serum containing media, however, they did not elicit detectable CNTF-triggered PY-STAT3 activation (Figure 4F)(Haura et al., 2005; Schaefer et al., 2000). Forced expression of BMPR1B in U251 cells did not induce modulation of STAT3 phosphorylation or STAT3 activity after CNTF challenge (Figure 4F). Furthermore, the in vitro growth kinetics and in vivo tumorigenic potential of U251 cells were not changed following overexpression of BMPR1B (data not shown), arguing against the importance of BMPR1B effects on tumor cell growth or differentiation in a non-stem cell setting.
Taken together, these data suggest that BMPR1B over-expression enhances the astrocytic differentiation potential of 0308 TICs in a STAT3-dependent, but not sufficient, manner.
BMPR1B overexpression decreases tumorigenicity of 0308 cells through induction of glial differentiation
Given the phenotypic and functional changes we observed in 0308-BMPR1B cells in vitro, we next tested whether BMPR1B overexpression affects the tumorigenicity and in vivo differentiation potential of 0308 cells by using a neonatal SCID mouse orthotropic tumor model (Figure 5). Mice undergoing intracerebral injection of 0308 cells die within 2–3 months of a diffusely infiltrating glioma histologically identical to human GBMs. By contrast, most mice (5 of 7) injected with 0308-BMPR1B cells did not develop detectable tumors even after 5 months of observations. The brains of mice injected with either control 0308 cells or 0308-BMPR1B cells were examined at various time points by immunohistochemical analysis (Figure 5A). Far fewer intracranial 0308-BMPR1B cells were seen compared to the parental 0308 cells at both 40 and 60 days following intracranial injection (Figure 5 A and B). Distinct tumor masses were detected near the ventricular walls and cortices of mice injected with 0308 cells as early as the 40 day time point (Figure 5A). By contrast, 0308-BMPR1B cells were mainly detected in the ventricles and corpus callosum, and did not form distinct tumor masses (Figure 5A). Immunohistochemical analysis using Sox2 and human specific antibodies revealed a significantly lower number of Sox2-positive human cells in brains injected with 0308-BMPR1B cells, compared to those injected with parental 0308 cells (Figure 5 A and B). In order to quantitatively evaluate the differentiation status of both 0308 and 0308-BMPR1B-derived tumors, we isolated individual intracerebral tumor cells by FACS sorting and determined their level of GFAP expression. Compared to the 0308 cells, a higher percentage of 0308-BMPR1B cells (about 1.7 fold) expressed GFAP and did so with a higher expression intensity (Figure 5C). These findings are consistent with a greater tendency for 0308-BMPR1B cells to undergo spontaneous differentiation in vivo compared to the parental 0308 cells thereby resulting in significantly prolonged survival in mice injected intracerebrally with 0308-BMPR1B cells compared to mice with the parental 0308 cells (Figure 5D).
Figure 5. Overexpression of BMPR1B in 0308 cells decreased tumorigenicity in vivo.

A) Representative immunohistochemistry of tumors derived from 0308 and 0308-BMPR1B cells at 40 days (a–c and f–h) and 60-days (d and e, i and j). Note that GFAP antibody recognizes both human and mouse GFAP proteins. Inset in (e) demonstrates immnuostaining of Sox2 (green) and GFAP (red). DAPI staining (blue) was used to reveal anatomic locations in brains. White bars represent 100 micron.
B) Quantitation of Sox2 positive cells in tumors derived from 0308 and 0308-BMPR1B cells. SVZ and CTX indicate subventricular zones and cortex, respectively. Error bars represent SD.
C) FACS analysis of GFAP expression in 0308 and 0308-BMPR1B-derived tumor cells.
D) Survival of animals injected with 0308 (n = 8) and 0308-BMPR1B cells (n = 7) (log-rank test: P < 0.001).
