

Abstract
We previously reported a genomewide linkage study for human longevity using 308 long-lived individuals (LLI) (centenarians or near-centenarians) in 137 sibships and identified statistically significant linkage within chromosome 4 near microsatellite D4S1564. This interval spans 12 million bp and contains ≈50 putative genes. To identify the specific gene and gene variants impacting lifespan, we performed a haplotype-based fine-mapping study of the interval. The resulting genetic association study identified a haplotype marker within microsomal transfer protein as a modifier of human lifespan. This same variant was tested in a second cohort of LLI from France, and although the association was not replicated, there was evidence for statistical distortion in the form of Hardy–Weinberg disequilibrium. Microsomal transfer protein has been identified as the rate-limiting step in lipoprotein synthesis and may affect longevity by subtly modulating this pathway. This study provides proof of concept for the feasibility of using the genomes of LLI to identify genes impacting longevity.
The ability to survive to old age is partially under genetic influence, and the heritability of average life expectancy has been estimated to be ≈25% (1–4). The genetic influence of achieving extreme old age might be even greater (5). Male and female siblings of centenarians have a 17- and 8-fold greater relative risk, respectively, of surviving to age 100 and about half the death rate from age 20 to age 100 of birth cohort-matched individuals (6).
Despite the challenges of studying complex traits such as lifespan, an increasing number of genetic studies are reporting genes influencing human longevity. These genes include ApoE, ApoB, and klotho (7–10), although only the ApoE association has been reproduced consistently. To achieve their extreme age, centenarians likely lack numerous gene variants that are associated with premature mortality, and they may be more likely to carry protective variants as well (11, 12).
We recently reported the results of a genomewide linkage scan using 308 extremely long-lived individuals (LLI) in 137 sibships. The results indicated linkage to exceptional longevity (defined as living beyond the 5% survival tail) at chromosome 4 near microsatellite D4S1564 with a maximum logarithm of odds score of 3.65 (P = 0.044 genomewide with nonparametric analysis) (13). No other chromosomal region achieved statistically significant linkage in this study. There are ≈50 putative genes in the 12 million bp spanning the 85% confidence interval of this linkage peak, and a priori it was difficult to exclude any of the genes based on functional considerations. In addition, it was possible that the polymorphism underlying the linkage was not within any of these 50 putative genes. Therefore, an unbiased, systematic fine-mapping of the region was desired.
In the current study, a systematic exploration of the chromosome 4 linkage peak has identified a single gene, microsomal transfer protein (MTP), as a modifier of lifespan in a cohort of LLI from the U.S. A follow-up study investigating LLI from France also demonstrated statistical distortion at MTP, but did not completely confirm the observations from the U.S. cohort.
Methods
Sample Ascertainment and Phenotyping. U.S. cohort. The study sample consists of 653 individuals (197 males and 456 females) 98 years and older (mean 100.8) recruited through Elixir Pharmaceuticals, Beth Israel Deaconess Medical Center, Children's Hospital of Boston, and the New England Centenarian Study previously of Beth Israel Deaconess Medical Center and now of Boston University Medical Center. Individuals were identified and recruited by a variety of methods including institutional web sites, direct mailings, and advertisements in newspapers targeting potential participants or organizations involved with the aging community. Physical and cognitive health were not used as participation criteria. All participants and/or their legally authorized representatives took part in the written informed consent process, as required by the Institutional Review Boards of the aforementioned institutions. Additional data collected included health and socio-demographic histories, proof of age, usually in the form of a birth certificate, a three-generation pedigree, and measures to assess functional independence and cognitive status. Anonymous controls, self-identified as “Caucasian” and <50 years of age (with an average age of 38.6 years), were obtained from several anonymous sources in the U.S.
French cohort. For the follow-up study, we accessed 564 French LLI who consented through the Chronos Project at the Foundation Jean Deausset. The cohort consisted of 92 men and 472 women, all Caucasian, and at least 99 years of age (mean 103.1) at the time of blood collection. Controls were selected from 564 unrelated Caucasians born in France and matched to the cohort with respect to geographic origin. The average age of this control group was 51.2 years (ages 18–70) at the time of sample collection.
