

Abstract
Telomeres are essential for preserving chromosome integrity during the cell cycle and have been specifically implicated in mitotic progression, but little is known about the signaling molecule(s) involved. The human telomeric repeat binding factor protein (TRF1) is shown to be important in regulating telomere length. However, nothing is known about its function and regulation during the cell cycle. The sequence of PIN2, one of three human genes (PIN1-3) we previously cloned whose products interact with the Aspergillus NIMA cell cycle regulatory protein kinase, reveals that it encodes a protein that is identical in sequence to TRF1 apart from an internal deletion of 20 amino acids; Pin2 and TRF1 may be derived from the same gene, PIN2/TRF1. However, in the cell Pin2 was found to be the major expressed product and to form homo- and heterodimers with TRF1; both dimers were localized at telomeres. Pin2 directly bound the human telomeric repeat DNA in vitro, and was localized to all telomeres uniformly in telomerase-positive cells. In contrast, in several cell lines that contain barely detectable telomerase activity, Pin2 was highly concentrated at only a few telomeres. Interestingly, the protein level of Pin2 was highly regulated during the cell cycle, being strikingly increased in G2+M and decreased in G1 cells. Moreover, overexpression of Pin2 resulted in an accumulation of HeLa cells in G2+M. These results indicate that Pin2 is the major human telomeric protein and is highly regulated during the cell cycle, with a possible role in mitosis. The results also suggest that Pin2/TRF1 may connect mitotic control to the telomere regulatory machinery whose deregulation has been implicated in cancer and aging.
Telomeres, the ends of eukaryotic chromosomes, are specific DNA-protein complexes that are essential for preserving the integrity of chromosomes during the cell division cycle (1, 2). Telomeres in most normal human cells undergo programmed shortening both in the soma and in culture following each cell division (3, 4). The striking correlation between telomere shortening and cellular aging led to the telomere hypothesis of aging (3). In contrast, in immortalized and transformed cells, telomeres are restored and maintained through the activity of telomerase, a reverse transcriptase-related enzyme that synthesizes the telomeric DNA repeat using an RNA template (1), as well as by other mechanisms such as recombination, with telomere length being regulated in part by telomeric DNA-binding proteins (5–7). These results point to an important role of telomere maintenance in controlling cell growth.
Interestingly, several observations suggest a link between telomeres and mitotic progression. Elimination of a telomere causes a Rad9p-mediated cell cycle arrest in G2 in budding yeast (8). Telomeres mediate the attachment of chromosomes to spindle bodies and lead chromosome movement in meiotic prophase in fission yeast (9). Furthermore, mutations in the yeast TEL1 gene result in shortened telomeres (10) and mutations in its human counterpart, the ATM gene, causes ataxia-telangiectasia in humans, displaying a wide range of abnormalities including tumors and premature aging that may be related to telomere dysfunction (11). More interestingly, cell lines derived from ataxia-telangiectasia patients have shortened telomere lengths (12) and a defect in G2/M checkpoint control (13, 14). Finally, mutations in the Tetrahymena telomeric DNA sequence cause a block in mitosis (15). Collectively, these results suggest that there is a telomere-mediated checkpoint that regulates progression through mitosis. However, little is known about the identity and function of the signaling molecule(s) involved in this process.
The NIMA (never-in-mitosis A) protein kinase is essential for mitosis in Aspergillus nidulans (16, 17) and a similar NIMA-like mitotic pathway also exists in vertebrate cells (18). Using a modified yeast two-hybrid screen, we have previously (19) cloned three human genes, PIN1–3, encoding proteins that not only interact with NIMA but also suppress the NIMA-induced lethal phenotype in yeast. Pin1 encodes a peptidyl-prolyl isomerase that is essential for mitosis and is conserved from yeast to man (19). Recent structural analysis of Pin1 (20) and characterization of Pin1 function and downstream targets (M.S., P. T. Stukenberg, M. W. Kirschner, and K.P.L., unpublished work; ref. 22) indicate that Pin1 is a sequence-specific and phosphorylation-dependent peptidyl-prolyl isomerase that specifically binds and regulates the activity of a subset of conserved mitotic phosphoproteins, including such important mitotic regulators as NIMA, Cdc25, Myt1, Wee1, Plk1, and Cdc27. Thus, Pin1 acts as a regulator of substrates for Cdc2 and other mitotic kinases.
Here we describe the characterization and cell cycle analysis of the PIN2 gene and its product. PIN2 (29) encodes a protein that was later found to be related to the human telomeric repeat binding factor protein (TRF1)/Orf1 (23, 24). Pin2 is identical to TRF1 except for a 20 amino acid internal deletion, suggesting that they may be generated by alternative splicing from the same gene, PIN2/TRF1. However, Pin2 is much more abundant than TRF1 in the cell. Although TRF1 has been shown to regulate telomere length in telomerase-positive cells (6), little is known about its regulation and cell cycle function. Furthermore, nothing is known about telomeric proteins in telomerase-negative cells. We found that Pin2 and TRF1 form homo- and heterodimers and that both forms are uniformly distributed at telomeres in telomerase-positive cells, but are highly concentrated only at a few telomeres in telomerase-negative cells. Interestingly, the levels of both endogenous and exogenously expressed Pin2 fluctuate during the cell cycle, being significantly elevated in G2 and M and decreased in G1. Furthermore, overexpression of Pin2 in HeLa cells results in accumulation of cells in G2+M. These results demonstrate for the first time that a major telomeric protein is tightly regulated during the cell cycle and suggest that Pin2/TRF1 may be important in mitotic regulatory events involving telomere integrity.