Expression of BMPR1B in 0308 TICs is silenced by hypermethylation of BMPR1B promoter and restored by demethylation or knockdown of EZH2
In an attempt to elucidate the mechanism responsible for the lack of BMPR1B expression in 0308 TICs, we sequenced both the BMPR1B cDNA and its corresponding exons in genomic DNA. No mutations were detected. We, therefore, explored a potential epigenetic mechanism for the diminished BMPR1B mRNA expression. DNA hypermethylation, especially in CpG islands, is a major epigenetic mechanism utilized by tumor cells to inactivate tumor suppressor genes and/or tumor susceptibility genes(Widschwendter et al., 2007). Intriguingly, it has been demonstrated that DNA methylation is a critical cell-intrinsic determinant of astrocytic differentiation of NSCs in the mouse fetal brain(Takizawa et al., 2001). Forced demethylation/hypomethylation of early embryonic neuroepithelial cells by treatment with 5-aza-2′-deoxycytidine (Aza-Cd) or genetic knockout of DNA methyl transferase 1 (DMNT1) has been shown to induce precocious astrocytic differentiation in the embryonic mouse brain(Fan et al., 2005; Takizawa et al., 2001). The human BMPR1B gene contains distinct CpG islands at the near 5′ transcription start site, which are conserved in other mammalian genomes including mouse. Thus, we explored the possibility of aberrant methylation of the BMPR1B promoter in 0308 cells. Bisulfite modification of genomic DNA and subsequent sequence analysis revealed that, in contrast to those in normal human NSCs and normal human fetal astrocytes, the CpG islands of BMPR1B gene were heavily methylated in 0308-TICs (Figure 6A). Treatment of 0308 cells with Aza-Cd caused demethylation of the BMPR1B promoter and significantly increased expression of BMPR1B mRNA (Figure 6 A and B). Additionally, within 2.5 days of Aza-Cd treatment, CNTF-mediated PY-STAT3 phosphorylation was restored to levels seen in normal NSCs leading to the increased astrocytic differentiation of 0308 TICs (Figure 6C). Similarly, mouse E11 neuroepithelial cells treated with Aza-Cd also demonstrated a dramatic increase in BMPR1B expression, acquired CNTF-mediated STAT3 activation, and underwent precocious astrocytic differentiation (Figure S8). Thus, the biological effects of demethylation seen in 0308 TICs are mirrored by those observed in E11 NSC.
Figure 6. Expression of BMPR1B in 0308-TICs is silenced by hypermethylation of BMPR1B promoter and restored by demethylation or knockdown of EZH2.

A) Methylation sequencing of BMPR1B promoter region in various cells.
a. The diagram depicts the predicted CpG islands near the BMPR1B promoter region (Li and Dahiya, 2002). The transcription start site of BMPR1B (indicated by arrow; designates as 1 in this diagram) is base pair 96036306 on chromosome 4 (Genome Browser). Each CpG nucleotide is depicted as a red vertical line.
b. Methylation status of CpG islands by bisulfite genomic sequencing.
Each circle represents a CpG nucleotide site. Filled circles indicate methylated CpG. Percentage of methylated clones (more than 25% of CpG sites are methylated) was calculated from 16 individual clones for each cell.
B) Real time RT-PCR analysis of BMPR1B expression in 0308 TICs and normal cells. Relative expression level of BMPR1B was evaluated by Cτ method. Error bars represent SD (performed in triplicate).
C) Increased astroglial differentiation of 0308 TICs treated with Aza-Cd.
a. Western blot analysis of PY-STAT3 and PS-SMAD1/5/8 in 0308 cells treated with Aza-Cd. GAPDH was used as a loading control.
b. Representative immunostaining microphotographs of 0308 cells with or withour Aza-Cd treatment. White bar represents 10 micron.
D) Chromatin immunoprecipitation of BMPR1B promoter region in 0308 and 0822-TICs. Antibodies used for immunoprecipitation are shown above. DNA products shown in the lower row served as PCR efficiency control.