Genotyping. Potential single-nucleotide polymorphisms (SNPs) were retrieved from the Human Genome Draft database. Assays were designed to be multiplexed up to five times with spectrodesigner software (Sequenom, San Diego).
SNP genotyping on the U.S. cohort was performed by Sequenom's chip-based matrix-assisted laser desorption/ionization time-of-flight MS (dna massarray) on PCR-based extension products from individual DNA samples. Cases and controls were always run on the same chip to avoid potential artifacts caused by chip-specific miss-calls.
Genotyping of the –493G/T and 95H markers for the French cohort was performed on the TaqMan platform (Applied Biosystems) based on PCR sequence amplification of genomic DNA followed by fluorometric detection using allele-specific TaqMan Minor Groove Binder fluorescence-labeled probes. Complete details of the protocol are available at http://home.appliedbiosystems.com, protocol 4332856.
Sequencing. Sequencing was performed on the AB 3100 by using a Big Dye termination (version 3) chemistry on RapXtract (Prolinx, Bothell, WA)-purified PCR products. The phred program (Codoncode, Dedham, MA) was used for quality scores, and sequencer (Gene Codes, Ann Arbor, MI) was used for sequence comparisons and SNP detection.
Haplotype Reconstruction. We genotyped 19 familial trios (mother, father, and offspring) at densely spaced SNP markers to create a haplotype map of the 12 million-bp region of interest. For each trio, we determined the parental origin of offspring alleles for all cases where phase could be resolved unambiguously. In cases where phase was ambiguous (i.e., triple heterozygotes), the data were treated as missing. By applying this method, four parental chromosomes were reconstructed, with intermittent missing allele data. A haplotype was defined as a contiguous region of DNA with little evidence (<2.5%) for meiotic recombination within the common genetic history of the individuals genotyped.
In situations where the boundaries were ambiguous, a second heuristic was applied that assigned boundaries in such a way as to minimize the size (i.e., base pairs) within each block. With haplotype boundaries assigned, haplotype frequencies were estimated for each haplotype allele by using an expectation maximization algorithm (14). Any haplotype that had a frequency of <2.5% was excluded from further analysis to avoid possible errors in either the genotyping or the estimation process. Within each haplotype block, between two and six common SNP-based haplotypes were observed, and each of these haplotypes could be used as genetic markers tested for association.
To reconstruct haplotypes for the case/control association studies, the haplotype boundaries and allele frequency estimates established in the trios were used as initial parameters to seed the haplotype allele frequency estimations for genotyping the cases and controls. In cases where haplotype data could not be estimated with >95% confidence, the haplotype allele was treated as missing.
Tests of Association. The G test with Williams correction (a statistic following a χ2 distribution) was used to test inferences about associating genetic markers (haplotype or SNP) with the longevity phenotype (15). For each allele, 2 × 2 contingency tables were constructed as ± allele vs. ± longevity. For tests where only one direction of allele frequency difference was tested, P values were divided by two.
Testing for Stratification. For the U.S. cohort, 60 random SNP markers were genotyped in all cases and controls, and χ2 values were calculated from the allele counts. Because these SNPs were selected at random, any differences in allele frequencies were inferred as representative of the differences in genetic backgrounds between cases and controls. If the genetic backgrounds of the two-armed study were perfectly matched, the mean χ2 of the G test statistics for these markers has an expected value of 1.0.
For the French cohort, 57 microsatellites, from the Applied Biosystems LMS-MD10 panel, located on six different chromosomes (chromosomes 2, 9, 10, 11, 17, and 18) were tested on all cases and controls and checked for stratification by the above method.
