

Abstract
In the present treatise, we provide evidence that the neuroprotective and mito-protective effects of estrogens are inexorably linked and involve the ability of estrogens to maintain mitochondrial function during neurotoxic stress. This is achieved by the induction of nuclear and mitochondrial gene expression, the maintenance of protein phosphatases levels in a manner that likely involves modulation of the phosphorylation state of signaling kinases and mitochondrial pro- and anti-apoptotic proteins, and the potent redox/antioxidant activity of estrogens. These estrogen actions are mediated through a combination of estrogens receptor (ER)-mediated effects on nuclear and mitochondrial transcription of protein vital to mitochondrial function, ER-mediated, non-genomic signaling and non-ER-mediated effects of estrogens on signaling and oxidative stress. Collectively, these multifaceted, coordinated action of estrogens leads to their potency in protecting neurons from a wide variety of acute insults as well as chronic neurodegenerative processes.
Keywords: Estrogens, 17β-Estradiol, Non-feminizing estrogens, Estrogen receptors, Protein phosphatases, Mitochondria, Neuroprotection
1. Introduction
Estrogens are potent, centrally neuroactive and neuromodulatory molecules. One important aspect of estrogens' effects on neurons is their neuroprotective activity. Estrogens are potent neuroprotectants against a variety of different toxicities including serum deprivation, oxidative stress, and amyloid β peptide (Aβ) induced toxicity as well as glutamate-induced excitotoxicity [65]. Further, both endogenous and exogenous estrogens as well as estrogen analogues have been shown to have neuroprotective effects against the pathological events seen in experimental animal models of neurodegeneration and cerebral ischemia [3,42,61–65,67,159,168,195]. The pathological mechanisms that are activated during stroke include oxidative stress, free radical activity, excitotoxicity, inflammatory response, mitochondrial dysfunction, and apoptosis, all of which are antagonized by estrogens. However, the mechanisms by which estrogens are protective remain elusive. It is clear that the effects of estrogens are complex and multifaceted. The mechanisms of the neuroprotective actions of estrogen are characterized by classical estrogen receptor (ER)-dependent genomic and non-genomic actions, the latter of which is expressed by rapid interactions with neuronal membranes and intracellular signal transduction pathways. In addition, estrogens have also been shown to have an intrinsic antioxidant structure that lies in the phenolic ring of the compounds, which provide the antioxidant/redox cycling activity in neurons [135,136] that complements other neuroprotective activities of estrogens.
As a result of the early cessation of the Women's Health Initiative (WHI) study due to increased risks of cardiovascular disease, stroke, blood clots, breast cancer, and dementia in women on either combined estrogen–progestin or estrogen alone therapy, the scientific community has struggled to reconcile the numerous epidemiological, experimental, and clinical studies that have shown hormone therapy (HT) to be protective against a wide variety of pathological diseased states such as Parkinson's Disease (PD) and Alzheimer's disease (AD), stroke, and cardiovascular diseases. However, recent re-evaluations of the WHI, other clinical trials, and cohort studies, such as the Nurses' Health Study, suggest that effects of HT are dependent on reproductive stage and the extent of preexisting neurodegenerative or cardiovascular disease at the onset of HT [44,68,71,73,105]. Specifically, emerging evidence indicates that younger (perimenopausal or early postmenopausal) women derive neuro- and cardiovascular protection from HT, whereas the initiation of such therapy in older individuals who are likely to have extensive preexisting conditions may be ineffectual or even detrimental [34,43,107,143,148,207].
2. Protein phosphatase 1, PP2A, and calcineurin in neurodegeneration
The brain is an extremely rich source of protein phosphatases (PPs). Historically, protein phosphatases are thought to be ubiquitous nonspecific scavengers of phosphoproteins, but it is becoming increasingly clear that protein phosphatases are important modulators of cellular activity. It is well documented that PP1, PP2A, and PP2B are essential modulators of LTP and LTD [78,79,114,169]. Cognitive declines observed in AD are related to the marked decrease in synaptic contact [38,172], where PP1 plays a central role since it is highly enriched in post-synaptic dendritic spines [123]. Not only is PP1 involved in the control of long-term depression (LTD) and synaptic plasticity [111], but also in aging-related memory defects [57]. PP2A and calcineurin-dependent mechanisms of learning and memory have also been shown [13,193,194,205]. Rats with reduced levels of PP1 and PP2A, due to chronic pharmacological inhibition, show memory impairment [4].
Hyperphosphorylation of tau is a hallmark of neurofibrillary tangles seen in AD brain. An imbalance between tau kinases and protein phosphatases is therefore proposed as a pathological mechanism of AD. There is a decreased calcineurin activity in the frontal cortex of human AD brains compared to age-matched controls, without a concominant change in calcineurin protein levels or regulatory proteins [100]. Moreover, the mRNA expression of DSCR1 (Adapt78), a calcineurin inhibitory gene, in AD brains is twice as high as control brains [47]. Immunoblotting analyses has also revealed that there is a significant reduction in the total amounts of PP2A in AD frontal and temporal cortices that matched the decrease in PP2A activity measured in the same brain homogenates [165]. Immunohistochemical studies also showed that neuronal PP2A expression levels are significantly and selectively decreased in AD-affected regions and in tangle-bearing neurons, but not in AD cerebellum and in non-AD dementias. The role of PP1 in the neurodegenerative diseases involves not only Aβ-deposition and hyperphosphorylated tau but also a decrease in cognitive function and memory formation. PP1 is a major tau phosphatase [139,206], and inhibition of PP1 stimulates the secretion of soluble amyloid precursor protein [37]. PP1 activity is also decreased in AD brains [58].
3. Protein phosphatases in estrogen-induced neuroprotection
We have recently shown that serine/threonine phosphatases are important players in the mechanism of estrogen-mediated neuroprotective effects against glutamate-induced oxidative stress and excitotoxicity in primary cortical neurons, an immortalized hippocampal cell line (HT-22), and C6-glioma cells [200–203].
3.1. Cytotoxicity induced by inhibition of serine/threonine phosphatases
Okadaic acid (OA) and Calyculin A (CA) are potent inhibitors of PP1 and PP2A as well as calcineurin at higher micromolar concentrations. Treatment of HT-22, C6-glioma, and primary cortical neurons with OA induced dose-dependent cell death with an LD50 of 81, 87, and 40 nM, respectively [201]. CA also caused a dose-dependent decrease in cell viability in HT-22 with a more typical dose-response that caused a 50% cell death at >500 pM. In addition, CA also showed a typical dose-dependent decrease in cell viability with an LD50 of 67 pM in primary cortical neurons. However, in glioma cells, an atypical dose-response was seen with a precipitous decrease in cell viability at a threshold concentration of 500 pM [201].
Inhibiting PP1, PP2A, or calcineurin individually did not induce cell death; however, when a combination of serine/threonine phosphatase activities are inhibited, neurons and glia are more vulnerable to cell death [203]. Any combination of two phosphatase inhibitors caused a mild insult which produces approximately 25–35% cell death, while inhibiting all three phosphatases caused 50% reduction in cell survival. This suggests that individual serine/threonine phosphatases are not essential for cell survival in and of itself but that phosphatases in general are necessary for normal cellular function, which is a theme often found in nature's redundancies.
3.2. Inhibition of protein phosphatases 1, 2A, and calcineurin abolishes the neuroprotective effects of estrogens
In the absence of PP inhibitors, 17β-estradiol and its analogues are neuroprotective in HT-22 immortalized neurons, C6-glioma, and primary cortical neurons against glutamate-induced cytotoxicity; however, in the presence of general serine/threonine phosphatase inhibition, the protective effects of estrogens against oxidative stress and/or excitotoxicity are lost (Fig. 1) [200–203]. Pretreatment of estrogens for 2 or 24 h does not alter the loss of estrogen-mediated neuroprotection against glutamate toxicity induced by phosphatase inhibition [202]. Specific inhibition of PP1, PP2A, or calcineurin only partially antagonizes the estrogen protection of primary cortical neurons against oxidative stress and/or excitotoxicity induced by glutamate [203], which is also the case in HT-22 and glial cells (Fig. 2). In addition, estrogens prevent glutamate-induced decrease in protein expression of serine/threonine phosphatases as well as their activities in HT-22 (Figs. 3 and 4), glial cells ([203], unpublished observations) as well as in primary cortical neurons [203].
Fig. 1.

Effects of calyculin A on estrogen-mediated neuroprotection from glutamate neurotoxicity. HT-22 cells or C6-glioma cells were seeded into 96-well plates at a density of 3500 cells/well. (A) HT-22 cells were treated simultaneously with 10 μM 17β-estradiol, varying concentrations of calyculin A and/or 10 mM glutamate. (B) C6-glioma cells were treated simultaneously with 10 μM 17β-estradiol, varying concentrations of calyculin A and/or 20 mM glutamate. Cell viability was determined by calcein AM assay after 24 h exposure to the various compounds. All data were normalized to percentage survival of vehicle control. Data are represented as mean ± SEM for n = 10. *P < 0.05 vs. control; †P < 0.05 vs. glutamate treated group.
Fig. 2.

Effects of PPI2, endothall, or cyclosporine A on 17β-estradiol mediated neuroprotection in HT-22 cells and C6-glioma. HT-22 (A–C) and C6-glioma (D–F) cells were seeded into 96-well plates at a density of 3500 cells/well. (A and D) Cells were treated simultaneously with with 200 nM PPI2, 10 mM glutamate, and/or 10 μM 17β-estradiol. (B and E) Cells were treated simultaneously with 9 μM endothall, 10 mM glutamate, and/or 10 μ 17β-estradiol. (C and F) Cells were treated simultaneously with 500 nM CsA, 10 mM glutamate, and/or 10 μ 17β-estradiol. Cell viability was determined by calcein AM assay (Molecular Probes, Eugene, OR) after 24 h exposure to the various compounds. All data were normalized to % survival of non-treated control. Depicted are mean ± SEM for 10 independent experiments with two replicates per experiment. *P < 0.05 vs. vehicle control; †P < 0.05 vs. glutamate treated group.
Fig. 3.

Time course of the effects 17β-estradiol, glutamate, and their combination on PP1 protein levels in HT-22. HT-22 cells were treated with either 10 mM glutamate (A) or 10 μM 17β-estradiol (B) and simultaneously with 10 mM glutamate and 10 μM 17β-estradiol (C). Cells were harvested at the times indicated for Western blot analysis of PP1. The graphs represent relative OD as a percentage of time 0 control and were normalized to β-actin (not shown). Data are represented as mean ± SEM for n = 3. *P < 0.05 versus time 0 control.
Fig. 4.

PP2A activity in HT-22 cells following treatment with glutamate and/or 17β-estradiol in the presence of specific inhibitors of PP1, PP2A, or calcineurin. HT-22 cells were seeded in 100 mm dishes at a density of 250,000 cells/ml. (A) Cells were treated simultaneously with 100 nM okadaic acid, 10 mM glutamate, and/or 10 μM 17β-estradiol. (B) Cells were treated simultaneously with 200 nM PPI2, 10 mM glutamate, and/or 10 μM 17β-estradiol. (C) Cells were treated simultaneously with 9 μM endothall, 10 mM glutamate, and/or 10 μM 17β-estradiol. (D) Cells were treated simultaneously with 500 nM CsA, 10 mM glutamate, and/or 10 μM 17β-estradiol. PP2A activity was determined using a serine/threonine phosphatase activity assay (Promega, Madison, WI) after 24 h exposure to the various compounds. All data were normalized to % survival of vehicle treated control. Depicted are mean ± SEM for six independent experiments with triplicates per experiment. *P < 0.05 vs. glutamate treated group.