E) Expression of BMPR1B and methylation status of its promoter in EZH2-siRNA treated 0308 TICs.
a. Western blot analysis of EZH2 expression in EZH2 siRNA treated 0308 cells. Three days after siRNA treatment, cells were harvested for analysis. Actin was used as a loading control.
b. Real time RT-PCR analysis of EZH2 and BMPR1B mRNA expression in EZH2 siRNA-treated 0308 TICs. Error bars represent SD (performed in triplicate).
c. Methylation status of the BMPR1B promoter region in EZH2 siRNA-treated 0308 cells as determined by a methylation-specific PCR assay. M, methylated; U, unmethylated; C, control PCR primers.
d. Bisulfite genomic sequencing analysis for CpG methylation following knockdown of EZH2. Each line is the result from an individual clone.
The data presented above support the hypothesis that at least one of the mechanisms responsible for the failure of 0308-TIC cells to express BMPR1B involves deregulated methylation/demethylation of the BMPR1B promoter. Interestingly, human embryonic stem (ES) cells also do not express BMPR1B due to Polycomb repressor complex (PRC)-mediated transcriptional repression(Lee et al., 2006b). In addition, recent papers have demonstrated that Enhancer of Zeste homolog 2 (EZH2), a key component of PRC2/3 complex, facilitates CpG methylation of EZH2-target promoters by recruiting DNA methyltransferases (DNMTs) and is highly expressed in various tumors as well as normal stem cells(Valk-Lingbeek et al., 2004; Vire et al., 2006). Indeed, we detected robust expression of EZH2 in all TICs we examined (Figure S9).
In order to determine whether BMPR1B expression in 0308 TICs is under the control of PRC-mediated transcriptional repression similar to human ES cells, we performed chromatin immunoprecipitation (Chip) analysis (Figure 6D). The significant association of RNA polymerase II with the BMPR1B promoter in 0822 cells, but not in 0308 cells, is consistent with the relative expression levels of BMPR1B mRNA in these two cell lines. BMPR1B promoter DNA from 0308 cells, but not from 0822 cells, was co-immunoprecipitated by both anti-EZH2 and anti-DNMT1 antibodies, implicating the possibility of physical association of PRC2/3 complexes and DNMTs with BMPR1B promoter DNA in 0308 cells (Figure 6D). Next, we examined whether this association contributes to the hypermethylation of the BMPR1B promoter and its low expression in 0308 cells by using small interference RNA (siRNA) against EZH2 (Figure 6E). Compared to control siRNA treated cells, EZH2 siRNA treated cells demonstrated a significant increase in BMPR1B expression. Furthermore, bisulfite genomic sequencing revealed that the CpG nucleotides in the BMPR1B promoter region were significantly less methylated in EZH2 siRNA treated 0308 cells compared to control cells, suggesting that the lack of BMPR1B expression in 0308 cells is due to Polycomb-mediated transcriptional repression via CpG methylation of the BMPR1B promoter.
Expression of BMPR1B and methylation status of its promoter in primary human glioblastoma tissues
The above data suggest that functional loss of BMPR1B expression in 0308 TICs may contribute to gliomagenesis through expansion of either a pre-TSC or TSC population via BMP-mediated proliferation and/or block to terminal differentiation through an aberrant Jak/STAT response to CNTF/LIF. In order to address whether this phenomenon occurs in primary glioblastomas in situ, we performed real-time RT-PCR for the purpose of determining the relative frequency of BMPR1B mRNA down-regulation in primary human GBMs (Figure S10). A total of 54 glioblastomas were evaluated, of which 10 (18.5%) demonstrated significant reduction (more than 3 fold) of BMPR1B mRNA expression compared to the non-tumor bearing cerebral cortex samples (n=6). By contrast, 12 samples (about 22 %) of primary tumors showed significant overexpression (more than 3 fold) of BMPR1B compared to normal cortex. We sequenced the BMPR1B cDNAs and corresponding genomic regions from these 12 glioblastomas that did not have BMPR1B down-regulation to order to determine whether any of these DNAs harbor mutations, but found none. Thus, it appears that BMPR1B down-regulation is found in approximately 20% of glioblastomas. Bisulfite genomic sequencing of the BMPR1B promoter region in a set of primary glioblastoma samples demonstrated significant hypermethylation of the promoter region in 6 of 7 tumors with significant reduction of BMPR1B mRNA (85 %). By contrast, non-tumor bearing brain tissues (n=4) and tumors without BMPR1B down-regulation (n=5) did not show any hypermethylation in CpG islands of BMPR1B gene (Figure 7A). Correlation of expression level of BMPR1B and methylation status of its promoter in the various glioblastomas was highly significant (p < 0.01). These data suggest that BMPR1B down-regulation seen in a subset of primary GBMs is mainly due to epigenetic silencing due to CpG methylation in BMPR1B promoter region as was seen in the 0308 TICs. Finally, the median expression of GFAP in glioblastoma specimens with low BMPR1B expression was significantly lower than that seen in all other glioblastomas consistent with a relative block to astroglial differentiation in situ in this subset of glioblastoma as demonstrated in 0308 TICs (Figures 7B and S11).