Proactive Sample Matching. Sixty random SNP markers (nonoverlapping with the stratification panel described above) were genotyped in 250 cases and 463 controls of the U.S. collection. Homozygotes for the minor allele were assigned the value –1, heterozygotes 0, and major allele homozygotes 1. Based on the multivariate means calculated from this coded data, a subgroup of the 250 controls was selected that minimized the Mahalanobis distance with respect to the case samples. The Mahalanobis distance is a measure of distance between two multivariate means that normalizes each dimension based on the covariance matrix:
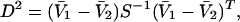 |
[1] |
where V̄1 is a vector representing the mean genotyping values of the cases, V̄2 is the mean vector for the controls, and S–1 is the inverse of the covariance matrix. No proactive sample matching was required for the French cohort as there was no detectable stratification between the cases and controls (see Results).
Results
We undertook fine-mapping of the chromosome 4 locus to identify specific gene variants associated with exceptional longevity in a U.S. population. With this aim, 2,000 SNPs (an average of 1 every 6 kb) within the longevity linkage locus were selected from the SNP consortium database. Based on our experience with an earlier pilot study, we expected that only a fraction of these markers would be useful in an association study. To screen this initial set of SNPs, 19 familial trios (mother, father, and offspring) acquired from the Centre d'Étude du Polymorphisme Humain repository were genotyped at all selected markers. Of these 2,000 markers, 1,494 had high confidence calls on the massarray platform. Of these markers, 990 had a minor allele frequency of at least 5%. SNPs of lower heterozygosity were excluded because of the reduced power of such markers with respect to mapping complex traits in association studies of limited sample size. Of the remaining SNPs, 113 were eliminated because the frequency distribution of the homozygotes and heterozygotes was not statistically compatible with Hardy–Weinberg equilibrium (HWE). These failures were attributed to systematic artifacts introduced by the genotyping platform. The use of familial trios allowed a Mendelian check on the validity of each SNP assay. If more than one Mendelian inheritance error per assay was detected within the 19 trios, the assay was judged unreliable.
From SNPs to Haplotypes. Haplotype-based approaches applied to smaller genomic regions have been successfully demonstrated by others (16–18), and advantages over single markers have been shown (19–21). In addition, the diversity of the genome can be effectively captured by reducing it to sequential segments of limited diversity (18, 22, 23). Once the common haplotypes within a block have been defined, SNPs within the same block redundant for discriminating between the different haplotypes can be omitted from subsequent typing and analysis. After removing the redundant SNPs, 875 validated SNPs and ≈700 “maximally informative SNPs” remained for the association studies.
Testing for Association. By densely genotyping across the 12 million-bp region previously identified through the linkage study, we were able to reconstruct a rough draft of the underlying haplotype structure. A total of 195 blocks were defined within the 12 million-bp region, with 2 to 15 SNPs per block. Approximately 75% of the mapped region was within regions of strong linkage disequilibrium. Using this carefully reconstructed assortment of SNP-based haplotype markers, we initiated a case/control association study between groups of unrelated LLI (age 98 and older) and a much younger control population (<50 years of age) from a U.S. cohort.
To reduce genotyping costs and increase the power by confirming the hypothesis in independent populations, the study was divided in two sequential tiers of samples, with the first tier comparing 190 LLI with 190 controls at SNP-based haplotype markers. The first tier was intended as a screen to generate the hypotheses tested in the second tier, thereby greatly reducing the statistical penalty for testing a large number of associations. Although several SNPs and haplotype markers were significant at P < 0.05, the marker showing the strongest association (P = 0.0005) was the SNP rs1553432, located 72 kb upstream from MTP. Other SNPs demonstrating statistical distortion include rs750032 (P = 0.0029), rs951085 (P = 0.0041), and rs752766 (P = 0.0058). The rs1553432 association provided a potentially interesting first hypothesis to pursue with dense genotyping and haplotype mapping of the nearby genes, although this result was not statistically significant after correcting for multiple testing.