MATERIALS AND METHODS
Yeast Two-Hybrid Screen and cDNA Isolation.
Out of thirteen positive clones that were identified from a HeLa cell cDNA library in a yeast two-hybrid screen using the Aspergillus nimA cDNA bait construct, three independent clones encoded the N-terminal fragment of PIN2 (19). The full-length PIN2 sequence was isolated from two HeLa cell cDNA libraries independently constructed by R. Fukunaga and S. Hanks (The Salk Institute). TRF1 was generated by inserting the missing 60 nucleotides into PIN2 by PCR, followed by sequencing confirmation.
Expression and Purification of Recombinant Pin2 Proteins.
To express (His)6 tagged and glutathione S-transferase fusion proteins, the full-length and the N-terminal 337 amino acids of Pin2 were subcloned into pET28a and pGEX-KG, respectively. Recombinant proteins were expressed in and purified from bacteria as described previously (19). To produce Pin2 protein in insect cells, His-Pin2 was subcloned into a baculoviral expression vector, pFastBacI and transfected into Sf9 cells; recombinant His-Pin2 protein was purified on a Ni2+-NTA agarose column, as described by the manufacturer (GIBCO).
Gel Retardation Assay.
The gel retardation assay for determining the binding between Pin2 and telomeric DNA was carried out as described (25). Briefly, a double-stranded oligonucleotide containing six repeats of TTAGGG was synthesized and labeled, followed by incubation with purified recombinant His-Pin2 in 20 μl of buffer containing 20 mM Hepes (pH 7.9), 150 mM KCl, 1 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 5% glycerol, 4% Ficoll, 2 μg poly dI:dC, RNA, or salmon sperm DNA. To some reactions casein (final concentration 1 mg/ml) and/or the unlabeled double-stranded oligonucleotides, DNA fragments, or RNA were added. The mixture was separated on a 5% polyacrylamide gel, followed by autoradiography.
Anti-Pin2 Antibody Production and Purification, Immunoprecipitation and Immunoblot Analysis.
To make antibodies against Pin2, rabbits were immunized with the purified glutathione S-transferase-Pin2 (1–337) fusion protein, as described (19). Anti-Pin2 antibodies were affinity purified on glutathione S-transferase-Pin2 coupled to glutathione-agarose using m-maleimidobenzoyl-N-hydroxysuccinimide ester as described (26). To detect protein-protein-interactions in the cell, tTA-1 cells were transfected using lipofectamine (GIBCO) with hemagglutinin (HA)-tagged Pin2 and FLAG-tagged TRF1 expression vectors and harvested 36 h after transfection. Cells were lysed in buffer containing 1% Triton X-100 and subjected to immunoprecipitation using mAb 12CA5, followed by immunoblot analysis using mAb M2, as described (19).
DNA Transfection and Immunofluorescence Microscopy.
To express epitope-tagged Pin2 or TRF1 in mammalian cells, PIN2 and TRF1 cDNAs were subcloned in pUHD-P1 or pUHD-P2, which contain a FLAG or HA tag, respectively, as described (18, 19). The Pin2(1–301) expression vector was the same as the full-length vector, but with a single bp deletion; Pin2(1–376) was generated by digesting Pin2/pUHD-P2 with BglII and XbaI, followed by self-ligation. To generate stable cell lines, HA-Pin2 was subcloned into pcDNA4 (Invitrogen) and transfected into a spontaneously immortalized human fibroblast cell lines MDAH041 and MDAH087 from different patients with the Li–Fraumeni cancer syndrome (27). Although most clones stopped growing after G418 selection, a few continuously growing clones were isolated. To determine the localization of Pin2 and TRF1 on mitotic chromosomes, cells were arrested in M with colcemid (5 μg/ml) for 1 h and mitotic cells were collected, followed by spreading chromosomes onto coverslips using cytocentrifugation and subsequent immunostaining. Transfection of the tTA-1 cell line and indirect immunofluorescence and confocal microscopy of the transfected cells were performed as described (18).
Determination of Pin2 Levels and the Effect of Pin2 on the Cell Cycle.
For analysis of Pin2 during the cell cycle, fibroblasts were incubated with 10 μM lovastatin to synchronize cells in G1, as described previously (28). After 24 h, the cells were released from the arrest by the addition of 1 mM mevalonic acid to fresh media. The effect of overexpression of Pin2 on tTA-1 cell cycle distribution was determined as described (18).
RESULTS
Identification of the Human PIN2 Gene.
PIN2, one of three classes of human cDNA clones identified in a yeast two-hybrid screen for proteins interacting with the NIMA mitotic kinase (19), encodes a 419 amino acid protein (Fig. 1A). The predicted amino acid sequence contains an N-terminal D+E-rich acidic domain, a C-terminal Myb-type helix-turn-helix (HTH) DNA-binding domain (29), a nuclear localization signal and a D-like motif (RIFGDPN) that is similar but not identical to destruction boxes (RXXXGDXXN) present in mitotic cyclins (ref. 30, Fig. 1). Pin2 was later found to be identical to TRF1 (23) with the exception that residues 296–316 in TRF1 are missing in Pin2 (Fig. 1B). Although the exact relationship between Pin2 and TRF1 remains to be determined by genomic sequencing, they could be generated by alternative splicing from the same gene PIN2/TRF1. In the following text, we have used Pin2 to refer to the smaller protein containing the internal 20 amino acid deletion and TRF1 to refer to the larger protein.