Figure 7. Methylation status of BMPR1B promoter in human GBMs.
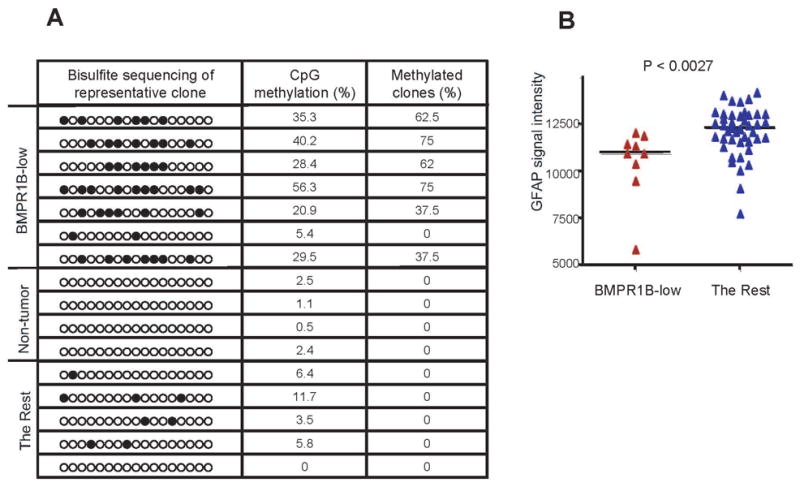
A) Methylation status of the BMPR1B promoter region in human GBMs. Each circle represents CpG nucleotides. The extent CpG methylation (%) and frequency of methylated clones were determined as shown in Figure 6. Eight clones for each sample were sequenced.
B) Correlation between the mRNA expression level of BMPR1B and GFAP in a subset of human GBMs. Expression levels of GFAP were compared between the BMPR1B-low mRNA expression group (defined as GBMs with more than 3 fold-reduction in median BMPR1B expression compared to all other GBMs) and all other GBM tissues. Median GFAP expression value was shown as a horizontal line. (two-tailed t test; p <0.0027).
DISCUSSION
A number of recent reports have identified clonogenic cancer cells with stem-like properties from various primary human epithelial tumors including those of the breast, colon, prostate, pancreas and brain(Liu et al., 2005; O’Brien et al., 2007; Ricci-Vitiani et al., 2007; Singh et al., 2004). These studies have shown that these stem-like cells have clonogenic and tumorigenic capacity as well as having phenotypes and functional properties similar to tissue-specific stem cells. Several studies have also demonstrated that these TSCs have genetic and gene expression patterns similar to those of the primary tumors(Lee et al., 2006a; Taylor et al., 2005). One recent report demonstrated that a few glioma-derived TICs differentiate in response to BMP, similar to what is seen in NSCs, adding to the growing data implicating a stem cell origin for malignant gliomas. Nevertheless, there have been few if any reports demonstrating exactly where along the developmental pathway of tissue-specific stem cell maturation and differentiation TSC arise and which, if any, of the intrinsic stem cell signaling pathways are perturbed in TSC remains largely unknown.