In the 250-kb region bracketing rs1553432, 60 SNPs were identified and assays were validated. Several of these densely spaced SNPs showed strong associations when analyzed in the set of 190 cases and controls used above; most of these markers were located near the 5′ end of MTP or just upstream of this gene and were particularly dense near the promoter region. All of the identified associations were in strong linkage disequilibrium with rs1553432 (e.g., they fell on the same “long-range” haplotype). With interest narrowing in on a single gene, all known SNP polymorphisms for MTP and its promoter were genotyped in the original 190 cases and 190 controls. After haplotype reconstruction of the area was completed, a single haplotype (see Fig. 1a, which is published as supporting information on the PNAS web site), underrepresented in the LLI (P = 0.0005, Table 1), could account for the majority of the variation at the locus. This risk haplotype was initially defined as the minor allele of rs2866164 paired with the major allele of MTP Q/H 95. Both of these SNP markers had been previously identified by other groups; rs2866164 is an MTP promoter mutation and MTP Q/H 95 is a semiconservative mutation in exon 3 of MTP (glutamine to histidine at amino acid 95). Evidence for association was present when each of these SNPs was analyzed separately (P = 0.034 for rs2866164, P = 0.071 for MTP 95Q/H, Table 2). Goodness-of-fit analysis indicated that neither SNP alone was likely to explain the statistical distortion between cases and controls as well as the two-SNP haplotype (P = 0.007). RS2866164 is perfectly correlated with another MTP promoter SNP, rs1800591 (also known as –493 G/T) that has been previously associated with several phenotypes including lipoprotein profiles, central obesity, and insulin resistance (see Discussion).
Table 1. Haplotype allele counts/frequencies for U.S. cohort.
| LLI
|
Controls
|
|||
|---|---|---|---|---|
| Allele | rs2866164-C | rs2866164-G | rs2866164-C | rs2866164-G |
| Q95 | 256 (74%) | 61 (18%) | 244 (67%) | 104 (29%) |
| H95 | 0 | 29 (8%) | 0 | 16 (4%) |
Shown are counts/frequencies for the two-SNP haplotype formed by combining alleles of these SNPs in cases (LLI) and controls. Only three of the four haplotypes were observed, fulfilling the criteria of no historic recombination between the two SNPs. As discussed in the text, the haplotype composed of the rs2866164-G allele and Q95 allele is underrepresented in LLI, suggesting this variant confers mortality risk. P = 0.0005.
Table 2. Risk SNP allele counts/frequencies for U.S.-based collection.
| SNP allele | Controls | LLI |
|---|---|---|
| rs2866164-C | 245 (66%) | 259 (74%) |
| rs2866164-G | 121 (34%) | 91 (26%) |
| MTP Q95 | 361 (95%) | 343 (92%) |
| MTP H95 | 19 (5%) | 31 (8%) |
Shown are counts/frequencies for rs2866164 and MTP Q/H. Note that the rs2866164 SNP has multiple “twins” displaying identical statistical behavior, including the -493G/T SNP. For rs2866164, P = 0.034, and for MTP Q/H, P = 0.071.
Genotyping an additional set of 190 cases and controls (referred to hereafter as the second tier; see Tables 3 and 4) strengthened the evidence for an association with longevity at the two-SNP haplotype (P = 0.0006, Table 4, one-sided G test). Because a single hypothesis was tested with this independent set of samples, there was no need to correct for multiple tested hypotheses. In the second tier of samples, rs2866164 was marginally less associated with lifespan (P = 0.0027, Table 3) than when analyzed as part of the two-SNP risk haplotype identified in the first tier. MTP Q/H 95 alone showed no association (P = 0.55, Table 3).
Table 3. Allele counts and frequencies for U.S. confirmatory sample set.
| SNP allele | Controls | LLI |
|---|---|---|
| rs2866164-C | 241 (67%) | 282 (76%) |
| rs2866164-G | 119 (33%) | 88 (24%) |
| MTP Q95 | 275 (94%) | 348 (94%) |
| MTP H95 | 19 (6%) | 24 (6%) |
See Table 2 for details. For rs2866164, P = 0.0027, and for MTP Q/H, P = 0.55.