Remarkably, all three protein phosphatases appear to respond similarly to 17β-estradiol, glutamate, OA and their combination in HT-22 cells, rat primary neuronal cultures, and C6-glioma cells. The robust decrease in these three protein phosphatases in response to glutamate in a model of oxidative stress (HT-22 cells) and to excitotoxicity (primary rat cortical neurons) suggests that protein phosphatase inhibition may be a common neurotoxic event for these insults. Similarly, the ability of estrogens to antagonize these glutamate-induced reductions in protein phosphatases suggests that this action is common to estrogen protection in both oxidative stress and excitotoxicity in both neurons and glia.
3.3. Inhibition of protein phosphatases induces oxidative stress and mitochondrial dysfunction
OA-mediated neuronal cell death is similar to glutamate induced neurotoxicity in that both compounds cause the production of reactive oxygen species (ROS), lipid peroxides, protein carbonyls, activation of caspase 3/7, and mitochondrial dysfunction [202]. Various experiments demonstrate the OA-induced increases in oxidative stress biomarkers such as lipid peroxidation products, GSH content, GSH-Px, catalase, glutathione transferase and glutathione reductase [152,154,176]. Mitochondrial dysfunction and the consequential induction of caspase 3/7 have also been observed with OA treatment [5,17,50,51,204]. In primary cortical neurons, mitochondrial swelling and decreased membrane potential as well as increased protein expression of caspase-3, Bad, Bax, and Bim are seen following OA treatment [204]. These events have been well established in glutamate induced excitoxicity and oxidative stress. Numerous reports show that estrogens are neuroprotective against glutamate-induced cell death by mitigating or abolishing the glutamate-induced lipid peroxidation, protein carbonylation, ROS production, activation of caspase 3/7, and mitochondrial dysfunction [19,116,129]. However, estrogens are not able to block or even partially attenuate the events induced by OA [202]. In addition, the presence of OA in non-lethal concentrations abolishes the estrogen-mediated attenuation of oxidative stress, mitochondrial dysfunction, and apoptosis induced by glutamate toxicity. Since protein phosphatases are important modulators of cellular function, it is not surprising that inhibition of protein phosphatases by OA leads to cellular dysfunction that ultimately ends with cell death.
4. MAPK pathway involvement in ER-independent, estrogen-induced neuroprotection and the role of serine/threonine phosphatases
A specific consideration of the effects of estrogens on signaling in the ERK1/2 pathway is informative. 17β-Estradiol causes a rapid, transient phosphorylation of ERK1/2 in neuroblastoma [188], non-neuronal cells [108], and cortical explants [163], as well as in the cortex of ER-α knock-out mice [162]. This transient phosphorylation of ERK1/2 is believed to mediate a neuroprotective signal while persistent phosphorylation of ERK1/2 is associated with apoptosis, likely through nuclear translocation and retention of phosphorylated ERK1/2 [87,91]. The transient nature of neuroprotective signaling indicates that dephosphorylation though activation of phosphatases is required. Indeed, pharmacological inhibition of ERK1/2 signaling is neuroprotective in a number of cell model systems [7,117], including the HT-22 cell oxidative stress model [150,166]. Similarly, in models of stroke, brain trauma, and neurodegenerative diseases, there are detrimental effects of persistent activation of ERK1/2 during oxidative neuronal injury as well as excitotoxic injury [2,52,72,164,166,184,186,212,213]. ERK1/2 is rapidly and persistently phosphorylated in response to oxidative and excitotoxic stresses caused by glutamate, and the presence of estrogens prevents this persistent phosphorylation [200]. These data suggests that prolonged phosphorylation of ERK1/2 is detrimental to cell survival, which is supported by the observations of aberrant neuronal expression of phosphorylated ERK1/2 and other MAPKs in AD brains in association with markers of oxidative stress [213]. Others have also shown that MAPK phosphorylation is associated with a variety of sporadic and familial neurodegenerative diseases characterized by tau deposits [52]. Patients with PD and other Lewy body diseases have increased phosphorylated ERK1/2 in neurons of substantia nigra and midbrain [212]. In addition, increased ERK1/2 phosphorylation has been noted in the vulnerable pneumbra following acute ischemic stroke in humans [164].
Accumulating evidence has shown that neuroprotective effects of 17β-estradiol involve activation of intracellular signaling pathways via G proteins [112,138], extracellularly regulated kinases (phosphoinositol 3-kinase and protein kinase B/AKT, ERK and p38 MAP kinases) [162,175,188], phosphorylation of the cAMP response element-binding protein [115,180,189,210,211], and alterations in intracellular calcium levels [15,77,132]. These actions suggest that there is a link between estrogen action at the cell membrane and discrete biological actions in the cell.
There is considerable evidence that 17β-estradiol acts on neurons through a variety of signal transduction pathways to induce rapid, but acute phosphorylation of signaling proteins [163,188,211]. Estradiol-induced phosphorylation of the adenylyl cyclase, Akt, PKA, PKC, and MAPK pathways have been reported [85,108,163,209]. Changes in the activity of these enzymes can regulate the phosphorylation of numerous intermediary signaling proteins such as Rsk, p38 and JNK, and nuclear transcriptional factors, cyclic AMP response element binding protein (CREB) and cfos/cjun, which may ultimately mediate cell survival changes (for review see [97]). Further, 17β-estradiol has been shown to block the persistent activation of both ERK and PKC in a variety of insults including ischemia and ethanol withdraw induced cytotoxicity [16,80,92,162,163,188].
Multiple, interactive death-inducing kinase pathways are likely activated simultaneously following glutamate or OA treatment. It has been observed that both glutamate and OA [89,204] persistently activate JNK, p38 MAPK and ERK1/2 pathways. Activation of the different MAPK pathways occurs simultaneously, and there is crosstalk between ERK1/2 and p38 that is mediated by various protein phosphatases [145,185]. Moreover, it has been demonstrated that PKC phosphorylates both ERK1/2 and p38 MAPK [76,86,153] providing a link between PKC and MAPK signaling. In the presence of functioning protein phosphatases, estrogens, via maintenance of protein phosphatase activity [200,201], or specific inhibitors of ERK1/2 or PKC [202] prevent glutamate death signaling. However, in the face of broad protein phosphatase inhibition by OA, profound activation of multiple death-inducing kinases cannot be overcome with either estrogens or specific kinase inhibitors. Therefore, it is possible that direct inhibition of death-inducing kinases is sufficient for neuronal survival, but inhibition of phosphatases leads to activation of multiple death signaling pathways that cannot be overcome by inhibition of ERK, p38, or PKC alone.
5. Mechanism by which estrogens-induce protein phosphatase expression and activity
There are numerous reports that cellular redox status plays an important role in the mechanisms to regulate the function of growth factors, serine/threonine phosphorylation-dependent and tyrosine phosphorylation-dependent signal transduction pathways [53,54,95,113,171,187]. Furthermore, reactive oxygen species such as H2O2 have been shown to be involved in growth factor signaling pathway [141,170], perhaps as a second messenger. Therefore, protein phosphatases appear to be logical targets of oxidative stress, leading to transient and reversible inactivation of phosphatase activity by oxidizing catalytic cysteine residue to sulfenic acid [39]. It has been reported that low levels of H2O2 (200 μM) robustly activate ERK1/2 [70,81,174,183] but only slightly activate p38 MAPK and c-Jun N-terminal kinase/stress-activated protein kinase in COS1 cells by inactivating endogenous MAPK phosphatases that dephosphorylate ERK [70]. It has also been shown that MAPK phosphatase activity in microsomal and soluble fractions of rat brain was attributable mainly to PP2A. Moreover, H2O2 and glutathione disulfide inhibited MAPK phosphatase activity by a dithiothreitol-reversible mechanism [55].
The mechanism by which estrogens increase protein phosphatase levels is not known. To date, we are not aware of an estrogen response element in the regulatory regions of the protein phosphatases under study. Therefore, we hypothesize that protein phosphatase signaling is through an alternative pathway. One intriguing possibility is that the signaling pathways activated by neurotoxic insults suppress phosphatase expression or enhance the proteolytic breakdown of these important proteins. Estrogens then could induce an increase in phosphatase production and/or reduce proteolysis thereby normalizing protein phosphatase levels and regulating signaling along phosphorylation-dependent pathways.
There are no previous experimental data suggesting a direct interaction between estrogens and PP. While PP2A has been shown to regulate ERα by mRNA stabilization [83] as well as direct interaction with ERα in the absence of estrogen [104], our data indicate that effects of estrogens on PPs occur without ER interactions since non-ER active estrogen analogues exert similar effects [200]. An interesting, but yet unexplained observation is that while 17β-estradiol [201] or ZYC3 [200], non-feminzing estrogen, alone have little effect on protein phosphatase levels, they cause a prompt and transient increase in protein phosphatase concentrations and a persistent resistance to glutamate-induced decline in serine/threonine phosphatase levels. The absence of effects of estrogens alone and the broad range of protein phosphatases affected by estrogens suggests that estrogens reduce the clearance of PPs that is activated by oxidative/excitotoxic insult, rather than causing expression of new protein. Our laboratory recently reported that estrogens attenuate ischemia induced increases in activities of MMP2 and MMP9, which belong to a class of proteases [101]. We have also shown that transient cerebral ischemia causes neurofibrillary tangle like tauopathy involving cdk5 [191]. This study showed that calpain II, a cytoplasmic cysteine protease, caused the cleavage of p35 to p25, which are co-activators of cdk5, and that the inhibition of calpain by MDL 28170 attenuated this cleavage, and prevented the activation of cdk5. It has also been shown that estrogens induce expression of secretory leukocyte protease inhibitor in the rat uterus [26]. Because estrogens have been shown to modify the expression or functions of various proteases, it is likely that 17β-estradiol protect cells by blocking the ubinquitination and/or degradation of protein phosphatases caused by oxidative or excitotoxic stresses.
Thus far, there is no evidence to suggest that estrogens interact physically with phosphatases to alter their activity. However, this is a possibility that cannot be ruled out. Previous work has shown a direct interaction of estrogens with a variety of proteins such as GSH, which contains a sulfhydryl group (redox sensitive cysteine groups). Estrogens are most likely to interact especially with those proteins that have sulfhydryls in their active domains, such as Na/K ATPase [157], ryanodine receptors [131], and GAPDH [22]. Further work needs to be done to determine if estrogens are interacting directly with the serine/threonine phosphatases as they do possess cysteine groups that are redox sensitive.
Since affected brains in AD have significantly decreased activities of PP1, PP2A, and calcineurin [59], this down-regulation of phosphatase activities and up-regulation of protein kinase activities is likely to trigger hyperphosphorylation of tau and activation of other pathological events involved in neurodegenerative diseases. In addition, these phosphatases are directly involved in synaptic plasticity and memory formation, which is affected in neurodegenerative diseases. The observation that estrogen-mediated neuroprotection involves maintaining the activities of these important phosphatases can provide a target for more clinically relevant postmenopausal hormone regime that would minimize the adverse effects of estrogens while maximizing the beneficial effects. Indeed, in our tMCAO animal model, we completely prevented the stroke-induced decrease in PP1, PP2A, and PP2B with 17β-estradiol [203].
6. Overview of mitochondrial structure and function
Mitochondria are the primary site of energy transduction and ATP generation in eukaryotes and play critical roles in life and death decisions for all cells. These organelles are composed of two distinctly different membranes, the outer membrane and inner membrane, which encloses a protein-rich matrix. The five enzyme complexes of the oxidative phosphorylation system are embedded in the mitochondrial inner membrane, and tricarboxylic acid cycle components are located in the matrix of mitochondria. In healthy cells, mitochondria supply the energy demand of cells by providing ATP through oxidative phosphorylation. In response to a wide variety of stress signals, including loss of growth factors, hypoxia, and oxidative stress, mitochondria are the most important regulators of cell death b both necrosis and apoptosis. Mitochondria can activate death programs by opening the mitochondrial permeability transition pore in the inner mitochondrial membrane, leading to the collapse of mitochondrial membrane potential, ATP depletion, and mitochondria and cell swelling, which ultimately result in necrosis. In addition, an increase in the permeability of the mitochondrial outer membrane and release of proapoptotic factors can trigger apoptosis.