Figure 1.
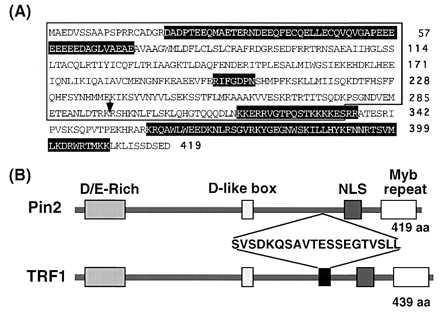
Amino acid sequence of Pin2 and its relationship with TRF1. (A) The predicted Pin2 peptide sequence is indicated in the one-letter code, with the arrow indicating the site at which the additional 20 amino acids are present in TRF1. Boxes with white letters from left to right indicate an acid (D/E)-rich domain, a D-like box, a bipartite nuclear localization signal and a Myb-type HTH DNA-binding domain. (B) Schematic presentation of the domain structure of the Pin2 and TRF1 proteins and sequence of the 20 amino acid insert in TRF1.
Pin2 Is More Abundant than TRF1 in the Cell.
To determine whether both Pin2 and TRF1 were expressed in the cell, and, if so, at what ratio, we measured their mRNA and protein levels. Using two sets of primers common to both PIN2 and TRF1 and RNAs isolated from several different cell lines as templates for reverse transcription–PCR reaction, we obtained two major PCR products, whose sizes and nucleotide sequences were the same as those predicted from the PIN2 and TRF1 cDNA sequences (Fig. 2A, and data not shown). Interestingly, the PIN2 PCR product was always much more abundant than TRF1 product in all cells we tested, with the ratio being ≈5–10 to 1. These results indicate that both Pin2 and TRF1 are indeed expressed in vivo and also suggest that Pin2 is expressed at a much higher level than TRF1. To confirm that Pin2 was indeed the major product in the cell, we raised polyclonal anti-Pin2 antibodies against the glutathione S-transferase-Pin2 (1–337) fusion protein. When used for immunoblot analysis after immunoprecipitation, these antibodies specifically recognized a protein doublet with apparent sizes of ≈61 and 63 kDa, respectively, that were not detected using preimmune serum (Fig. 2B). Again, the level of the 61-kDa form was 5–10-fold higher than that of the 63-kDa protein in several other human cell lines (Fig. 2B and data not shown). The small ≈2 kDa difference in the size of the two proteins in the doublet is consistent with the 20 amino acid difference between Pin2 and TRF1. This was further confirmed by transfecting HeLa cells with Pin2 and TRF1 cDNAs, followed by immunoblot analysis (Fig. 2C). These results collectively indicate that Pin2 is much more abundant in the cell than TRF1.
Figure 2.
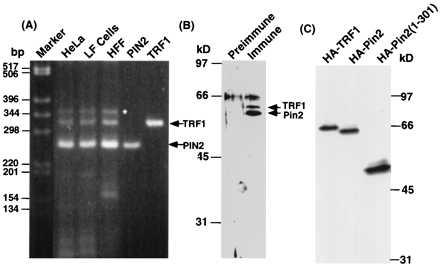
Comparison of Pin2 and TRF1 proteins and their expression levels in the cell. (A) The mRNA level was determined by reverse transcription–PCR using total RNAs isolated from various cell lines as indicated and a set of primers that are common for both PIN2 and TRF1 (outside the deleted amino acid residues 296–316 of TRF1). The two major PCR products were confirmed by direct sequencing to be a fragment of PIN2 and TRF1. The minor larger PCR product (∗) was found to be the same as the TRF1 fragment, but with a tandem-duplicated primer at the 3′ end. (B) The levels of Pin2 and TRF1 protein were determined by immunoprecipitation with the anti-Pin2 serum or preimmune serum, followed immunoblot analysis using anti-Pin2 antibodies. (C) PIN2 and TRF1 expression constructs with an N-terminal HA tag were transiently transfected into tTA-1 cells for 24 h. One hundred micrograms of lysates were subjected to immunoblot analysis using anti-Pin2 antibodies.
Pin2 Binds Telomeric DNA and Forms Homodimers and Heterodimers with TRF1.
To determine whether Pin2 is a telomeric DNA-binding protein, we tested for direct binding of Pin2 protein to human telomeric repeat DNA (Fig. 3A). When purified His-Pin2 protein was incubated with a 32P-labeled double-stranded hexameric telomeric repeat oligonucleotide, Pin2 bound the oligonucleotide, as shown by gel shift analysis. This binding was completely abolished by a 30-fold excess of either unlabeled telomeric DNA repeat oligonucleotide or a 780-bp human telomeric DNA fragment (31), but not by RNA (Fig. 3A, and data not shown). Furthermore, the ability of Pin2 to bind the oligonucleotide was significantly enhanced by including casein in the reaction (Fig. 3A), as is the case for TRF1 (23). These results, together with localization of Pin2 at telomeres (Fig. 4), indicate that Pin2 is a telomeric DNA-binding protein.
Figure 3.