Table 4. Allele counts and frequencies for U.S. confirmatory sample set: Two-SNP haplotype.
| LLI
|
Controls
|
|||
|---|---|---|---|---|
| MTP | rs2866164-C | rs2866164-G | rs2866164-C | rs2866164-G |
| Q95 | 280 (77%) | 63 (17%) | 241 (67%) | 98 (27%) |
| H95 | 0 | 23 (6%) | 0 | 21 (6%) |
P = 0.0006. See Table 1 for details.
Genetic Stratification and Controlling Type I Error. Genetic association studies have been plagued with false positive or otherwise irreproducible results (24, 25). A recognized problem affecting genetic association studies is the failure to adequately match the genetic backgrounds of the cases and controls, a phenomenon called stratification. Our association studies comparing individuals born decades apart can be potentially vulnerable to this confounder because the geographic distribution of ethnicities has changed in the U.S. over the past 100 years. Specifically, our case population reflects the ethnic distribution of the U.S. near the beginning of the last century, whereas our control population was sampled from more recent generations. To minimize this problem, we only used DNA from people who identified themselves as Caucasian, but this is obviously a diverse group.
Consequently, our cases and controls would differ not only with respect to the longevity phenotype but also have ethnicity as an uncontrolled confounder. If the effect is strong enough, associations will be found that reflect these ethnic differences rather than differences in lifespan. The mean χ2 for randomly selected SNP markers (representing differences in genetic background) for the 190 cases and controls used for the first screening showed absence of stratification (χ2 mean of 1.07 compared with an expected value of 1.0). Unfortunately, the second set of 190 LLI and controls used to replicate the hypothesis showed moderate stratification (χ2 mean of 1.7). Although modest, any amount of stratification is undesirable, and the methods of correcting for this potential confounder have not been well validated empirically.
To avoid correcting for the hundreds of partially independent hypotheses tested with the original sample set and to simultaneously eliminate stratification as a problem, 250 cases were proactively matched (see Methods) against a new set of 463 controls. Using the approach discussed in Methods, a subgroup of 250 controls was selected that best matched the cases with respect to genetic background. The mean χ2 for this group of samples (measured using a second, independent set of 60 SNPs) was 0.92, indicating a very high level of genetic balance. It should be emphasized that none of these samples were used in tier I to generate the single inference that the risk allele was underrepresented in LLI. The association at rs2866164 was confirmed with this well-matched group of cases and controls (P = 0.0144 by one-sided G test, relative risk = 0.69, Table 5). The two-SNP haplotype also demonstrated an association (P = 0.0124, Table 6).
Table 5. Risk allele frequency differences in proactively matched LLI and controls (U.S.-based).
| SNP allele | Controls | LLI |
|---|---|---|
| rs2866164-C | 316 (67%) | 365 (76%) |
| rs2866164-G | 146 (34%) | 123 (24%) |
The minor allele of rs2866164 is underrepresented in LLI compared to controls (P = 0.0144). There is no evidence in this sample set that the haplotype explains the association better than rs2866164 alone.
Table 6. Risk allele frequency differences in proactively matched LLI and controls (U.S.-based): Two-SNP haplotype.
| LLI
|
Controls
|
|||
|---|---|---|---|---|
| MTP | rs2866164-C | rs2866164-G | rs2866164-C | rs2866164-G |
| Q95 | 365 (75%) | 94 (19%) | 316 (68%) | 117 (25%) |
| H95 | 0 | 29 (6%) | 0 | 29 (6%) |
P = 0.0124. The two-SNP haplotype is underrepresented in LLI compared to controls.
MTP Resequencing. Although the two-SNP haplotype was sufficient to account for all of the association at the locus, it is imprudent to conclude that this polymorphism was causative with respect to longevity. In particular, a few “twins” (SNPs whose alleles are perfectly correlated) of –493 G/T and rs2866164 were identified that could equally explain the data.
To search for additional SNPs that could explain the association as well as or better than rs2866164, we sequenced 12 DNA samples within the 12-kb risk block and the 72-kb block of DNA extending up to the initial rs1553432 SNP. In addition, all 18 exons of MTP were sequenced in a group of 50 LLI to search for rare functional polymorphisms that would not fall on welldefined haplotypes. Altogether, 104 SNPs were identified, although none explained the association better than rs2866164. Sixty-one of these SNPs (named MTP001, MTP002 etc.), were novel to our study (i.e., they were not included within the December 2001 Human Genome Draft). After adding the additional SNPs to the map, a new block structure was defined with significant changes (Fig. 1b).