Our data demonstrate that 0308 TICs, although isolated from adult patient, are significantly more similar to early embryonic NSCs (i.e. E11 mouse NSCs) than to later embryonic or adult brain-derived NSCs. These similarities include a transient mitogenic response to BMP ligands, lack of BMPR1B expression secondary to promoter hypermethylation, little or no PY-STAT3 activation following CNTF/LIF exposure, and delayed astroglial differentiation. In contrast to early embryonic NSCs, however, 0308-TICs do not spontaneously acquire BMPR1B expression nor develop responsiveness to CNTF/LIF-mediated STAT3 phosphorylation over time and/or serial passages in vitro. Thus, 0308 TICs appear be perpetually stuck at a developmental point similar to that of an early embryonic NSCs, at least in part, due to aberrant genomic methylation as is seen in other tumors(Bernstein et al., 2007; Jones and Baylin, 2007; Schlesinger et al., 2007). The precise reasons why 0308–TICs cannot proceed through their methylation-associated BMPR1B-dependent developmental block in vitro (and presumably in vivo) is not fully clear. Possibilities include aberrations in oncogenic pathways implicated in both developmental and glioma biology (e.g. EGFR, PTEN, and mTOR) or other stem cell pathways (Kamakura et al., 2004). Potential interactions between these fundamental pathways clearly warrant further investigation. Regardless of the mechanism, however, lack of BMPR1B expression appears to be mechanistically responsible for the failure of 0308 TICs to efficiently differentiate. Support for this contention is based on our observations that over-expression of BMPR1B cDNA in 0308 cells leads to decreased BMP2-mediated mitogenic effects, STAT3 responsiveness to CNTF/LIF exposure, increased astroglial differentiation and loss of TIC tumorigenicity in vivo.
CNTF signaling pathways are believed to be restricted to astroglial differentiation in brain cells. As has been previously reported and demonstrated here, early embryonic NSCs are less responsive to CNTF/LIF-mediated STAT3 activation. Although there is debate regarding the relative amount of STAT3 activation in early NSCs(He et al., 2005; Molne et al., 2000; Takizawa et al., 2001), a clear increase in the amount of STAT3 activation either by modulation of DNA demethylation, down-regulation of specific neurogenic transcription factors, and/or a feed-forward STAT3 activation signaling loop seems to be required for the onset of astrogenesis. We have shown that CNTF/LIF-mediated STAT3 activation is impaired in 0308-TICs and can be restored by overexpression of BMPR1B, suggesting a causal role of BMPR1B induction in the Jak/STAT pathways and astroglial differentiation. Further studies are ongoing in order to elucidate the underlying molecular mechanisms between BMP and Jak/STAT signaling in early embryonic NSCs.
STAT3 activation is critical for maintenance of stem cell function, differentiation, and survival(Levy and Darnell, 2002). Over-activation of STAT3 has been reported to be oncogenic in various model systems and is frequently observed in various tumor types(Bromberg et al., 1999). For example, tyrosine phosphorylation-mediated STAT activation can be induced by EFGR/Src, CNTF/LIF, and ligand-independent Jak2 activation(Bromberg et al., 1998). Notch and mTOR pathways also have been reported to activate this pathway(Hu et al., 2005; Kamakura et al., 2004). By contrast, our data suggest that although not sufficient, a relative increase in the level and/or timing of STAT3 activation by CNTF is necessary for astroglial differentiation of 0308 TICs. Thus, STAT3 activation can be cytostatic as well as pro-proliferative. The likely explanation for this seemingly paradoxical behavior of STAT3 activation is that the STAT pathway is context-dependent and various intracellular as well as environmental cues play a pivotal role in determining the outcome of pathway activation. In addition, a recent report has demonstrated that STAT3 serine phosphorylation in addition to, or in lieu of, tyrosine phosphorylation is critical to stem cell survival, further emphasizing the complexity of this pathway(Androutsellis-Theotokis et al., 2006). Finally, previous studies have largely relied on traditionally grown and established cancer cell lines where STAT3 and other signaling pathways may have very different functions than they do in primary human tumor-derived TICs and NSCs(Schaefer et al., 2000).