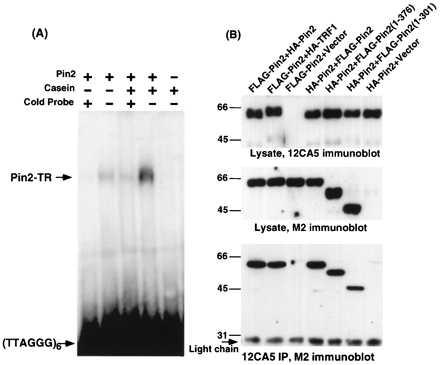
Pin2 is a telomeric DNA-binding protein that forms homo- and heterodimers with TRF1. (A) The purified recombinant Pin2 protein was incubated with a [32P]-labeled double-stranded oligonucleotide containing six human telomeric DNA repeats, in the presence or absence of a 30-fold excess of unlabeled oligonucleotide and 1 mg/ml casein. The samples were separated on a 5% polyacrylamide gel, followed by autoradiography. (B) tTA-1 cells were cotransfected for 36 h with two different epitope-tagged Pin2 and/or TRF1 expression constructs, as indicated. Cells were lysed in lysis buffer containing 1% Triton X-100. Aliquots of the cellular proteins were directly subjected to SDS/PAGE, followed by immunoblot analysis using the tag-specific 12CA5 or M2 mAb. The remainder of the lysates were first immunoprecipitated with the 12CA5 mAb and then subjected to immunoblot analysis using the M2 mAb.
Figure 4.
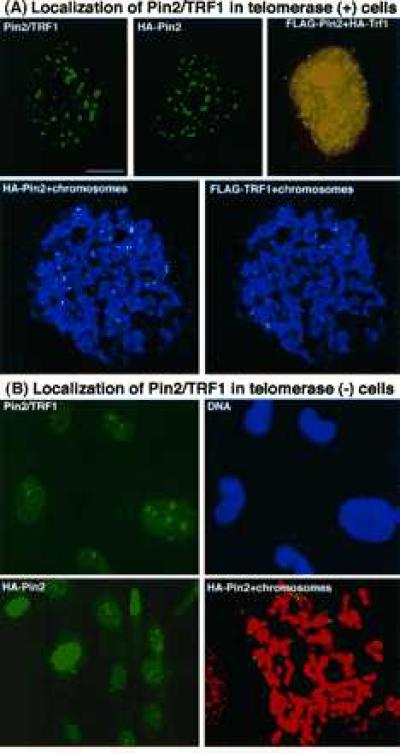
Differential localization of Pin2 is in telomerase-positive and -negative cells. (A) Localization of Pin2 protein in telomerase-positive HeLa cells. Pin2/TRF1, localization of the endogenous Pin2/TRF1 detected by immunostaining with anti-Pin2 antibodies; HA-Pin2, localization of the ectopically expressed Pin2 detected by staining with the 12CA5 mAb; HA-Pin2 and FLAG-TRF1, colocalization of expressed Pin2 and TRF1 detected by staining doubly transfected cells with 12CA5 and M2 mAbs, with the yellow image indicating a colocalization produced by superimposing the green Pin2 and the red TRF1 images; HA-Pin2/FLAG-TRF1+chromosomes, colocalization of expressed Pin2 and TRF1 evenly at telomeres detected by staining mitotic chromosomes from doubly transfected cells with 12CA5 and M2 mAbs. Green, HA-Pin2; red, FLAG-TRF1; blue, chromosomes. (B) Localization of Pin2 protein in telomerase-negative MDAH087 cells. Pin2/TRF1, localization of the endogenous Pin2/TRF1 detected by staining with anti-Pin2 antibodies (green) and the DNA dye Hoechst (blue); HA-Pin2, localization of the ectopically expressed Pin2 detected by staining a single stably HA-Pin2-expressing cell line (clone 6) with the 12CA5 mAb; HA-Pin2+chromosomes, localization of expressed Pin2 at telomeres detected by staining clone 6 mitotic chromosomes with the 12CA5 mAb. Green, HA-Pin2; red, chromosomes.
The crystal structure of the yeast telomeric protein Rap1p reveals that both its HTH domains interact with the telomeric DNA (32). In contrast, Pin2 and TRF1 contain only a single HTH domain. One way to provide two HTH domains would be through dimerization. To examine this possibility, we examined in vivo protein-protein interaction using coimmunoprecipitation and colocalization. The Pin2 and TRF1 cDNAs were tagged with different epitopes and transfected into HeLa cells. Immunoprecipitation and immunoblot analysis showed that Pin2 was present in TRF1 immunoprecipitates, and vice versa (Fig. 3B). Furthermore, immunostaining showed that the expressed Pin2 was colocalized with TRF1 in both interphase and mitotic HeLa cells (Fig. 4A). These results indicate that Pin2 interacts with TRF1 at telomeres in the cell. In addition, in tTA-1 cells transiently coexpressing two different tagged forms of Pin2 or TRF1, HA-Pin2 was detected in FLAG-Pin2 immunoprecipitates and HA-TRF1 was detected in FLAG-TRF1 immunoprecipitates, indicating homooligomerization (Fig. 3B and data not shown). Deletion analysis revealed that the oligomerization domain is located at the N terminus, and minimally contains residues 1–301 (Fig. 3B). His-Pin2 protein expressed in insect cells was found to be a dimer by both glutaraldehyde crosslinking and gel filtration (data not shown). Collectively, these results indicate that Pin2 and TRF1 form homo- and heterodimers.
Differential Localization of Pin2/TRF1 in Telomerase-Positive and -Negative Cells.