It is clear that mitochondria play a central role in estrogen-induced cell protection. Protective effects of estrogens on mitochondrial have been demonstrated including protection from insult-induced ATP depletion, decreased reactive oxygen species production, attenuated mitochondrial Ca2+ influx, and prevention of mitochondrial membrane potential collapse (see reviews [20,21,158,160,161]). Indeed, our recent structure–activity relationship studies indicated a correlation between the protective action of estrogens against glutamate-induced cell death and Ca2+-loading [45] and H2O2-induced mitochondrial membrane potential collapse (Fig. 5). The following sections focus on mechanisms underlying the mito-protective effects of estrogens.
Fig. 5.
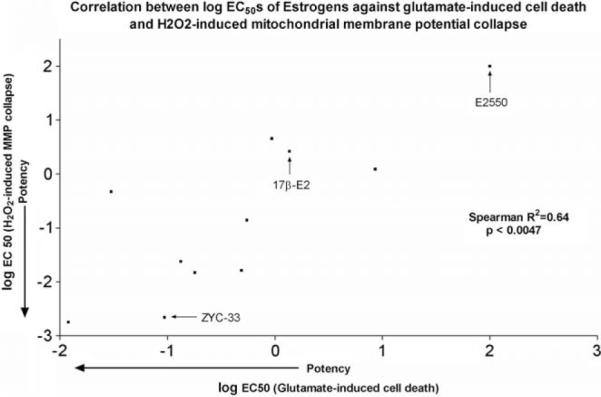
Relationship between neuroprotective and mitoprotective potency of estrogens. Eleven estrogens were selected from our library of compounds and tested for potency in protection from glutamate-induced cells death (x-axis) as well as their potency in protection from H2O2-induced collapse of mitochondrial member potential (y-axis). Depicted are the EC50 values for each assay. Denoted with arrows are a potent synthetic estrogen (ZYC-33), 17βb-estradiol (17β-E2) and an inactive estrogen (E2550).
7. Mechanisms mediating the mito-protective actions of estrogens
A variety of studies indicate that mitochondria are a convergence point for mechanisms underlying estrogen-induced cell protection. Given the unique role of mitochondria in energy generation, homeostasis maintenance, free radical production, and cell death determination, it is not surprising that the neuroprotective effects of estrogens are linked with the mitochondrial protection. Accumulating evidence suggests that estrogens play a critical role in the mitochondrial function directly and indirectly, through both genomic and non-genomic mechanisms. Genomic actions of estrogens on mitochondria are through transcriptional regulation of both nuclear and mitochondrial DNA-encoded mitochondrial proteins. Non-genomic actions of estrogens are exerted directly on mitochondrial through redox cycling of estrogens [135,136]. In addition, estrogens could affect mitochondrial function through indirect non-classical ER action, such as membrane estrogen receptor.
7.1. Genomic actions
Mitochondria are thought to have originated from a free-living prokaryotic organism, which explains the presence of a compact mammalian mitochondrial genome in mammalian cells. During its evolution into the present-day organelle of the eukaryotic cell, many of its essential genes have been transferred to the nuclear chromosomes. Most proteins involved in mitochondrial functions are synthesized in cytoplasm through transcriptional regulation of the nuclear genome. Nevertheless, mitochondria still carry hallmarks of its bacterial ancestry, and the essential proteins involved in mitochondrial electron transport and oxidative phosphorylation are synthesized within mitochondria through encoding in the mitochondrial genome. Therefore, eukaryotic mitochondrial biogenesis is coordinated by the expression of numerous genes encoded in two different genetic compartments: the nucleus and mitochondria. The nuclear genes encode the enzyme systems responsible for energetic metabolism, mtDNA replication and transcription, while the mitochondrial genome encodes for key subunits of the electron transport chain and RNA components needed for mitochondrial translation. Estrogens could regulate mitochondrial function through both nuclear- and mitochondrial-dependent genomic actions.
7.2. Nuclear genomic actions
Estrogens regulate mitochondrial protein expression encoded by nuclear genes. Estrogens not only increase expression of glucose transporter subunits and increase glucose transport, and thereby promoting glucose transport into the brain [155,156], but also increase glycolytic enzyme expression and activities of hexokinase, phosphofructokinase, phosphoglycerate kinase [88], and the E2 and E3 component of pyruvate dehydrogenase complex [119]. Each of these glycolytic enzymes are encoded by nuclear genes and are closely associated with mitochondrial outer (hexokinae), or localize in the mitochondrial matrix (E2 and E3 components of pyruvate dehydrogenase). Moreover, estrogens regulate enzymes involved in TCA cycle such as aconitase 2 and malate dehydrogenase [119]. Estrogens enhance glycolytic activity that is coupled with an increase in glutamate turnover, evidenced by the increased expression of glutamate dehydrogenase and glutamate oxaloacetate transaminase-2 [119]. The latter can impact generation of neurotoxic free ammonium and reduce excitotoxic free glutamate [125]. Consistent with their actions on glycolytic metabolism, estrogens also enhance the expression of F1 subunits of ATP synthase [119]. All these findings indicated that estrogens promote utilization of glucose through nuclear genomic actions.
Mitochondria integrate cellular apoptotic signals and amplify apoptotic responses [90]. The anti-apoptotic actions of estrogens have been well established in different cell types, including cancer cells, cardiomyocytes, and neurons. This ubiquitous effect of estrogens could be partially due to estrogens' regulatory effects on the expression of mitochondrial oxidative phosphorylation components. Enhanced oxidative phosphorylation has been found to be associated with decreased apoptosis whereas reduced oxidative phosphorylation associate with increase in apoptosis [182,192]. In addition, estrogens can directly regulate the expression of mitochondrial stress responsive factors encoded by nucleus. The Bcl-2 family of proteins are important regulators of the mitochondrial pathway of apoptosis. These proteins determine whether the mitochondria initiate the cell death program by releasing proapoptotic factors such as cytochrome c. The family of Bcl-2 proteins consists of anti- and pro-apoptotic members and plays a key role in regulating apoptosis in cells [69]. Estrogens increase anti-apoptotic proteins, Bcl-2 and Bcl-xL, which prevent activation of permeability transition pore [118]. In primary neuronal cultures, estrogens upregulate expression of antiapoptotic Bcl-w and downregulate expression of proapoptotic bim in an ER-dependent manner [199].
Estrogen-induced enhancement of energetic efficiency and anti-apoptotic action is paralleled by an increase in free radical defense systems. The antioxidant effect of estrogens has been well documented [12,20,160]. Since estrogens are comparatively poor scavenger of reactive oxygen species [173], the antioxidant effect of estrogens is unlikely due to the direct interaction of estrogens and free radicals. Rather, estrogens appear to strengthen the free radical defense system. Estrogens increase the expression and activity of glutaredoxin, gamma-glutamylcysteine synthetase, and MnSOD [40,60,128,167,177]. In addition, an increase in peroxiredoxin-V was also found upon estrogen treatment [119]. The action of estrogens on glutaredoxin, gamma-glutamylcysteine synthetase, and MnSOD expression and activation are inhibited by the ER antagonist, ICI182780, suggesting an ER-dependent mechanism.
7.3. Mitochondrial genomic actions
In contrast to nuclear genes, which encode the enzyme systems responsible for energetic metabolism, mtDNA replication and transcription, mitochondrial genome encodes for key subunits of the electron transport chain and RNA components needed for mitochondrial translation. Mitochondrial DNA only encodes 13 of the ~90 different proteins present in the respiratory chain of mammalian mitochondria. The structure and gene organization of mtDNA is highly conserved among mammals. All 13 proteins are the essential components of the enzyme complexes of the oxidative phosphorylation system. The mammaliam mitochondrial genome is a closed-circular, double-stranded DNA molecule of about 16.6 kb, that is maternally inherited because mitochondria in mammalian sperm are destroyed in the fertilized oocyte. The strands of the DNA duplex can be distinguished as heavy (H) and light (L) strand based upon the G+T base composition which results in different buoyant densities of each stand. Most information is encoded on the H strand, with genes for two fRNAs, 14tRNAs, and 12 proteins. The L strand codes the other eight tRNAs and a single protein. Different from the nuclear genome, mtDNA is continuously turned over and replicated during the entire cell cycle, displaying no strict phase specificity [48].
The compact mammalian mitochondrial genome lacks introns, and the only substantial noncoding region, the D-loop, contains the origin of H strand replication and promoters for bidirectional transcription from opposing H and L strand [33,48]. In human cells, a single promoter region has been identified in each strand for transcriptional initiation, the light-strand promoter (LSP) and the heavy-strand promoter (HSP). Transcription from the mitochondrial promoters produce polycistronic precursor RNAs, encompassing all of the genetic information encoded in each of the specific strands.
Interestingly, many of the electron transport chain components encoded by mitochondrial genome have been found to be regulated by estrogens. Van Itallie and Dannies [178] found a 16-fold increase of cytochrome c oxidase subunit II mRNA upon 17β-estradiol treatment in rat pituitary tumor cells. An estrogen-induced increase of cytochrome c oxidase subunit III transcript was also observed [14]. The estrogen-regulated mitochondrial encoded transcripts have been extended to all three subunits of the complex IV and subunits 6 and 8 of ATP synthase [27,28]. More recently, Nilsen et al. [119] identified 4 of the 7 subunits of complex I encoded by mitochondrial genome were regulated by 17β-estradiol. Given the single promoter for each strand of mtDNA and the broad range of estrogen-regulated mitochondrial transcripts, the action of estrogens on mitochondrial transcription seems universal, not specific to any single gene.
It is not clear how estrogens regulate mitochondrial gene expression. Studies have shown that the enhancement of mitochondrial transcripts by estrogens can be blocked by ER antagonist, ICI182780, suggesting an ER-dependent mechanism [29,30]. This notion is further supported by the newly identified mitochondrial localization of ERs, specially ERb [29,197]. An up-regulation of mitochondrial complex IV by ERβ selective ligand, diarylpropionitrile (DPN), has been demonstrated [74]. The crystal structure of ERβ has been well described. ERβ shares a highly conserved structure with other nuclear receptors such as ERα. Although ERα and ERβ have nearly identical DNA-binding domain, increasing evidence indicates that they regulate the expression of a distinct set of genes [82,121]. Most studies have been focused on the nuclears transcription regulation. Consistently, most of the genes modified in ERβ knock-out mice are mitochondrial structural proteins related to oxidative phosphorylation [121]. This distinction could be partly due to different compartmentation of ERα and ERβ. In addition to its broad distribution among neuronal organelles, ERβ is localized in the mitochondrial matrix, hence enabling its access to the mitochondrial genome [31]. Therefore, both the ERβ structure and matrix localization provide ERβ the capacity to regulate mitochondrial gene expression. Indeed, recent studies have found that ERβ could directly interact with mitochondrial genes to modulate cytochrome c oxidase subunits expression [31].
It remains unclear how ERβ interacts with mtDNA promoter to regulate mitochondrial gene expression. Classically, ERs bind to estrogen response elements (ERE) in target genes and recruit coactivator complexes that mediate stimulation of transcription. Alternatively, ERs also activate transcription at activator protein 1(AP-1) sites [93,94]. Putative ERE sequence has been found in mtDNA [29]. In addition, although a completely identical core nucleotide sequence for recognizing AP-1 was not found anywhere in mitochondrial DNA, approximately 10 sites with sequences similar to the AP-1 site have been found in the noncoding region of mitochondrial DNA [122]. Therefore, the mitochondrial localization of ERβ and the putative ERE and AP-1 bind sites in the mtDNA enable ERβ to mediate the action of estrogens on mitochondrial transcriptional regulation.