One unresolved, yet intriguing, question is precisely how and to what extent the BMPR1B-associated developmental block contributes to tumorigenesis. It had been once believed, based largely on the leukemia model of cancer, that all tumor cells were dedifferentiated cells with complete clonogenic capacity. A block in differentiation was, therefore, essentially the cause of the cancer resulting in uncontrolled proliferation of each and every cell (i.e. the tumor was essentially a mass of accumulating tumor stem/initiating cells). It is increasingly clear, however, that in epithelial cancers like glioblastoma, TSC/TICs generally represent a minor component of the bulk of tumor tissue with the majority of tumor consisting of more differentiated progeny of TSC/TICs with limited clonogenicity (as was the case for the tumor from which the 0308 cells were derived from; data not shown). Thus, a relative block in capacity for TSC differentiation, such as seen in the 0308 cells, could contribute to tumorigenesis through the maintenance of a non-differentiated and hence self-renewing TSC population readily available to generate progeny that make up the bulk of the tumor under conditions that would otherwise lead to differentiation of that TSC. An alternative explanation, however, is that the inhibition of BMPR1B expression in 0308 TICs and a subset of primary glioblastomas may have less to do with blocking terminal differentiation and more to do with expansion of the clonogenic stem cell population through the mitogenic effects of BMP2/4, as we have demonstrated in 0308 TICs. This is certainly plausible given the well-documented presence of BMP 2/4 throughout the normal and injured adult brain. This is consistent with work by others and us demonstrating that malignant gliomas express higher levels of BMP2/4 than lower grade gliomas or normal cerebral tissue, thereby, potentially setting up an autocrine feedback loop for BMP-mediated TSC/TIC proliferation (Liang et al., 2005)(data not shown). Further work is required to determine the relative contribution of these two, non-mutually exclusive, mechanisms of TSC/TIC-mediated tumorigenesis.
Epigenetic changes such as hypermethylation of CpG islands in promoter regions and altered profiles of histone modification are now recognized to be likely major contributors to tumorigenesis in a number of different cancers(Schlesinger et al., 2007; Trojer and Reinberg, 2006). Polycomb group (PcG) proteins are essential components by which stem cells reversibly repress genes related to differentiation(Sparmann and van Lohuizen, 2006). Recently, it has been reported that PcG target genes are much more likely to have promoter hypermethylation in cancer than non-target genes(Widschwendter et al., 2007). We have shown that hypermethylation of the BMPR1B gene in 0308 cells and a subset of primary glioblastomas is mechanistically linked to EZH2, a key component of PRC2/3 complex. Interestingly, the BMPR1B gene is a PRC target gene and is transcriptionally repressed in undifferentiated human ES cells. These observations, taken together with the data presented here, lead us to hypothesize that 0308 TICs and a corresponding subset of primary glioblastomas have an aberrant NSC developmentally regulated demethylation program that results in the failure to induce BMPR1B expression and leads to a selective block in TSC/TIC/stem cell differentiation. This is entirely consistent with the idea that the normal, developmentally-regulated, reversible repression of differentiation-inducing genes is replaced by their permanent silencing in cancers, thereby favoring a perpetual stem cell state(Widschwendter et al., 2007).