Testing in a New Cohort. Because of the potential unpredictable bias that affects association studies, we attempted to confirm the observation in a group of matched LLI from the Centre d'Étude du Polymorphisme Humain centenarian cohort. Because an effort was made to collect geographically matched controls, stratification was not evident in this study. Testing 57 microsatellite markers, the χ2 mean between cases and controls was 1.00.
Tables 7 and 8 show the genotyping results for rs2866164 and the haplotype composed of rs2866164 and MTP Q/H 95. Neither the minor allele of rs2866164 nor the rs2866164-G/MTP-H95 haplotype is significantly underrepresented in the French LLI. Interestingly, as shown in Table 9, the rs2866164-G/MTP-H95 haplotype (HI) is out of HWE with the other haplotypes (HII, collectively) in the LLI (P = 0.0164) but not in controls (P = 0.5923). The departure from HWE observed in the LLI can be entirely explained by an excess of HI/HI homozygotes. Allele frequencies out of HWE are suggestive of either genotyping artifact or selective pressure (26). A genotyping artifact was ruled out by typing the –493G/T polymorphism. As expected, this marker was perfectly correlated with rs2866164, arguing against marker-specific systematic error. The modest departure from HWE in the cases and lack of statistical distortion in the controls argue in favor of selective pressure at this locus that manifests itself as a population ages.
Table 7. rs2866164 frequency differences in French LLI cohort and controls.
| SNP allele | Controls | LLI |
|---|---|---|
| rs2866164-C | 812 (73.6%) | 835 (74.7%) |
| rs2866164-G | 292 (26.4%) | 283 (25.3%) |
The minor allele of rs2866164 is underrepresented in LLI compared to controls, but the effect is not statistically significant (P = 0.54).
Table 8. Two-SNP haplotype frequency differences in French LLI cohort and controls.
| LLI
|
Controls
|
|||
|---|---|---|---|---|
| MTP | rs2866164-C | rs2866164-G | rs2866164-C | rs2866164-G |
| Q95 | 826 (73.2%) | 228 (20.2%) | 835 (74.0%) | 221 (19.6%) |
| H95 | 0 | 72 (6.3%) | 0 | 65 (5.8%) |
Table 9. Haplotype-based counts and frequencies from the French LLI cohort and controls.
| Haplotype-based genotype | Controls | LLI |
|---|---|---|
| HII/HII | 360 | 369 |
| HI/HII | 177 | 159 |
| HI/HI | 24 | 33 |
| HWE P(χ2) value | 0.93 | 0.02 |
HI represents the rs2866164-G/MTP-H95 haplotype, and HII collectively represents the other two haplotypes.
Multiple Associations? After fully characterizing the MTP finding, there remained the possibility that one or more additional genes associated with longevity could be contributing to the linkage peak. To address this, any SNP or SNP-based haplotype genotyped in the first tier of samples associated at P < 0.05 was genotyped in the second tier of 190 cases and controls. None of these putative associations was replicated.
How adequately did the maximally informative SNPs cover the area under the linkage peak? There were inevitable gaps in the haplotype map, because of incomplete sequences of the human genome in this region, areas with few documented SNPs, and regions where our genotyping was not successful. Attempting anything near 100% statistical coverage of the region would have been prohibitively expensive and time consuming given the extensive sequencing and genotyping required. As a compromise, we attempted to genotype, in 190 cases and controls (first tier), at least five SNPs near all of the well-characterized genes under the locus (including the alcohol dehydrogenase cluster and NF-κB), which involved assaying an additional 55 SNP markers. This effort yielded no additional associations, leaving MTP as the lone candidate to explain the original linkage result. We recognize that a study with denser genotyping, larger sample sizes, or more extreme ages may have produced additional candidates.