In addition to the ERβ-dependent mechanism, estrogens could also regulate mitochondrial transcription through its genomic action independent of classic ER activation. Membrane sites of estrogen activation, which activate the PI3K/Src/ERK signaling pathway, activating CREB, have been identified to mediate the protective action of estrogens [24,98,106]. CREB is a widely expressed transcription factor whose role in neuronal protection is now well established. It has been suggested that CREB is present in the mitochondrial matrix of neurons and binds directly to cyclic AMP response elements (CREs) found within the mitochondrial genome [96]. Therefore, estrogens could also regulate mitochondrial transcription through a ER-independent mechanism.
7.4. Non-genomic actions
In addition to genomic action, growing evidence also indicates non-genomic actions of estrogens. In contrast to genomic action, non-genomic actions are principally characterized by their rapid onset of action and insensitivity to inhibitors of transcription and protein synthesis [49]. These rapid effects are distinct both physiologically and pharmacologically from those of the genomic actions. For example, we have very recently demonstrated that estrogens rapidly (~500 ms) potentiate L-type voltage-gated calcium channels at extremely low concentrations independent of ER activation [149]. We and others have also demonstrated that estrogens exert protective effects against ischemic stroke in both acute and post-treatment paradigms [196]. This suggests that rapid non-genomic actions of estrogens can play critical role in the neuroprotective effects of estrogens.
Distinct from the classical genomic actions of estrogens through nuclear ERs, the rapid non-genomic actions of estrogens are transmitted via specific membrane receptors. Many steroid receptors, including ERs, have been found to translocate to the plasma membrane [98,99]. Recent studies have identified a highly conserved motif in the ligand binding domains of steroid receptor which enable this translocation [127]. Further, it has been demonstrated that the plasma membrane pool of ERs plays functions distinct from the nuclear ERs [126]. Another recently identified membrane receptor that has been claimed as an estrogen binding protein is the orphan receptor, GPCR30, which to mediates PI3 K and calcium signaling [144]. Regardless of the receptor mediator of estrogen's effects, the action of estrogens initiated at the cell membrane activates a variety of intracellular signaling pathways, including but not limited to PKC, cyclic AMP, PKA, MAPK, and the tyrosine kinases [46]. The activation of these intracellular signaling pathways results primarily in phosphorylation/dephosphorylation of signaling proteins and produces a variety of cellular responses (see review [35]). Given the critical function of mitochondria under normal physiological conditions as well as oxidative stress, it is not surprising that many of these signaling pathways converge on the mitochondria. For example, the PI3K signaling pathway could directly interact with mitochondria by phosphorylation of Bad and inhibition of apoptosis inducing factor (AIF) translocation [124]. Protein kinase C epsilon, which has been found to play important role in the neuroprotective action of estrogens [120], has been classified as a novel type of PKC that is involved in many cellular events regulating mitochondrial function [1]. Further, the rapid non-genomic intracellular cascades may also lead to a transcriptional activation. Therefore, estrogens induce both non-genomic and genomic actions via the integration of its action on membrane associated and nuclear receptors [99].
8. The role of serine/threonine phosphatases in mitochondrial function
Phosphorylation events on the outer membrane of the mitochondria are well defined. Bcl-2 can be modulated by dimerization with proapoptotic family members (i.e. Bax, BAD, Bid) and by phosphorylation. The dynamic phosphorylation and dephosphorylation of Bcl-2 causes conformational change within the protein and has been suggested to serve as survival sensor during stress stimuli [147]. Bcl-2 phosphorylation by a variety of kinases, such as PKC, ERK, Akt, PI-3 K, and others, is a cell survival signal; while dephosphorylation by PP2A and/or PP1 is associated with cell death [146]. BAD, a BH3-proapoptotic Bcl-2 family member, acts at a key nodal point in the mitochondrial apoptotic pathway. Unphosphorylated BAD binds and inactivates antiapoptotic Bcl-2 homologues. This provokes release of cytochrome c from mitochondria and consequent activation of the apoptotic pathway. PP1, PP2A, and calcineurin has been shown to dephosphorylate and activate BAD [6,32,181] and BAX [25,75,208]. In addition, upon dephosphorylation by PP1 and/or PP2A, BAX is thought to insert into the outer mitochondrial membrane to disrupt membrane stability and release cytochrome c.
9. Evidence of ER-independent intracellular signaling actions of estrogens
The antioxidant activity of estrogens observed at high (non-physiological) concentrations (10−5 M) is dependent upon the presence of a phenolic A ring in the steroid structure and is independent of activation of the ERs [10–12,36,110]. A large body of evidence has demonstrated that estradiol possesses antioxidant properties and suppresses oxidative stress in neurons and neuronal cell lines induced by hydrogen peroxide, superoxide anions and other pro-oxidants [8–12,18,23,151]. Both 17α- and 17β-estradiol have similar antioxidant effects [11,12,18,61], suggesting that the neuroprotective effects of estrogens are in part ER-independent.
Strong evidence from our laboratory and others indicate that many of the neuroprotective effects of 17β-estradiol are ER receptor-independent since 17α-estradiol, the enantiomer of 17β-estradiol (ENT E2), and other non-ER binding estrogen analogues show potent protection against a variety of neurotoxicities [11,61,66,102,129,151]. The finding that 17α- and 17β-estradiol protect SK-N-SH human neuroblastoma cells from serum deprivation and the effect is only partially reversed by the ER antagonist tamoxifen suggests that estrogens may have ER-independent neuroprotective effects [61]. 17α- and 17β-estradiol also protect against beta-amyloid peptide toxicity in a murine immortalized neuronal cell line lacking functional estrogen receptors [64]. Additionally, it has also been demonstrated that treatment with either 17α-estradiol or ENT E2 markedly reduces ischemic brain damage produced by middle cerebral artery occlusion in ovariectomized rats [159,195,198], and that estradiol administration reduces secondary ischemic damage and mortality following subarachnoid hemorrhage in vivo [195]. ZYC3, a non-receptor-binding estrogen analogue, possesses both neuroprotective and vasoactive effects, which offers the possibility of clinical application for stroke without the side effects of estrogens [103]. ER antagonists do not block the neuroprotective effects of estradiol against NMDA-induced neuronal death in rat hippocampal cultures, glutamate neurotoxicity in mesencephalic cultures and pro-oxidants in rodent neuronal cultures [12,110,151,190]. Also, estrogens can exert their neuroprotective action even in the presence of transcription and translation inhibitors [142,151]. Estrogens may decrease the production of cytokines in astroglial cells exposed to Aβ1–40, lipopolysaccharides [41] or decrease the inflammatory response after brain injury [56] by decreasing the activation of NF-κB, a potent immediate-early transcriptional regulator of numerous proinflammatory genes, through anti-oxidative mechanism. Estradiol interact with estrogen binding sites in the plasma membrane [140] or mitochondrial membrane [109] and may have many different rapid effects on neuronal excitability and neuronal signal transduction.
Estrogens are potent inhibitors of lipid peroxidation. 17β-Estradiol inhibits iron-induced lipid peroxidation of membrane and fatty acids in rat brain homogenates, rat cortical synaptosomes, hippocampal HT-22 cells and primary neocortical cultures [84,179]. Further, 17α-estradiol, ENT E2, and ZYC3 potently inhibit of glutamate-induced lipid peroxidation, protein carbonylation, and ROS production, events that cause mitochondrial dysfunction followed by apoptosis [200]. ZYC3 is more potent in a number of cell types than 17β-estradiol, 17α-estradiol or ENT E2 in protecting neurons against glutamate toxicity [66,103,129].
Studies of structure–activity relationships (SAR) and mitochondrial function have led to a mechanistic model in which estrogens intercalate into cell membranes where they block lipid peroxidation reactions, and are in turn recycled. Indeed, the parental estrogens and novel analogs stabilize mitochondria under Ca2+ loading otherwise sufficient to collapse membrane potential [45]. Also, for a series of estrogens that were chosen for their wide differences in potency in neuorprotection against H2O2 toxicity, we observed a strong correlation between neuroprotective activity and protection from mitochondrial membrane potential collapse (Fig. 5). This correlation between neuroprotective and mito-protective potencies suggests that these compounds prevent cell death in large measure by maintaining functionally intact mitochondria.
In these SAR studies, the driving relationship appears to be the antioxidant potential of the estrogen analogue [133,134,137]. These observations lead us to assess the possibility that estrogens intercalate into membrane, including the mitochondrial members, and there participate in a redox cycle where the estrogens can be oxidized and thereby stop the cascade of lipid oxidation during damage to membranes. We observed that estrogens were indeed oxidized to a 10-hydroxy-quinols, the preferred oxidative product in both liver and brain [135,136]. Intriguingly, the oxidized form of estrogens can be rapidly converted to the parent estrogen in the brain [135]. This redox activity of estrogens can explain their potent antioxidant activity, despite the fact that estrogens are poor free radical scavengers [130].
10. Summary and conclusions
This treaties has provided evidence that the neuroprotective effects of estrogens are inexorably linked to their ability to protect mitochondria. The mechanisms by which estrogens protect mitochondria are multifaceted and complex. These include the ER-mediated transcription of nuclear genes involved in glucose transport into the brain, bioenergetics and the production of anti-apoptotic proteins. Second, localization of ERβ to the mitochondrial matrix positions this ER to mediate mitochondrial transcription of gene involved in oxidative phosphorylation. Third, signaling of estrogens from plasma membrane ERs as well as non-ER proteins activate a number of signaling pathways that are believed to provide vital information to the neuron leading to activation of survival mechanisms. Forth, estrogens have remarkable effects on protein phophatases that maintain their activity during neuronal stress, preserving the delicate balance between protein phosphorylation and dephosphorylation that is so vital to neuronal homeostasis. Finally, estrogens are potent lipid peroxidation inhibitors, most likely through their ability to stop propagation of lipid damage by engaging in a redox cycle and thereby tapping into other antioxidant molecules and applying them to member phospholipids.
A challenge for future research on estrogen neuroprotection is to decipher if these many actions of estrogens are organized hierarchically and are temporally sequenced, or simply represent the many characteristics of a very intriguing class of molecules. Which ever turn out to be the case, the targeting of the various neuroprotective actions of estrogens for protection from acute brain injury as well as chronic neurodegenerative diseases is a useful endeavor. Through chemical modification of naturally occurring estrogens as well as the synthesis of compounds that mimic the neuroprotective effects of estrogens, we may be able to produce more selective compounds that provide the benefits of estrogens in the brain while avoiding their side effects.
Acknowledgments
Original data presented were supported by P01 AG10485, P01 AG22550 and P01 AG027956.