We propose that lack of BMPR1B expression in a tumor stem cell population contributes to the differentiation block in a subset of primary human glioblastomas. We hypothesize that other glioblastomas likely have different aberrant differentiation/proliferation pathways unique to their corresponding TIC populations. Therefore, we do not necessarily believe that BMPR1B-deficient GBMs are “more tumorigenic” than other GBMs with normal or high levels of BMPR1B or that BMPR1B expression has a particular prognostic significance. Rather, the identification of a subset of glioblastomas that has this particular differentiation block will be of great utility for further understanding glioma pathophysiology, cancer stem cell biology and for ultimately designing rationale therapeutic strategies targeted to tumors with the appropriate biology. One example is our finding that Aza-Cd-mediated demethylation of the BMPR1B promoter leads to up-regulation of BMPR1B and terminal astrocytic differentiation of 0308 TICs. Since Aza-Cd is currently being evaluated in clinical trials of various tumors, as are other demethylating and chromatin modulating agents, our data demonstrate that such agents may be particularly useful for patients with glioblastomas harboring hypermethylation-mediated transcriptional repression of BMPR1B. These observations therefore identify BMPR1B as a promising molecular therapeutic target in a subset of glioblastomas worthy of further development. Likewise, the suggestion that BMP2/4 might be explored as a potential therapeutic agent, through its ability to induce differentiation in some glioma-derived TICs, must be viewed with caution given our demonstration of the mitogenic effects of BMPs in tumors with repressed BMPR1B expression(Piccirillo et al., 2006).
In summary, we have identified a human glioblastoma TSC/TIC, representative of a subset of primary human GBMs, that appears to be very similar to a very early embryonic NSC but is apparently blocked from further stem cell development and differentiation though an aberration in the BMP signaling pathway. Forced expression of methylated-promoter-repressed BMPR1B restores normal differentiation capacity of the TSC/TICs halting further proliferation and inducing terminal differentiation of the TSC/TICs. We provide here an example of a temporally deregulated and aberrantly fixed normal stem cell developmental block to differentiation contributing to the pathogenesis of a human tumor. Not only will such insights pave the way for a more thorough understanding of TSC biology, but they also identify BMPR1B as a promising molecular target and open the potential for targeted therapeutic approaches for agents that can induce TSC terminal differentiation.
MATERIALS AND METHODS
Human and mouse tissues
Following informed consent, tumor samples classified as glioblastoma based on World Health Organization (WHO) criteria were obtained from patients undergoing surgical treatment at the NIH in accordance with the appropriate Institutional Review Boards(Kleihues et al., 2002). Procedures regarding derivation of TICs were described previously(Lee et al., 2006a). Time-pregnant Swiss-Webster mice were used to isolate NSCs from various gestational stages. NSCs isolated from E10 to E11 embryos were cultured in the presence of FGF2 (10 ng/ml) without EGF. All mice experiments were performed according to NIH guidelines of the Animal Use and Care Committees.
Antibodies for immunohistochemistry and Western blot analysis
The following antibodies were used: GFAP, S100β (Dako), TuJ1 (Covance), BrdU (Roche), STAT3 (Upstate), PY705-STAT3, PS727-STAT3, PS-SMAD1/5/8, SMAD1 (Cell signaling), BMPR1A, BMPR1B, Sox2 (R&D systems), EZH2 (Cell signaling and Upstate), β-actin (Santa Cruz Biotechnology), and Human ribonucleoprotein (1281, Chemicon).
Intracranial tumor cell injection into SCID mice
An intracranial orthotropic model was utilized for evaluation of TIC tumorigenicity(Uchida et al., 2000). 0308 cells or 0308-BMPR1B-cells were dissociated and resuspended in 2 μl of HBSS. One hundred thousand cells, as determined by trypan blue dye exclusion, were injected stereotactically into the lateral ventricles of cryoanesthetized neonatal SCID mice at postnatal day 1. Following injection, neonatal mice were returned to their mother and allowed to grow to adulthood. There was no injection procedure-related animal lethality. The animals were killed at given time points for the analysis of tumor histology and immunohistochemistry. Brains were perfused with 4% paraformaldehyde by cardiac perfusion and further fixed at 4°C overnight.
Chromatin immunoprecipitation
Chip assays were performed by manufacturer’s protocol (Chip-It Express, Active Motif) with enzymatic DNA shearing. Antibodies used for Chip assay include EZH2 (Upstate) and DNMT1 (Active Motif). For negative and positive control of Chip assay, a RNA polymerase II antibody and a negative control IgG antibody were used (Chip-IT control kit).