We examined the subcellular localization of endogenous Pin2 and ectopically expressed HA-Pin2 using affinity-purified anti-Pin2 antibodies and the 12CA5 mAb. Both proteins displayed an evenly speckled, exclusively nuclear staining pattern in interphase HeLa cells (Fig. 4A), which was not observed with antibodies preincubated with the cognate antigen (data not shown). Although Pin2 antibodies also reacted with TRF1, the staining pattern of the endogenous proteins should mainly represent Pin2, because Pin2 was expressed at a 5–10-fold higher level than TRF1 (Fig. 2). These results indicate that both endogenous and exogenous Pin2 proteins are primarily concentrated at distinct subnuclear loci.
To confirm that the Pin2 speckles truly reflect a telomeric localization, HeLa cells were cotransfected with HA-Pin2 and FLAG-TRF1, followed by staining with tag-specific mAbs (19). HA-Pin2 was colocalized with FLAG-TRF1 in an evenly speckled pattern in interphase cells and uniformly at all telomeres on mitotic chromosomes of HeLa cells (Fig. 4A). A similar Pin2 distribution pattern was also observed in 293 cells, and MDAH041 cells derived from a patient with the Li-Fraumeni cancer syndrome (data not shown). All these cells contain readily detectable telomerase activity and have chromosomes with rather uniform telomere length (data not shown). Thus, Pin2, like TRF1 (23), is evenly distributed at telomeres in telomerase-positive cells.
Many immortalized cells contain no detectable telomerase activity and their telomeres are highly heterogeneous in length (33–36). However, nothing is known about where Pin2 or TRF1 is localized in these cells. To address this question, we used three human diploid fibroblast cell lines, MDAH087 (27), KMST6 (37), and SUSM-1 (38), immortalized by different procedures. These cells contain heterogeneous telomeres with a few being abnormally long, and have undetectable telomerase as determined by the TRAP assay (ref. 36; C.H., O. Glebov, and M.V., unpublished data). The staining pattern of Pin2/TRF1 in these three cell lines was very similar, being highly concentrated only at a few (3–6) spots (Fig. 4B), and was strikingly different from that in telomerase-positive cells (Fig. 4A)
To confirm that these distinct immunostaining patterns in telomerase-positive and -negative cells indeed represent Pin2, we isolated clones from two representative cell lines, MDAH041 (telomerase-positive)(2 clones) and MDAH087 (telomerase-negative) (3 clones), that stably expressed HA-Pin2 driven by a cytomegalovirus promoter at levels ≈5-fold higher than the endogenous Pin2 level. Stably expressed HA-Pin2 displayed an evenly speckled pattern in MDAH041 cells, but was highly concentrated at only a few nuclear spots in MDAH087 cells (Fig. 4B, and data not shown). The localization of HA-Pin2 in mitotic chromosomes from MDAH087 cells further confirmed that Pin2 was highly concentrated at only a few telomeres, although it was detectable at most telomeres (Fig. 4B). These results indicate that in sharp contrast to telomerase-positive cells, in these telomerase-negative cells Pin2 is abnormally concentrated only at a few telomeres. These results are consistent with the idea that Pin2 coats the telomeric repeats, which have been shown to be homogeneous in length in telomerase-positive, but highly heterogeneous in telomerase-negative cells (34–36).
Cell Cycle-Specific Regulation of Pin2 Protein Levels.
As indicated above, Pin2 contains a D-like motif similar, albeit not identical, to the destruction box present in many mitotic proteins (30). During our immunostaining analysis of Pin2, we consistently observed that the immunostaining intensity of endogenous Pin2/TRF1 proteins was highly variable among cells. Pin2 staining intensity was strikingly stronger in cells containing a large late G2-like nucleus, compared with those with a small G1/S-like nucleus (Fig. 4B and data not shown). To rule out the possibility that the anti-Pin2 antibodies used might only immunoreact with certain subpopulations of the Pin2 protein, we immunostained HA-Pin2 using 12CA5 mAb and again observed the same variation among cells within the individual stable HA-Pin2-expressing clones (Fig. 4B). These results suggest that the level of Pin2 protein might be increased during the later part of the cell cycle. To check this we analyzed the level of exogenously expressed HA-Pin2 during the cell cycle. HA-Pin2-expressing MDAH087 cells were synchronized in G1 by lovastatin treatment and then released to enter the cell cycle (28, 39). HA-Pin2 protein level remained relatively low during G1 and S, but was significantly increased when cells progressed through G2 and M phases at the 20 h point (Fig. 5A). The level decreased when cells moved into the next G1 (24 h point), but remained elevated if cells were prevented from completing mitosis by adding nocodazole at the 16 h point (Fig. 5A). Furthermore, immunostaining also showed that the Pin2 staining signal was low in G1/S cells, but dramatically increased in G2/M cells (data not shown). In addition, Pin2 levels were higher in nocodazole-induced mitotic cells than those in G1 or asynchronous cells (data not shown). These results demonstrate that Pin1 levels are tightly regulated during the cell cycle, with a maximal level observed in G2/M cells.
Figure 5.