References
- [1].Akita Y, Kawasaki H, Imajoh-Ohmi S, Fukuda H, Ohno S, Hirano H, Ono Y, Yonekawa H. Protein kinase C epsilon phosphorylates keratin 8 at Ser8 and Ser23 in GH4C1 cells stimulated by thyrotropin-releasing hormone. FEBS J. 2007;274:3270–3285. doi: 10.1111/j.1742-4658.2007.05853.x. [DOI] [PubMed] [Google Scholar]
- [2].Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–165. doi: 10.1161/01.str.29.1.159. discussion 166. [DOI] [PubMed] [Google Scholar]
- [4].Arendt T, Holzer M, Fruth R, Bruckner MK, Gartner U. Paired helical filament-like phosphorylation of tau, deposition of beta/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience. 1995;69:691–698. doi: 10.1016/0306-4522(95)00347-l. [DOI] [PubMed] [Google Scholar]
- [5].Arias C, Sharma N, Davies P, Shafit-Zagardo B. Okadaic acid induces early changes in microtubule-associated protein 2 and tau phosphorylation prior to neurodegeneration in cultured cortical neurons. J. Neurochem. 1993;61:673–682. doi: 10.1111/j.1471-4159.1993.tb02172.x. [DOI] [PubMed] [Google Scholar]
- [6].Ayllon V, Martinez AC, Garcia A, Cayla X, Rebollo A. Protein phosphatase 1alpha is a Ras-activated Bad phosphatase that regulates interleukin-2 deprivation-induced apoptosis. EMBO J. 2000;19:2237–2246. doi: 10.1093/emboj/19.10.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001;21:129–145. doi: 10.1002/1098-1128(200103)21:2<129::aid-med1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- [8].Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog. Neurobiol. 1999;57:301–323. doi: 10.1016/s0301-0082(98)00055-0. [DOI] [PubMed] [Google Scholar]
- [9].Behl C, Holsboer F. The female sex hormone oestrogen as a neuroprotectant. Trends Pharmacol. Sci. 1999;20:441–444. doi: 10.1016/s0165-6147(99)01392-9. [DOI] [PubMed] [Google Scholar]
- [10].Behl C, Manthey D. Neuroprotective activities of estrogen: an update. J. Neurocytol. 2000;29:351–358. doi: 10.1023/a:1007109222673. [DOI] [PubMed] [Google Scholar]
- [11].Behl C, Skutella T, Lezoualc'h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol. Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- [12].Behl C, Widmann M, Trapp T, Holsboer F. 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem. Biophys. Res. Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- [13].Bennett PC, Zhao W, Ng KT. Concentration-dependent effects of protein phosphatase (PP) inhibitors implicate PP1 and PP2A in different stages of memory formation. Neurobiol. Learn Mem. 2001;75:91–110. doi: 10.1006/nlme.1999.3959. [DOI] [PubMed] [Google Scholar]
- [14].Bettini E, Maggi A. Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus. J. Neurochem. 1992;58:1923–1929. doi: 10.1111/j.1471-4159.1992.tb10070.x. [DOI] [PubMed] [Google Scholar]
- [15].Beyer C, Raab H. Nongenomic effects of oestrogen: embryonic mouse midbrain neurones respond with a rapid release of calcium from intracellular stores. Eur. J. Neurosci. 1998;10:255–262. doi: 10.1046/j.1460-9568.1998.00045.x. [DOI] [PubMed] [Google Scholar]
- [16].Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc. Natl. Acad. Sci. USA. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boe R, Gjertsen BT, Vintermyr OK, Houge G, Lanotte M, Doskeland SO. The protein phosphatase inhibitor okadaic acid induces morphological changes typical of apoptosis in mammalian cells. Exp. Cell Res. 1991;195:237–246. doi: 10.1016/0014-4827(91)90523-w. [DOI] [PubMed] [Google Scholar]
- [18].Bonnefont AB, Munoz FJ, Inestrosa NC. Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase–amyloid complexes. FEBS Lett. 1998;441:220–224. doi: 10.1016/s0014-5793(98)01552-x. [DOI] [PubMed] [Google Scholar]
- [19].Brecht S, Gelderblom M, Srinivasan A, Mielke K, Dityateva G, Herdegen T. Caspase-3 activation and DNA fragmentation in primary hippocampal neurons following glutamate excitotoxicity. Brain Res. Mol. Brain Res. 2001;94:25–34. doi: 10.1016/s0006-8993(01)02767-6. [DOI] [PubMed] [Google Scholar]
- [20].Brinton RD. Estrogen regulation of glucose metabolism and mitochondrial function: therapeutic implications for prevention of Alzheimer's disease. Adv. Drug Deliv. Rev. 2008;60:1504–1511. doi: 10.1016/j.addr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Brinton RD. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends Neurosci. 2008;31:529–537. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brune B, Mohr S. Protein thiol modification of glyceraldehyde-3-phosphate dehydrogenase and caspase-3 by nitric oxide. Curr. Protein Pept. Sci. 2001;2:61–72. doi: 10.2174/1389203013381206. [DOI] [PubMed] [Google Scholar]
- [23].Calderon FH, Bonnefont A, Munoz FJ, Fernandez V, Videla LA, Inestrosa NC. PC12 and neuro 2a cells have different susceptibilities to acetylcholinesterase-amyloid complexes, amyloid25–35 fragment, glutamate, and hydrogen peroxide. J. Neurosci. Res. 1999;56:620–631. doi: 10.1002/(SICI)1097-4547(19990615)56:6<620::AID-JNR8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- [24].Carlstrom L, Ke ZJ, Unnerstall JR, Cohen RS, Pandey SC. Estrogen modulation of the cyclic AMP response element-binding protein pathway. Effects of long-term and acute treatments. Neuroendocrinology. 2001;74:227–243. doi: 10.1159/000054690. [DOI] [PubMed] [Google Scholar]
- [25].Chatfield K, Eastman A. Inhibitors of protein phosphatases 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem. Biophys. Res. Commun. 2004;323:1313–1320. doi: 10.1016/j.bbrc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- [26].Chen D, Xu X, Cheon YP, Bagchi MK, Bagchi IC. Estrogen induces expression of secretory leukocyte protease inhibitor in rat uterus. Biol. Reprod. 2004;71:508–514. doi: 10.1095/biolreprod.103.024919. [DOI] [PubMed] [Google Scholar]
- [27].Chen J, Delannoy M, Odwin S, He P, Trush MA, Yager JD. Enhanced mitochondrial gene transcript, ATP, bcl-2 protein levels, and altered glutathione distribution in ethinyl estradiol-treated cultured female rat hepatocytes. Toxicol. Sci. 2003;75:271–278. doi: 10.1093/toxsci/kfg183. [DOI] [PubMed] [Google Scholar]
- [28].Chen J, Gokhale M, Li Y, Trush MA, Yager JD. Enhanced levels of several mitochondrial mRNA transcripts and mitochondrial superoxide production during ethinyl estradiol-induced hepatocarcinogenesis and after estrogen treatment of HepG2 cells. Carcinogenesis. 1998;19:2187–2193. doi: 10.1093/carcin/19.12.2187. [DOI] [PubMed] [Google Scholar]
- [29].Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am. J. Physiol. Endocrinol. Metab. 2004;286:E1011–E1022. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- [30].Chen JQ, Yager JD. Estrogen's effects on mitochondrial gene expression: mechanisms and potential contributions to estrogen carcinogenesis. Ann. NY Acad. Sci. 2004;1028:258–272. doi: 10.1196/annals.1322.030. [DOI] [PubMed] [Google Scholar]
- [31].Chen JQ, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim. Biophys. Acta. 2005;1746:1–17. doi: 10.1016/j.bbamcr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- [32].Chiang CW, Harris G, Ellig C, Masters SC, Subramanian R, Shenolikar S, Wadzinski BE, Yang E. Protein phosphatase 2A activates the proapoptotic function of BAD in interleukin-3-dependent lymphoid cells by a mechanism requiring 14-3-3 dissociation. Blood. 2001;97:1289–1297. doi: 10.1182/blood.v97.5.1289. [DOI] [PubMed] [Google Scholar]
- [33].Clayton DA. Transcription and replication of mitochondrial DNA. Hum. Reprod. 2000;15(suppl 2):11–17. doi: 10.1093/humrep/15.suppl_2.11. [DOI] [PubMed] [Google Scholar]
- [34].Coker LH, Hogan PE, Bryan NR, Kuller LH, Margolis KL, Bettermann K, Wallace RB, Lao Z, Freeman R, Stefanick ML, Shumaker SA. Postmenopausal hormone therapy and subclinical cerebrovascular disease: the WHIMS-MRI study. Neurology. 2009;72:125–134. doi: 10.1212/01.wnl.0000339036.88842.9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cornil CA, Ball GF, Balthazart J. Functional significance of the rapid regulation of brain estrogen action: where do the estrogens come from? Brain Res. 2006;1126:2–26. doi: 10.1016/j.brainres.2006.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Culmsee C, Vedder H, Ravati A, Junker V, Otto D, Ahlemeyer B, Krieg JC, Krieglstein J. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: evidence for a receptor-independent antioxidative mechanism. J. Cereb. Blood Flow Metab. 1999;19:1263–1269. doi: 10.1097/00004647-199911000-00011. [DOI] [PubMed] [Google Scholar]
- [37].da Cruz e Silva OA, Fardilha M, Henriques AG, Rebelo S, Vieira S, da Cruz e Silva EF. Signal transduction therapeutics: relevance for Alzheimer's disease. J. Mol. Neurosci. 2004;23:123–142. doi: 10.1385/JMN:23:1-2:123. [DOI] [PubMed] [Google Scholar]
- [38].DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- [39].Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- [40].Diwakar L, Kenchappa RS, Annepu J, Saeed U, Sujanitha R, Ravindranath V. Down-regulation of glutaredoxin by estrogen receptor antagonist renders female mice susceptible to excitatory amino acid mediated complex I inhibition in CNS. Brain Res. 2006;1125:176–184. doi: 10.1016/j.brainres.2006.10.015. [DOI] [PubMed] [Google Scholar]
- [41].Dodel RC, Du Y, Bales KR, Gao F, Paul SM. Sodium salicylate and 17beta-estradiol attenuate nuclear transcription factor NF-kappaB translocation in cultured rat astroglial cultures following exposure to amyloid A beta(1–40) and lipopolysaccharides. J. Neurochem. 1999;73:1453–1460. doi: 10.1046/j.1471-4159.1999.0731453.x. [DOI] [PubMed] [Google Scholar]
- [42].Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J. Cereb. Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- [43].Dubey RK, Imthurn B, Barton M, Jackson EK. Vascular consequences of menopause and hormone therapy: importance of timing of treatment and type of estrogen. Cardiovasc. Res. 2005;66:295–306. doi: 10.1016/j.cardiores.2004.12.012. [DOI] [PubMed] [Google Scholar]
- [44].Dumas J, Hancur-Bucci C, Naylor M, Sites C, Newhouse P. Estradiol interacts with the cholinergic system to affect verbal memory in postmenopausal women: evidence for the critical period hypothesis. Horm. Behav. 2008;53:159–169. doi: 10.1016/j.yhbeh.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dykens JA, Simpkins JW, Wang J, Gordon K. Polycyclic phenols, estrogens and neuroprotection: a proposed mitochondrial mechanism. Exp. Gerontol. 2003;38:101–107. doi: 10.1016/s0531-5565(02)00162-6. [DOI] [PubMed] [Google Scholar]
- [46].Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Ann. Rev. Physiol. 2005;67:335–376. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- [47].Ermak G, Morgan TE, Davies KJ. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J. Biol. Chem. 2001;276:38787–38794. doi: 10.1074/jbc.M102829200. [DOI] [PubMed] [Google Scholar]
- [48].Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Ann. Rev. Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- [49].Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M. Multiple actions of steroid hormones – a focus on rapid, nongenomic effects. Pharmacol. Rev. 2000;52:513–556. [PubMed] [Google Scholar]
- [50].Fernandez-Sanchez MT, Garcia-Rodriguez A, Diaz-Trelles R, Novelli A. Inhibition of protein phosphatases induces IGF-1-blocked neurotrophin-insensitive neuronal apoptosis. FEBS Lett. 1996;398:106–112. doi: 10.1016/s0014-5793(96)01192-1. [DOI] [PubMed] [Google Scholar]