Bisulfite DNA modification and PCR sequencing
Bisulfite DNA modification was essentially performed by manufacturer’s instruction (Chemicon and Active Motif). After PCR using chemically modified DNA as templates, PCR products were ligated into TOPO PCR cloning vector (Invitrogen) and then used for transformation of E. Coli. The following PCR primers were used: 5′-GGAGGTTTTTTGGAAATTTTTTTAA-3′ and 5′-CCTCTCCAACTCCCACTAATAATTA-3′ (for human BMPR1B); 5′-AGAGTTTTGAGTAAGTGTGAGGGTTA-3′ and 5′-AAAAAAAATTTCCTTTTAAAACAAAAATAC-3′ (for mouse BMPR1B).
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We are grateful to Dr. Eugene Major for providing human fetal neural progenitor cells, and Dr. David Panchision and Dr. James Darnell for providing BMPR1B and STAT3C plasmids. We also thank NINDS sequencing facility and NINDS light image facility for their valuable contributions to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol. 1998;18:2553–2558. doi: 10.1128/mcb.18.5.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Chen HL, Panchision DM. Concise review: bone morphogenetic protein pleiotropism in neural stem cells and their derivatives--alternative pathways, convergent signals. Stem Cells. 2007;25:63–68. doi: 10.1634/stemcells.2006-0339. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer Stem Cells--Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, et al. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development. 2005;132:3345–3356. doi: 10.1242/dev.01912. [DOI] [PubMed] [Google Scholar]
- Hall AK, Miller RH. Emerging roles for bone morphogenetic proteins in central nervous system glial biology. J Neurosci Res. 2004;76:1–8. doi: 10.1002/jnr.20019. [DOI] [PubMed] [Google Scholar]
- Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol. 2005;2:315–324. doi: 10.1038/ncponc0195. [DOI] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Oishi K, Yoshimatsu T, Nakafuku M, Masuyama N, Gotoh Y. Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat Cell Biol. 2004;6:547–554. doi: 10.1038/ncb1138. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. discussion 226–219. [DOI] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006a;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006b;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- Liang Y, Diehn M, Watson N, Bollen AW, Aldape KD, Nicholas MK, Lamborn KR, Berger MS, Botstein D, Brown PO, Israel MA. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A. 2005;102:5814–5819. doi: 10.1073/pnas.0402870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Zhang ZY, Song YC, Qiu KJ, Zhang KC, An N, Zhou Z, Cai WQ, Yang H. SVZa neural stem cells differentiate into distinct lineages in response to BMP4. Exp Neurol. 2004;190:109–121. doi: 10.1016/j.expneurol.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci. 2000;22:74–85. doi: 10.1159/000017429. [DOI] [PubMed] [Google Scholar]
- Molne M, Studer L, Tabar V, Ting YT, Eiden MV, McKay RD. Early cortical precursors do not undergo LIF-mediated astrocytic differentiation. J Neurosci Res. 2000;59:301–311. doi: 10.1002/(sici)1097-4547(20000201)59:3<301::aid-jnr3>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 2001;15:2094–2110. doi: 10.1101/gad.894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Schaefer LK, Menter DG, Schaefer TS. Activation of stat3 and stat1 DNA binding and transcriptional activity in human brain tumour cell lines by gp130 cytokines. Cell Signal. 2000;12:143–151. doi: 10.1016/s0898-6568(99)00077-7. [DOI] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell. 2001;104:365–376. doi: 10.1016/s0092-8674(01)00224-0. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell. 2001;1:749–758. doi: 10.1016/s1534-5807(01)00101-0. [DOI] [PubMed] [Google Scholar]
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125:213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Viti J, Feathers A, Phillips J, Lillien L. Epidermal growth factor receptors control competence to interpret leukemia inhibitory factor as an astrocyte inducer in developing cortex. J Neurosci. 2003;23:3385–3393. doi: 10.1523/JNEUROSCI.23-08-03385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.