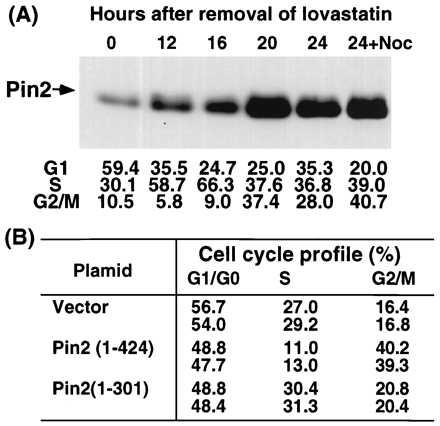
Pin2 protein level is tightly regulated during the cell cycle and induces accumulation of cells at G2+M when overexpressed. (A) HA-Pin2-expressing clone 6 MDAH087 cells were incubated with 10 μM lovastatin to synchronize cells in G1 (time 0). At the times indicated following release, cells were harvested and subjected to flow cytometric analysis to examine the cell cycle status or to immunoprecipitation and immunoblot analysis to determine HA-Pin2 levels using 12CA5 mAb. (B) The tTA-1 HeLa cells were transfected for 48 h with vectors expressing the indicated HA epitope tagged Pin2 proteins, followed by staining first with 12CA5 mAb and then with fluorescein isothiocyanate-conjugated secondary antibodies and propidium iodide. Pin2- and Pin2(1–301)-expressing cells were selected and their cell cycle profiles were determined to compare with that for the vector-only transfected cells.
Overexpression of Pin2 Induces an Accumulation of HeLa Cells in G2+M.
The above results suggested that Pin2 protein levels might be important for cell cycle progression. To test this possibility, we examined the effect of overexpressing Pin2 on cell cycle progression. tTA-1 HeLa cells were transfected with an HA-Pin2 expression vector, followed by analyzing the cell cycle profile in HA-Pin2-positive cells (18, 19). The percentage of cells with G2+M DNA content in cells overexpressing Pin2 was more than double that in cells not overexpressing Pin2 within the same transfected cell population or in cells transfected with a control vector (Fig. 5B). At the same time, the percentage of S phase cells was also significantly reduced when compared with that of the control cells (Fig. 5B), suggesting that there was also a delay in the G1/S transition. A similar accumulation of cells in G1 and G2+M is observed when growing cells are irradiated with UV to induce DNA damage (40). In contrast, overexpression of a C-terminally truncated form of Pin2 lacking the DNA-binding domain, Pin2(1–301) had no obvious effect on the cell cycle distribution (Fig. 5B). The mutant protein was expressed at least as well as wild-type, but was not localized in a speckled telomeric pattern, consistent with the removal of the DNA-binding domain, even though it was nuclear (Fig. 2C and data not shown). These results suggest that the Pin2 protein level is likely to be a potential regulatory factor in cell cycle progression.
DISCUSSION
In this study we have characterized human Pin2 and studied its cell cycle regulation and function. PIN2 encodes a protein that was later found to be the same as TRF1 except for a small internal deletion, which may be a splicing variant of the PIN2/TRF1 gene. In the cell, Pin2 is the major expressed product and forms homo- or heterodimers with TRF1; both dimers are localized at telomeres. Significantly, Pin2/TRF1 was uniformly distributed at telomeres in telomerase-positive cells, but was highly concentrated only at a few telomeres in telomerase-negative cells. Interestingly, the level of both endogenous and exogenously expressed Pin2 fluctuated strikingly during the cell cycle, reaching a maximal level in G2+M cells. Moreover, overexpression of Pin2 in HeLa cells resulted in an accumulation of cells with a G2+M DNA content, suggesting a potential role for telomere-binding proteins like Pin2/TRF1 in mitotic regulation.
Telomeric DNA-binding proteins have been isolated from several different species. In contrast to Rap1p, which has two HTH domains that coordinate specific binding to telomeric DNA (32), most of these proteins contain a single HTH domain (7, 23, 24). It is unclear how the single HTH domain binds the telomeric DNA. We have shown that Pin2 and TRF1 form homo- and heterodimers via the N-terminal domain and that both forms are localized at telomeres. Similar dimerization results have recently been reported for TRF1 in vitro, where the minimal dimerization domain was identified as residues 65–376 (21). Therefore, we propose that dimerization of these proteins provides a mechanism to bring two single C-terminal HTH domains together, allowing them to bind the telomeric DNA repeat. Furthermore, at least one other Pin2/TRF1-like molecule has been isolated (24), and it is possible that all three proteins will form homo- and heterodimers with slightly different properties. In this regard, both Pin2 and TRF1 are phosphoproteins in vivo (K.P.L and T.H., unpublished data), and the 20 additional residues in TRF1, SVSDKQSAVTESSEGTVSLL, contain several potential phosphorylation sites that might allow differential regulation of TRF1 and Pin2 by phosphorylation.
Telomeric proteins have been shown to regulate telomere length in budding and fission yeast and telomerase-positive human cells (5–7). Our results demonstrate that Pin2/TRF1 is highly concentrated on only a few telomeres in telomerase-negative cells, with a staining pattern similar to the large telomere hybridization signals obtained by fluorescence in situ hybridization in other telomerase-negative cells (35). It would be interesting to determine the role of Pin2/TRF1 in maintaining telomeres in telomerase-negative cells.