- [51].Fernandez MT, Zitko V, Gascon S, Novelli A. The marine toxin okadaic acid is a potent neurotoxin for cultured cerebellar neurons. Life Sci. 1991;49:PL157–PL162. doi: 10.1016/0024-3205(91)90398-u. [DOI] [PubMed] [Google Scholar]
- [52].Ferrer I, Friguls B, Dalfo E, Planas AM. Early modifications in the expression of mitogen-activated protein kinase (MAPK/ERK) stress-activated kinases SAPK/JNK and p38 and their phosphorylated substrates following focal cerebral ischemia. Acta Neuropathol. (Berl.) 2003;105:425–1437. doi: 10.1007/s00401-002-0661-2. [DOI] [PubMed] [Google Scholar]
- [53].Ferri A, Gabbianelli R, Casciati A, Paolucci E, Rotilio G, Carri MT. Calcineurin activity is regulated both by redox compounds and by mutant familial amyotrophic lateral sclerosis-superoxide dismutase. J. Neurochem. 2000;75:606–613. doi: 10.1046/j.1471-4159.2000.0750606.x. [DOI] [PubMed] [Google Scholar]
- [54].Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Rad. Biol. Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]
- [55].Foley TD, Armstrong JJ, Kupchak BR. Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochem. Biophys. Res. Commun. 2004;315:568–574. doi: 10.1016/j.bbrc.2004.01.096. [DOI] [PubMed] [Google Scholar]
- [56].Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Prog. Neurobiol. 2001;63:29–60. doi: 10.1016/s0301-0082(00)00025-3. [DOI] [PubMed] [Google Scholar]
- [57].Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418:970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- [58].Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J. Neurochem. 1995;65:732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- [59].Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993;61:921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- [60].Gottipati S, Cammarata PR. Mitochondrial superoxide dismutase activation with 17 beta-estradiol-treated human lens epithelial cells. Mol. Vis. 2008;14:898–905. [PMC free article] [PubMed] [Google Scholar]
- [61].Green PS, Bishop J, Simpkins JW. 17 alpha-Estradiol exerts neuroprotective effects on SK-N-SH cells. J. Neurosci. 1997;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Green PS, Gordon K, Simpkins JW. Phenolic A ring requirement for the neuroprotective effects of steroids. J. Steroid Biochem. Mol. Biol. 1997;63:229–235. doi: 10.1016/s0960-0760(97)00124-6. [DOI] [PubMed] [Google Scholar]
- [63].Green PS, Gridley KE, Simpkins JW. Estradiol protects against beta-amyloid (25–35)-induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci. Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- [64].Green PS, Gridley KE, Simpkins JW. Nuclear estrogen receptor-independent neuroprotection by estratrienes: a novel interaction with glutathione. Neuroscience. 1998;84:7–10. doi: 10.1016/s0306-4522(97)00595-2. [DOI] [PubMed] [Google Scholar]
- [65].Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int. J. Dev. Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- [66].Green PS, Yang SH, Nilsson KR, Kumar AS, Covey DF, Simpkins JW. The nonfeminizing enantiomer of 17beta-estradiol exerts protective effects in neuronal cultures and a rat model of cerebral ischemia. Endocrinology. 2001;142:400–406. doi: 10.1210/endo.142.1.7888. [DOI] [PubMed] [Google Scholar]
- [67].Gridley KE, Green PS, Simpkins JW. Low concentrations of estradiol reduce beta-amyloid (25–35)-induced toxicity, lipid peroxidation and glucose utilization in human SK-N-SH neuroblastoma cells. Brain Res. 1997;778:158–165. doi: 10.1016/s0006-8993(97)01056-1. [DOI] [PubMed] [Google Scholar]
- [68].Grodstein F, Manson JE, Stampfer MJ. Hormone therapy and coronary heart disease: the role of time since menopause and age at hormone initiation. J. Womens Health (Larchmt) 2006;15:35–44. doi: 10.1089/jwh.2006.15.35. [DOI] [PubMed] [Google Scholar]
- [69].Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am. J. Physiol. Cell Physiol. 2007:C45–C51. doi: 10.1152/ajpcell.00229.2006. [DOI] [PubMed] [Google Scholar]
- [70].Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- [71].Harman SM. What do hormones have to do with aging? What does aging have to do with hormones? Ann NY Acad. Sci. 2004;1019:299–308. doi: 10.1196/annals.1297.051. [DOI] [PubMed] [Google Scholar]
- [72].Harper SJ, Wilkie N. MAPKs: new targets for neurodegeneration. Expert Opin. Ther. Targets. 2003;7:187–200. doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- [73].Hodis HN, Mack WJ, Lobo RA. Randomized controlled trial evidence that estrogen replacement therapy reduces the progression of subclinical atherosclerosis in healthy postmenopausal women without preexisting cardiovascular disease. Circulation. 2003;108:e5. doi: 10.1161/01.CIR.0000080080.76333.38. author reply e5. [DOI] [PubMed] [Google Scholar]
- [74].Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J. Mol. Cell Cardiol. 2006;41:511–521. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- [75].Hsiung SC, Tin A, Tamir H, Franke TF, Liu KP. Inhibition of 5-HT1A receptor-dependent cell survival by cAMP/protein kinase A: role of protein phosphatase 2A and Bax. J. Neurosci. Res. 2008;86:2326–2338. doi: 10.1002/jnr.21676. [DOI] [PubMed] [Google Scholar]
- [76].Hu Y, Kang C, Philp RJ, Li B. PKC delta phosphorylates p52ShcA at Ser29 to regulate ERK activation in response to H2O2. Cell Signal. 2007;19:410–418. doi: 10.1016/j.cellsig.2006.07.017. [DOI] [PubMed] [Google Scholar]
- [77].Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc. Natl. Acad. Sci. USA. 1999;96:4686–4691. doi: 10.1073/pnas.96.8.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jouvenceau A, Dutar P. A role for the protein phosphatase 2B in altered hippocampal synaptic plasticity in the aged rat. J. Physiol. Paris. 2006;99:154–161. doi: 10.1016/j.jphysparis.2005.12.009. [DOI] [PubMed] [Google Scholar]
- [79].Jouvenceau A, Hedou G, Potier B, Kollen M, Dutar P, Mansuy IM. Partial inhibition of PP1 alters bidirectional synaptic plasticity in the hippocampus. Eur. J. Neurosci. 2006;24:564–572. doi: 10.1111/j.1460-9568.2006.04938.x. [DOI] [PubMed] [Google Scholar]
- [80].Jung ME, Gatch MB, Simpkins JW. Estrogen neuroprotection against the neurotoxic effects of ethanol withdrawal: potential mechanisms. Exp. Biol. Med. (Maywood) 2005;(23):8–22. doi: 10.1177/153537020523000102. [DOI] [PubMed] [Google Scholar]
- [81].Kanterewicz BI, Knapp LT, Klann E. Stimulation of p42 and p44 mitogen-activated protein kinases by reactive oxygen species and nitric oxide in hippocampus. J. Neurochem. 1998;70:1009–1016. doi: 10.1046/j.1471-4159.1998.70031009.x. [DOI] [PubMed] [Google Scholar]
- [82].Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation: estrogen receptor alpha and estrogen receptor beta: regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Keen JC, Zhou Q, Park BH, Pettit C, Mack KM, Blair B, Brenner K, Davidson NE. Protein phosphatase 2A regulates estrogen receptor alpha (ER) expression through modulation of ER mRNA stability. J. Biol. Chem. 2005;280:29519–29524. doi: 10.1074/jbc.M505317200. [DOI] [PubMed] [Google Scholar]
- [84].Keller JN, Germeyer A, Begley JG, Mattson MP. 17Beta-estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J. Neurosci. Res. 1997;50:522–530. doi: 10.1002/(SICI)1097-4547(19971115)50:4<522::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- [85].Kelly MJ, Wagner EJ. Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol. Metab. 1999;10:369–374. doi: 10.1016/s1043-2760(99)00190-3. [DOI] [PubMed] [Google Scholar]
- [86].Kim JY, Yang MS, Oh CD, Kim KT, Ha MJ, Kang SS, Chun JS. Signalling pathway leading to an activation of mitogen-activated protein kinase by stimulating M3 muscarinic receptor. Biochem. J. 1999;337(Pt 2):275–280. [PMC free article] [PubMed] [Google Scholar]
- [87].Kins S, Kurosinski P, Nitsch RM, Gotz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am. J. Pathol. 2003;163:833–843. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kostanyan A, Nazaryan K. Rat brain glycolysis regulation by estradiol-17 beta. Biochim. Biophys. Acta. 1992;1133:301–306. doi: 10.1016/0167-4889(92)90051-c. [DOI] [PubMed] [Google Scholar]
- [89].Kraft CA, Efimova T, Eckert RL. Activation of PKCdelta and p38delta MAPK during okadaic acid dependent keratinocyte apoptosis. Arch. Dermatol. Res. 2007;299:71–83. doi: 10.1007/s00403-006-0727-4. [DOI] [PubMed] [Google Scholar]
- [90].Kroemer G, Reed JC. Mitochondrial control of cell death. Nat. Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- [91].Kuperstein F, Yavin E. ERK activation and nuclear translocation in amyloid-beta peptide- and iron-stressed neuronal cell cultures. Eur. J. Neurosci. 2002;16:44–54. doi: 10.1046/j.1460-9568.2002.02056.x. [DOI] [PubMed] [Google Scholar]
- [92].Kuroki Y, Fukushima K, Kanda Y, Mizuno K, Watanabe Y. Putative membrane-bound estrogen receptors possibly stimulate mitogen-activated protein kinase in the rat hippocampus. Eur. J. Pharmacol. 2000;400:205–209. doi: 10.1016/s0014-2999(00)00425-8. [DOI] [PubMed] [Google Scholar]
- [93].Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R, Webb P, Greene G. Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp. 2000;230:20–26. doi: 10.1002/0470870818.ch3. discussion 27–40. [DOI] [PubMed] [Google Scholar]
- [94].Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- [95].Leclerc P, de Lamirande E, Gagnon C. Regulation of protein–tyrosine phosphorylation and human sperm capacitation by reactive oxygen derivatives. Free Rad. Biol. Med. 1997;22:643–656. doi: 10.1016/s0891-5849(96)00379-6. [DOI] [PubMed] [Google Scholar]
- [96].Lee J, Kim CH, Simon DK, Aminova LR, Andreyev AY, Kushnareva YE, Murphy AN, Lonze BE, Kim KS, Ginty DD, Ferrante RJ, Ryu H, Ratan RR. Mitochondrial cyclic AMP response element-binding protein (CREB) mediates mitochondrial gene expression and neuronal survival. J. Biol. Chem. 2005;280:40398–40401. doi: 10.1074/jbc.C500140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Lee SJ, McEwen BS. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Ann. Rev. Pharmacol. Toxicol. 2001;41:569–591. doi: 10.1146/annurev.pharmtox.41.1.569. [DOI] [PubMed] [Google Scholar]
- [98].Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J. Appl. Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- [99].Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Mol. Endocrinol. 2005;19:1951–1959. doi: 10.1210/me.2004-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp. Neurol. 2001;167:158–165. doi: 10.1006/exnr.2000.7534. [DOI] [PubMed] [Google Scholar]
- [101].Liu H, Liu K, Bodenner DL. Estrogen receptor inhibits interleukin-6 gene expression by disruption of nuclear factor kappaB transactivation. Cytokine. 2005;31:251–257. doi: 10.1016/j.cyto.2004.12.008. [DOI] [PubMed] [Google Scholar]
- [102].Liu R, Wen Y, Perez E, Wang X, Day AL, Simpkins JW, Yang SH. 17beta-Estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res. 2005;1060:55–61. doi: 10.1016/j.brainres.2005.08.048. [DOI] [PubMed] [Google Scholar]
- [103].Liu R, Yang SH, Perez E, Yi KD, Wu SS, Eberst K, Prokai L, Prokai-Tatrai K, Cai ZY, Covey DF, Day AL, Simpkins JW. Neuroprotective effects of a novel non-receptor-binding estrogen analogue: in vitro and in vivo analysis. Stroke. 2002;33:2485–2491. doi: 10.1161/01.str.0000030317.43597.c8. [DOI] [PubMed] [Google Scholar]
- [104].Lu Q, Surks HK, Ebling H, Baur WE, Brown D, Pallas DC, Karas RH. Regulation of estrogen receptor alpha-mediated transcription by a direct interaction with protein phosphatase 2A. J. Biol. Chem. 2003;278:4639–4645. doi: 10.1074/jbc.M210949200. [DOI] [PubMed] [Google Scholar]
- [105].Maki PM. Potential importance of early initiation of hormone therapy for cognitive benefit. Menopause. 2006;13:6–7. doi: 10.1097/01.gme.0000194822.76774.30. [DOI] [PubMed] [Google Scholar]
- [106].Mannella P, Brinton RD. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J. Neurosci. 2006;26:9439–9447. doi: 10.1523/JNEUROSCI.1443-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, Kuller LH, Cochrane BB, Hunt JR, Ludlam SE, Pettinger MB, Gass M, Margolis KL, Nathan L, Ockene JK, Prentice RL, Robbins J, Stefanick ML. Estrogen therapy and coronary-artery calcification. New Engl. J. Med. 2007;356:2591–2602. doi: 10.1056/NEJMoa071513. [DOI] [PubMed] [Google Scholar]
- [108].Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO. J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- [109].Monje P, Boland R. Subcellular distribution of native estrogen receptor alpha and beta isoforms in rabbit uterus and ovary. J. Cell Biochem. 2001;82:467–479. doi: 10.1002/jcb.1182. [DOI] [PubMed] [Google Scholar]
- [110].Moosmann B, Behl C. Antioxidants as treatment for neurodegenerative disorders. Exp. Opin. Investig. Drugs. 2002;11:1407–1435. doi: 10.1517/13543784.11.10.1407. [DOI] [PubMed] [Google Scholar]
- [111].Morishita W, Connor JH, Xia H, Quinlan EM, Shenolikar S, Malenka RC. Regulation of synaptic strength by protein phosphatase 1. Neuron. 2001;32:1133–1148. doi: 10.1016/s0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- [112].Moss RL, Gu Q. Estrogen: mechanisms for a rapid action in CA1 hippocampal neurons. Steroids. 1999;64:14–21. doi: 10.1016/s0039-128x(98)00092-0. [DOI] [PubMed] [Google Scholar]
- [113].Muller JM, Cahill MA, Rupec RA, Baeuerle PA, Nordheim A. Antioxidants as well as oxidants activate c-fos via Ras-dependent activation of extracellular-signal-regulated kinase 2 and Elk-1. Eur. J. Biochem. 1997;244:45–52. doi: 10.1111/j.1432-1033.1997.00045.x. [DOI] [PubMed] [Google Scholar]
- [114].Munton RP, Vizi S, Mansuy IM. The role of protein phosphatase-1 in the modulation of synaptic and structural plasticity. FEBS Lett. 2004;567:121–128. doi: 10.1016/j.febslet.2004.03.121. [DOI] [PubMed] [Google Scholar]
- [115].Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc. Natl. Acad. Sci. USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- [117].Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr. Drug Targets CNS Neurol. Disord. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- [119].Nilsen J, Irwin RW, Gallaher TK, Brinton RD. Estradiol in vivo regulation of brain mitochondrial proteome. J. Neurosci. 2007;27:14069–14077. doi: 10.1523/JNEUROSCI.4391-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Novotny JL, Simpson AM, Tomicek NJ, Lancaster TS, Korzick DH. Rapid estrogen receptor-alpha activation improves ischemic tolerance in aged female rats through a novel protein kinase C epsilon-dependent mechanism. Endocrinology. 2009;150:889–896. doi: 10.1210/en.2008-0708. [DOI] [PubMed] [Google Scholar]
- [121].O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol. Endocrinol. 2007;21:1281–1296. doi: 10.1210/me.2006-0497. [DOI] [PubMed] [Google Scholar]
- [122].Ogita K, Okuda H, Kitano M, Fujinami Y, Ozaki K, Yoneda Y. Localization of activator protein-1 complex with DNA binding activity in mitochondria of murine brain after in vivo treatment with kainate. J. Neurosci. 2002;22:2561–2570. doi: 10.1523/JNEUROSCI.22-07-02561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Ouimet CC, da Cruz e Silva EF, Greengard P. The alpha and gamma 1 isoforms of protein phosphatase 1 are highly and specifically concentrated in dendritic spines. Proc. Natl. Acad. Sci. USA. 1995;92:3396–3400. doi: 10.1073/pnas.92.8.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Parcellier A, Tintignac LA, Zhuravleva E, Hemmings BA. PKB and the mitochondria: AKTing on apoptosis. Cell Signal. 2008;20:21–30. doi: 10.1016/j.cellsig.2007.07.010. [DOI] [PubMed] [Google Scholar]
- [125].Parihar MS, Brewer GJ. Mitoenergetic failure in Alzheimer disease. Am. J. Physiol. Cell Physiol. 2007;292:C8–C23. doi: 10.1152/ajpcell.00232.2006. [DOI] [PubMed] [Google Scholar]
- [126].Pedram A, Razandi M, Kim JK, O'Mahony F, Lee EY, Luderer U, Levin ER. Developmental phenotype of a membrane only estrogen receptor {alpha} (MOER) mouse. J. Biol. Chem. 2009;284:3488–3495. doi: 10.1074/jbc.M806249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J. Biol. Chem. 2007;282:22278–22288. doi: 10.1074/jbc.M611877200. [DOI] [PubMed] [Google Scholar]
- [128].Pedram A, Razandi M, Wallace DC, Levin ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol. Biol. Cell. 2006;17:2125–2137. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Perez E, Liu R, Yang SH, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005;1038:216–222. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- [130].Perez E, Cai Zu Yun, Covey Douglas F., Simpkins James W. Neuroprotective effects of estratriene analogs: structure–activity relationships and molecular optimization. Drug Develop. Res. 2005;66:78–92. [Google Scholar]
- [131].Pessah IN, Kim KH, Feng W. Redox sensing properties of the ryanodine receptor complex. Front Biosci. 2002;7:a72–a79. doi: 10.2741/A741. [DOI] [PubMed] [Google Scholar]
- [132].Pozzo-Miller LD, Inoue T, Murphy DD. Estradiol increases spine density and NMDA-dependent Ca2+ transients in spines of CA1 pyramidal neurons from hippocampal slices. J. Neurophysiol. 1999;81:1404–1411. doi: 10.1152/jn.1999.81.3.1404. [DOI] [PubMed] [Google Scholar]
- [133].Prokai-Tatrai K, Perjesi P, Rivera-Portalatin NM, Simpkins JW, Prokai L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids. 2008;73:280–288. doi: 10.1016/j.steroids.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Prokai L, Oon SM, Prokai-Tatrai K, Abboud KA, Simpkins JW. Synthesis and biological evaluation of 17beta-alkoxyestra-1,3,5(10)-trienes as potential neuroprotectants against oxidative stress. J. Med. Chem. 2001;44:110–114. doi: 10.1021/jm000280t. [DOI] [PubMed] [Google Scholar]
- [135].Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez EJ, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc. Natl. Acad. Sci. USA. 2003;100:11741–11746. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Simpkins JW. Quinol-based metabolic cycle for estrogens in rat liver microsomes. Drug Metab. Dispos. 2003;31:701–704. doi: 10.1124/dmd.31.6.701. [DOI] [PubMed] [Google Scholar]
- [137].Prokai L, Simpkins JW. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol. Ther. 2007;114:1–12. doi: 10.1016/j.pharmthera.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Raap DK, DonCarlos L, Garcia F, Muma NA, Wolf WA, Battaglia G, Van de Kar LD. Estrogen desensitizes 5-HT(1A) receptors and reduces levels of G(z), G(i1) and G(i3) proteins in the hypothalamus. Neuropharmacology. 2000;39:1823–1832. doi: 10.1016/s0028-3908(99)00264-6. [DOI] [PubMed] [Google Scholar]
- [139].Rahman A, Grundke-Iqbal I, Iqbal K. Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem. Res. 2005;30:277–287. doi: 10.1007/s11064-005-2483-9. [DOI] [PubMed] [Google Scholar]
- [140].Ramirez VD, Zheng J. Membrane sex-steroid receptors in the brain. Front Neuroendocrinol. 1996;17:402–439. doi: 10.1006/frne.1996.0011. [DOI] [PubMed] [Google Scholar]
- [141].Rao GN. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene. 1996;13:713–719. [PubMed] [Google Scholar]
- [142].Regan RF, Guo Y. Estrogens attenuate neuronal injury due to hemoglobin, chemical hypoxia, and excitatory amino acids in murine cortical cultures. Brain Res. 1997;764:133–140. doi: 10.1016/s0006-8993(97)00437-x. [DOI] [PubMed] [Google Scholar]
- [143].Resnick SM, Espeland MA, Jaramillo SA, Hirsch C, Stefanick ML, Murray AM, Ockene J, Davatzikos C. Postmenopausal hormone therapy and regional brain volumes: the WHIMS-MRI study. Neurology. 2009;72:135–142. doi: 10.1212/01.wnl.0000339037.76336.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- [145].Rice AB, Ingram JL, Bonner JC. p38 mitogen-activated protein kinase regulates growth factor-induced mitogenesis of rat pulmonary myofibroblasts. Am. J. Respir. Cell Mol. Biol. 2002;27:759–765. doi: 10.1165/rcmb.2002-0070OC. [DOI] [PubMed] [Google Scholar]
- [146].Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J. Biol. Chem. 1999;274:20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- [147].Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia. 2001;15:515–522. doi: 10.1038/sj.leu.2402090. [DOI] [PubMed] [Google Scholar]
- [148].Salpeter SR, Walsh JM, Greyber E, Salpeter EE. Brief report: Coronary heart disease events associated with hormone therapy in younger and older women. A meta-analysis. J. Gen. Int. Med. 2006;21:363–366. doi: 10.1111/j.1525-1497.2006.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA. 2008;105:15148–15153. doi: 10.1073/pnas.0802379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Satoh T, Nakatsuka D, Watanabe Y, Nagata I, Kikuchi H, Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci. Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- [151].Sawada H, Ibi M, Kihara T, Urushitani M, Akaike A, Shimohama S. Estradiol protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J. Neurosci. Res. 1998;54:707–719. doi: 10.1002/(SICI)1097-4547(19981201)54:5<707::AID-JNR16>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [152].Schmidt KN, Traenckner EB, Meier B, Baeuerle PA. Induction of oxidative stress by okadaic acid is required for activation of transcription factor NF-kappa B. J. Biol. Chem. 1995;270:27136–27142. doi: 10.1074/jbc.270.45.27136. [DOI] [PubMed] [Google Scholar]
- [153].Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel and atypical protein kinase C isotypes. Mol. Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Schreck R, Baeuerle PA. Assessing oxygen radicals as mediators in activation of inducible eukaryotic transcription factor NF-kappa B. Methods Enzymol. 1994;234:151–163. doi: 10.1016/0076-6879(94)34085-4. [DOI] [PubMed] [Google Scholar]
- [155].Shi J, Simpkins JW. 17 beta-Estradiol modulation of glucose transporter 1 expression in blood-brain barrier. Am. J. Physiol. 1997;272:E1016–E1022. doi: 10.1152/ajpendo.1997.272.6.E1016. [DOI] [PubMed] [Google Scholar]
- [156].Shi J, Zhang YQ, Simpkins JW. Effects of 17beta-estradiol on glucose transporter 1 expression and endothelial cell survival following focal ischemia in the rats. Exp. Brain Res. 1997;117:200–206. doi: 10.1007/s002210050216. [DOI] [PubMed] [Google Scholar]
- [157].Shivakumar BR, Kolluri SV, Ravindranath V. Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J. Pharmacol. Exp. Ther. 1995;274:1167–1173. [PubMed] [Google Scholar]
- [158].Simpkins JW, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Brain Res. Rev. 2008;57:421–430. doi: 10.1016/j.brainresrev.2007.04.007. [DOI] [PubMed] [Google Scholar]
- [159].Simpkins JW, Rajakumar G, Zhang YQ, Simpkins CE, Greenwald D, Yu CJ, Bodor N, Day AL. Estrogens may reduce mortality and ischemic damage caused by middle cerebral artery occlusion in the female rat. J. Neurosurg. 1997;87:724–730. doi: 10.3171/jns.1997.87.5.0724. [DOI] [PubMed] [Google Scholar]