The levels of both endogenous and ectopically expressed Pin2 are tightly regulated during the cell cycle, being dramatically increased in G2+M. Since exogenous Pin2 was expressed under the control of the constitutively active cytomegalovirus promoter, the cell cycle fluctuation of Pin2 protein level must be regulated at the posttranscriptional level. Interestingly, Pin2 contains a motif related to the destruction box that mediates degradation of many mitotic proteins (30). Thus, the most likely mechanism for the fluctuation of Pin2 protein is an increase in protein stability in G2 and a decrease in stability in G1. The degradation of Pin2 as cells enter G1 is reminiscent of the degradation of other cell cycle regulatory proteins, such as the mitotic cyclins (30). In these cases, degradation is required for exit from M, and it is possible that degradation of Pin2/TRF1 (possibly unbound Pin2/TRF1) is also needed for cells to exit from M phase. The biological significance of the increase in Pin2 during G2+M is supported by the phenotype induced by overexpression of Pin2, which caused a significant accumulation of cells in G2+M, with a decrease in the percent of S phase cells. The increase in G2+M cells is consistent with Pin2/TRF1 regulating progression through mitosis; the very high level of Pin2 may overwhelm the cell’s ability to degrade it, thus blocking cells in G2+M. The decrease in S phase cells caused by Pin2 overexpression could reflect a delay in the G1/S transition; degradation of Pin2/TRF1 may also be needed before this transition can occur. Therefore, the level of Pin2/TRF1 proteins may be an important regulatory factor in cell cycle progression.
It is worth noting that van Steensel and de Lange (6) did not observe any changes in the growth rate of their TRF1-overexpressing cell lines. However, the induced TRF1 expression levels in their stable cell lines are not very high (9), compared with that obtained by transient transfection where we observed cell cycle arrest. Moreover, we have now shown that the endogenous degradation machinery is sufficient to regulate exogenous Pin2 if expressed at a low level, and this could explain why a cell cycle arrest phenotype was not observed in their study (6).
Our results raise the possibility that Pin2/TRF1 is involved in a telomere-mediated G2/M checkpoint that regulates mitotic progression. Pin2 was originally isolated as a protein that binds and suppresses the essential mitotic kinase NIMA in budding yeast (19). Pin2 is a phosphoprotein that is likely phosphorylated on a potential NIMA phosphorylation site; mutation of this site affects association between Pin2 and telomeres (M.S. and K.P.L., unpublished data). These results suggest that Pin2 and TRF1 may be potential downstream substrates for a NIMA-like kinase. Since telomere length is sensed by the concentration of bound telomeric proteins, as shown in the case of Rap1p (5), a high concentration of bound Pin2/TRF1 could be a signal that the telomeres are long enough for cells to continue dividing. Conversely, a high concentration of unbound Pin2/TRF1 could indicate that the telomeres are too short for the cell to divide. The idea that there is a telomere checkpoint is consistent with reports linking telomeres to mitotic regulation (3, 4, 12–15). Further functional studies of Pin2/TRF1 and characterization of the proteins it interacts with will be crucial to the understanding of the relationship between cell cycle progression and telomere function.
Acknowledgments
K.P.L. thanks B. Neel and L. Cantley for their assistance in setting up his new laboratory at Harvard Institutes of Medicine. Special thanks goes to J. Noel and M. Verdecia for pointing out a putative D-like box in Pin2, and M. Kirschner for improving the quality of the paper. We are grateful to M. Tainsky, X.-D. Fu, G. Hannon, D. Beach, S. Elledge, T. de Lange, R. Fukunaga, and H. Bujard for various reagents. We thank D. Chambers for the fluorescence-activated cell sorter analysis, and V. Edelman and J. Roberts for assistance with confocal microscopy, which was carried out in the San Diego Microscopy and Imaging Resource (supported by U.S. Public Health Service Grant RR04050) and in the Cell Biology Confocal Facility at Harvard Medical School. T.H. is an American Cancer Society Research Professor. The studies were supported by U.S. Public Health Service Grants CA14195 and CA39780 to T.H., CA13608 to M.V., and Leukemia Society fellowship and U.S. Public Health Service Grant GM56230 to K.P.L.
ABBREVIATIONS
- HTH domain
Myb-type helix-turn-helix DNA-binding domain
- Pin
protein interacting with NIMA
- TRF
telomeric repeat binding factor
- HA
hemaglutinin
Footnotes
Data deposition: the sequence reported in this paper has been deposited in the GenBank database (accession no. U74382).