- [160].Simpkins JW, Wang J, Wang X, Perez E, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr. Drug Targets CNS Neurol. Disord. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- [161].Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria-physiological and pathological implications. Mol. Cell Endocrinol. 2008;290:51–59. doi: 10.1016/j.mce.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Singh M, Setalo G, Jr., Guan X, Frail DE, Toran-Allerand CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. J. Neurosci. 2000;20:1694–1700. doi: 10.1523/JNEUROSCI.20-05-01694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Singh M, Setalo G, Jr., Guan X, Warren M, Toran-Allerand CD. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J. Neurosci. 1999;19:1179–1188. doi: 10.1523/JNEUROSCI.19-04-01179.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Slevin M, Krupinski J, Slowik A, Rubio F, Szczudlik A, Gaffney J. Activation of MAP kinase (ERK-1/ERK-2), tyrosine kinase and VEGF in the human brain following acute ischaemic stroke. Neuroreport. 2000;11:2759–2764. doi: 10.1097/00001756-200008210-00030. [DOI] [PubMed] [Google Scholar]
- [165].Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL., 3rd Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004;63:1080–1091. doi: 10.1093/jnen/63.10.1080. [DOI] [PubMed] [Google Scholar]
- [166].Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- [167].Strehlow K, Rotter S, Wassmann S, Adam O, Grohe C, Laufs K, Bohm M, Nickenig G. Modulation of antioxidant enzyme expression and function by estrogen. Circ. Res. 2003;93:170–177. doi: 10.1161/01.RES.0000082334.17947.11. [DOI] [PubMed] [Google Scholar]
- [168].Sudo S, Wen TC, Desaki J, Matsuda S, Tanaka J, Arai T, Maeda N, Sakanaka M. Beta-estradiol protects hippocampal CA1 neurons against transient forebrain ischemia in gerbil. Neurosci. Res. 1997;29:345–354. doi: 10.1016/s0168-0102(97)00106-5. [DOI] [PubMed] [Google Scholar]
- [169].Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. 2003;118:1175–1182. doi: 10.1016/s0306-4522(02)00697-8. [DOI] [PubMed] [Google Scholar]
- [170].Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- [171].Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Rad. Biol. Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- [172].Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- [173].Thibodeau PA, Kachadourian R, Lemay R, Bisson M, Day BJ, Paquette B. In vitro pro- and antioxidant properties of estrogens. J. Steroid Biochem. Mol. Biol. 2002;81:227–236. doi: 10.1016/s0960-0760(02)00067-5. [DOI] [PubMed] [Google Scholar]
- [174].Todd JL, Tanner KG, Denu JM. Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J. Biol. Chem. 1999;274:13271–13280. doi: 10.1074/jbc.274.19.13271. [DOI] [PubMed] [Google Scholar]
- [175].Toran-Allerand CD, Singh M, Setalo G., Jr. Novel mechanisms of estrogen action in the brain: new players in an old story. Front Neuroendocrinol. 1999;20:97–121. doi: 10.1006/frne.1999.0177. [DOI] [PubMed] [Google Scholar]
- [176].Tunez I, Munoz Mdel C, Feijoo M, Munoz-Castaneda JR, Bujalance I, Valdelvira ME, Montilla Lopez P. Protective melatonin effect on oxidative stress induced by okadaic acid into rat brain. J. Pineal. Res. 2003;34:265–268. doi: 10.1034/j.1600-079x.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- [177].Urata Y, Ihara Y, Murata H, Goto S, Koji T, Yodoi J, Inoue S, Kondo T. 17beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J. Biol. Chem. 2006;281:13092–13102. doi: 10.1074/jbc.M601984200. [DOI] [PubMed] [Google Scholar]
- [178].Van Itallie CM, Dannies PS. Estrogen induces accumulation of the mitochondrial ribonucleic acid for subunit II of cytochrome oxidase in pituitary tumor cells. Mol. Endocrinol. 1988;2:332–337. doi: 10.1210/mend-2-4-332. [DOI] [PubMed] [Google Scholar]
- [179].Vedder H, Anthes N, Stumm G, Wurz C, Behl C, Krieg JC. Estrogen hormones reduce lipid peroxidation in cells and tissues of the central nervous system. J. Neurochem. 1999;72:2531–2538. doi: 10.1046/j.1471-4159.1999.0722531.x. [DOI] [PubMed] [Google Scholar]
- [180].Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23:48–53. doi: 10.1016/s0166-2236(99)01500-3. [DOI] [PubMed] [Google Scholar]
- [181].Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+ induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- [182].Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc. Natl. Acad. Sci. USA. 2001;98:4038–4043. doi: 10.1073/pnas.061038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Wang X, Martindale JL, Liu Y, Holbrook NJ. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem. J. 1998;333(Pt 2):291–300. doi: 10.1042/bj3330291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Wang X, Wang H, Xu L, Rozanski DJ, Sugawara T, Chan PH, Trzaskos JM, Feuerstein GZ. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: exploration of potential mechanism associated with apoptosis. J. Pharmacol. Exp. Ther. 2003;304:172–178. doi: 10.1124/jpet.102.040246. [DOI] [PubMed] [Google Scholar]
- [185].Wang Z, Yang H, Tachado SD, Capo-Aponte JE, Bildin VN, Koziel H, Reinach PS. Phosphatase-mediated crosstalk control of ERK and p38 MAPK signaling in corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 2006;47:5267–5275. doi: 10.1167/iovs.06-0642. [DOI] [PubMed] [Google Scholar]
- [186].Wang ZQ, Chen XC, Yang GY, Zhou LF. U0126 prevents ERK pathway phosphorylation and interleukin-1beta mRNA production after cerebral ischemia. Chin. Med. Sci. J. 2004;19:270–275. [PubMed] [Google Scholar]
- [187].Watson RW, Rotstein OD, Nathens AB, Dackiw AP, Marshall JC. Thiol-mediated redox regulation of neutrophil apoptosis. Surgery. 1996;120:150–157. doi: 10.1016/s0039-6060(96)80282-0. discussion 157–158. [DOI] [PubMed] [Google Scholar]
- [188].Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- [189].Watters JJ, Dorsa DM. Transcriptional effects of estrogen on neuronal neurotensin gene expression involve cAMP/protein kinase A-dependent signaling mechanisms. J. Neurosci. 1998;18:6672–6680. doi: 10.1523/JNEUROSCI.18-17-06672.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Weaver CE, Jr., Park-Chung M, Gibbs TT, Farb DH. 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- [191].Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta. 2006 doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- [192].Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. Mitochondrial respiratory chain inhibitors induce apoptosis. FEBS Lett. 1994;339:40–44. doi: 10.1016/0014-5793(94)80380-3. [DOI] [PubMed] [Google Scholar]
- [193].Woo NH, Nguyen PV. “Silent” metaplasticity of the late phase of long-term potentiation requires protein phosphatases. Learn Mem. 2002;9:202–213. doi: 10.1101/lm.498402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Yamashita T, Inui S, Maeda K, Hua DR, Takagi K, Fukunaga K, Sakaguchi N. Regulation of CaMKII by alpha4/PP2Ac contributes to learning and memory. Brain Res. 2006;1082:1–10. doi: 10.1016/j.brainres.2006.01.101. [DOI] [PubMed] [Google Scholar]
- [195].Yang SH, He Z, Wu SS, He YJ, Cutright J, Millard WJ, Day AL, Simpkins JW. 17-beta Estradiol can reduce secondary ischemic damage and mortality of subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2001;21:174–181. doi: 10.1097/00004647-200102000-00009. [DOI] [PubMed] [Google Scholar]
- [196].Yang SH, Liu R, Perez EJ, Wang X, Simpkins JW. Estrogens as protectants of the neurovascular unit against ischemic stroke. Curr. Drug Targets CNS Neurol. Disord. 2005;4:169–177. doi: 10.2174/1568007053544174. [DOI] [PubMed] [Google Scholar]
- [197].Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Yang SH, Shi J, Day AL, Simpkins JW. Estradiol exerts neuroprotective effects when administered after ischemic insult. Stroke. 2000;31:745–749. doi: 10.1161/01.str.31.3.745. discussion 749–750. [DOI] [PubMed] [Google Scholar]
- [199].Yao M, Nguyen TV, Pike CJ. Estrogen regulates Bcl-w and Bim expression: role in protection against beta-amyloid peptide-induced neuronal death. J. Neurosci. 2007;27:1422–1433. doi: 10.1523/JNEUROSCI.2382-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Yi KD, Cai ZY, Covey DF, Simpkins JW. Estrogen receptor-independent neuroprotection via protein phosphatase preservation and attenuation of persistent extracellular signal-regulated kinase 1/2 activation. J. Pharmacol. Exp. Ther. 2008;324:1188–1195. doi: 10.1124/jpet.107.132308. [DOI] [PubMed] [Google Scholar]
- [201].Yi KD, Chung J, Pang P, Simpkins JW. Role of protein phosphatases in estrogen-mediated neuroprotection. J. Neurosci. 2005;25:7191–7198. doi: 10.1523/JNEUROSCI.1328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Yi KD, Covey DF, Simpkins JW. Mechanism of okadaic acid-induced neuronal death and the effect of estrogens. J. Neurochem. 2009;108:732–740. doi: 10.1111/j.1471-4159.2008.05805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Yi KD, Simpkins JW. Protein phosphatase 1, protein phosphatase 2A, and calcineurin play a role in estrogen-mediated neuroprotection. Endocrinology. 2008 doi: 10.1210/en.2008-0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Yoon SY, Choi JE, Yoon JH, Huh JW, Kim DH. BACE inhibitor reduces APP-beta-C-terminal fragment accumulation in axonal swellings of okadaic acid-induced neurodegeneration. Neurobiol. Dis. 2006;22:435–444. doi: 10.1016/j.nbd.2005.12.013. [DOI] [PubMed] [Google Scholar]
- [205].Yu DY, Luo J, Bu F, Song GJ, Zhang LQ, Wei Q. Inhibition of calcineurin by infusion of CsA causes hyperphosphorylation of tau and is accompanied by abnormal behavior in mice. Biol. Chem. 2006;387:977–983. doi: 10.1515/BC.2006.121. [DOI] [PubMed] [Google Scholar]
- [206].Zambrano CA, Egana JT, Nunez MT, Maccioni RB, Gonzalez-Billault C. Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Rad. Biol. Med. 2004;36:1393–1402. doi: 10.1016/j.freeradbiomed.2004.03.007. [DOI] [PubMed] [Google Scholar]
- [207].Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, Breitner JC. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA. 2002;288:2123–2129. doi: 10.1001/jama.288.17.2123. [DOI] [PubMed] [Google Scholar]
- [208].Zecchin KG, Seidinger AL, Chiaratti MR, Degasperi GR, Meirelles FV, Castilho RF, Vercesi AE. High Bcl-2/Bax ratio in Walker tumor cells protects mitochondria but does not prevent H2O2-induced apoptosis via calcineurin pathways. J. Bioenerg. Biomembr. 2007;39:186–194. doi: 10.1007/s10863-007-9076-z. [DOI] [PubMed] [Google Scholar]
- [209].Zhang L, Rubinow DR, Xaing G, Li BS, Chang YH, Maric D, Barker JL, Ma W. Estrogen protects against beta-amyloid-induced neurotoxicity in rat hippocampal neurons by activation of Akt. Neuroreport. 2001;12:1919–1923. doi: 10.1097/00001756-200107030-00030. [DOI] [PubMed] [Google Scholar]
- [210].Zhou Q, Gedrich RW, Engel DA. Transcriptional repression of the c-fos gene by YY1 is mediated by a direct interaction with ATF/CREB. J. Virol. 1995;69:4323–4330. doi: 10.1128/jvi.69.7.4323-4330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Zhou Y, Watters JJ, Dorsa DM. Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology. 1996;137:2163–2166. doi: 10.1210/endo.137.5.8612562. [DOI] [PubMed] [Google Scholar]
- [212].Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am. J. Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Zhu X, Lee HG, Raina AK, Perry G, Smith MA. The role of mitogen-activated protein kinase pathways in Alzheimer's disease. Neurosignals. 2002;11:270–281. doi: 10.1159/000067426. [DOI] [PubMed] [Google Scholar]