References
- 1.Greider C W. Annu Rev Biochem. 1996;65:337–365. doi: 10.1146/annurev.bi.65.070196.002005. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V A. Science. 1995;270:1601–1607. doi: 10.1126/science.270.5242.1601. [DOI] [PubMed] [Google Scholar]
- 3.Harley C B, Futcher A B, Greider C W. Nature (London) 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 4.Hastie N D, Dempster M, Dunlop M G, Thompson A M, Green D K, Allshire R C. Nature (London) 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 5.Marcand S, Gilson E, Shore D. Science. 1997;275:986–990. doi: 10.1126/science.275.5302.986. [DOI] [PubMed] [Google Scholar]
- 6.van Steensel B, de Lange T. Nature (London) 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- 7.Cooper J P, Nimmo E R, Allshire R C, Cech T R. Nature (London) 1997;385:744–747. doi: 10.1038/385744a0. [DOI] [PubMed] [Google Scholar]
- 8.Sandell L L, Zakian V A. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- 9.Chikashige Y, Ding D Q, Funabiki H, Haraguchi T, Mashiko S, Yanagida M, Hiraoka Y. Science. 1994;264:270–273. doi: 10.1126/science.8146661. [DOI] [PubMed] [Google Scholar]
- 10.Greenwell P W, Kronmal S L, Porter S E, Gassenhuber J, Obermaier B, Petes T D. Cell. 1995;82:823–829. doi: 10.1016/0092-8674(95)90479-4. [DOI] [PubMed] [Google Scholar]
- 11.Savitsky K, Bar S A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 12.Pandita T K, Pathak S, Geard C R. Cytogenet Cell Genet. 1995;71:86–93. doi: 10.1159/000134069. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph N S, Latt S A. Mutat Res. 1989;211:31–41. doi: 10.1016/0027-5107(89)90104-8. [DOI] [PubMed] [Google Scholar]
- 14.Beamish H, Khanna K K, Lavin M F. Radiat Res. 1994;138:S130–S133. [PubMed] [Google Scholar]
- 15.Kirk K E, Harmon B P, Reichardt I K, Sedat J W, Blackburn E H. Science. 1997;275:1478–1481. doi: 10.1126/science.275.5305.1478. [DOI] [PubMed] [Google Scholar]
- 16.Osmani A H, McGuire S L, Osmani S A. Cell. 1991;67:283–291. doi: 10.1016/0092-8674(91)90180-7. [DOI] [PubMed] [Google Scholar]
- 17.Lu K P, Means A R. EMBO J. 1994;13:2103–2113. doi: 10.1002/j.1460-2075.1994.tb06486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu K P, Hunter T. Cell. 1995;81:413–424. doi: 10.1016/0092-8674(95)90394-1. [DOI] [PubMed] [Google Scholar]
- 19.Lu K P, Hanes S D, Hunter T. Nature (London) 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 20.Ranganathan R, Lu K P, Hunter T, Noel J P. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 21.Bianchi A, Smith S, Chong L, Elias P, de Lange T. EMBO J. 1997;16:1785–1794. doi: 10.1093/emboj/16.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaffe, M. B., Schutkowski, M., Shen, M., Zhou, X. Z., Stukenberg, P. T., Rahfeld, J., Xu, J., Kuang, J., Kirschner, M. W., Fischer, G., Cantley, L. C. & Lu, K. P. (1997) Science, in press. [DOI] [PubMed]
- 23.Chong L, Van Steensel B, Broccoli D, Erdjument B H, Hanish J, Tempst P, de Lange T. Science. 1995;270:1663–1667. doi: 10.1126/science.270.5242.1663. [DOI] [PubMed] [Google Scholar]
- 24.Bilaud T, Koering C E, Binet B E, Ancelin K, Pollice A, Gasser S M, Gilson E. Nucleic Acids Res. 1996;24:1294–1303. doi: 10.1093/nar/24.7.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong Z, Shiue L, Kaplan S, de Lange T. Mol Cell Biol. 1992;12:4834–4843. doi: 10.1128/mcb.12.11.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harlow E, Lane D. Antibodies: A Laboratory Mannual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 27.Bischoff F Z, Yim S O, Pathak S, Grant G, Siciliano M J, Giovanella B C, Strong L C, Tainsky M A. Cancer Res. 1990;50:7979–7984. [PubMed] [Google Scholar]
- 28.Jakobisiak M, Bruno S, Skierski J S, Darzynkiewicz Z. Proc Natl Acad Sci USA. 1991;88:3628–3632. doi: 10.1073/pnas.88.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogata K, Morikawa S, Nakamura H, Sekikawa A, Inoue T, Kanai H, Sarai A, Ishii S, Nishimura Y. Cell. 1994;79:639–648. doi: 10.1016/0092-8674(94)90549-5. [DOI] [PubMed] [Google Scholar]
- 30.King R W, Deshaies R J, Peters J M, Kirschner M W. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- 31.de Lange T. EMBO J. 1992;11:717–724. doi: 10.1002/j.1460-2075.1992.tb05104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konig P, Giraldo R, Chapman L, Rhodes D. Cell. 1996;85:125–136. doi: 10.1016/s0092-8674(00)81088-0. [DOI] [PubMed] [Google Scholar]
- 33.Kim N W, Piatyszek M A, Prowse K R, Harley C B, West M D, Ho P L, Coviello G M, Wright W E, Weinrich S L, Shay J W. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 34.Bryan T M, Englezou A, Gupta J, Bacchetti S, Reddel R R. EMBO J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henderson S, Allsopp R, Spector D, Wang S S, Harley C. J Cell Biol. 1996;134:1–12. doi: 10.1083/jcb.134.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugihara S, Mihara K, Marunouchi T, Inoue H, Namba M. Hum Genet. 1996;97:1–6. doi: 10.1007/BF00218823. [DOI] [PubMed] [Google Scholar]
- 37.Namba M, Nishitani K, Hyodoh F, Fukushima F, Kimoto T. Int J Cancer. 1985;35:275–280. doi: 10.1002/ijc.2910350221. [DOI] [PubMed] [Google Scholar]
- 38.Bai L, Mihara K, Kondo Y, Honma M, Namba M. Int J Cancer. 1993;53:451–456. doi: 10.1002/ijc.2910530317. [DOI] [PubMed] [Google Scholar]
- 39.Keyomarsi K, Sandoval L, Band V, Pardee A B. Cancer Res. 1991;51:3602–3609. [PubMed] [Google Scholar]
- 40.Poon R Y C, Jiang W, Toyoshima H, Hunter T. J Biol Chem. 1996;271:13283–13291. doi: 10.1074/jbc.271.22.13283. [DOI] [PubMed] [Google Scholar]