Discussion
We have shown a statistically significant underrepresentation of a risk allele in a group of LLI compared with younger controls in a U.S.-based longevity study. Although there was evidence of statistical distortion at this locus in a French-based longevity study, there was no significant difference in allele frequencies with respect to the risk haplotype identified from the former study. Although apparently a discrepancy (a significant and replicated finding not validated in a third sample), the most likely explanation is a lack of power of individual studies. If one examines the odds ratios estimated for the two unstratified U.S. studies [combined OR 0.72 (0.58, 0.89)] and the French study [OR 0.94 (0.78, 1.14)] we see their confidence bounds overlap significantly (suggesting no discrepancy necessarily exists). Taking a plausible intermediate genetic model to explain this association to exceptional longevity consistently (e.g., frequency = 0.70, genotype relative risk = 0.83), we find that even a sample as large as the French replication sample of 500 cases and 500 controls would have only 50% power to reach nominal statistical significance (P < 0.05) given the modest individual effect of this allele. Thus, even if we assume the effect is true and present equally in all study samples, half of all studies of this magnitude will record a nominally “negative” result (P > 0.05). This calculation underscores the general observation that association studies of complex phenotypes are likely to require extremely large samples to convincingly elucidate the modest genetic influences involved.
Although not necessary to explain the data, it is worth noting that other factors could very likely contribute to inconsistency among the studies. Among the possibilities relevant to this particular study are two worth noting:
An observed covariate may be modulating the impact of the risk allele in the different populations. Diet is one obvious possibility; the culinary tastes of Europeans and Americans may place different stresses on equivalent genetic backgrounds. The interaction between the ApoE gene, diet, genetic background, and other covariates has now been well documented (27, 28), and it is reasonable to assume that much of the variance in human longevity will be explained by complex interactions between environment and genetics.
The American and French samples have important phenotypic differences. For example, the average age of the American controls was much younger than the French controls. Second, the French study involved a smaller fraction of male centenarians compared with the American study. Because the ascertainment strategies were different for the two studies, this may at least partially explain this discrepancy.
MTP Biology and Previous Associations. The gene product of MTP has been well characterized since the mid-1980s for its role in lipoprotein assembly and is an investigational target for treating combined hyperlipidemia and obesity (29, 30). MTP is thought to be the rate-limiting step in the production of lipoproteins (31), making it a particularly appealing target for next-generation lipid-lowering drugs. Structurally, the protein dimerizes with the ubiquitous protein disulfide isomerase and resides on the luminal surface of the endoplasmic reticulum where it facilitates the proper assembly of very low-density lipoprotein and chylomicron particles. Functionally, MTP is directly involved in the packaging of ApoB and triglyceride into these particles, and MTP and ApoB are thought to directly bind one another during this assembly (32). Rare humans with two nonfunctioning copies of the gene suffer from abetalipoproteinemia and are characterized by the near absence of ApoB particles in serum (33).
Drugs that inhibit MTP activity have been shown to improve lipoprotein profiles (34). Several food products have also been shown to reduce MTP activity, including garlic (35), ethanol (36), and citric flavanoids (37). One study found that the MTP promoter allele –493T up-regulated MTP expression by 2-fold (38), consistent with our finding that –493T is underrepresented in long-lived populations.
Other groups have found genetic associations between MTP and several phenotypes including lipoprotein profiles, insulin resistance, and fat distribution, and most of these studies focused on the –493G/T marker (38–44). Like so many other mapping studies of complex traits, the literature surrounding MTP has been complex and often contradictory, although as a general trend the –493T allele has been associated with detrimental phenotypes.
The known activity of MTP, as a rate-limiting step in lipid metabolism, lends functional credibility to our association of MTP with human longevity. Coronary artery disease and other vasculopathies attributed to unfavorable lipid profiles (peripheral vascular disease, renal-vascular disease, and stroke) account for a large percentage of human mortality. Common genetic variants that impact the function of lipid metabolism should be expected to impact human lifespan; for example, the offspring of centenarians have higher levels of high-density lipoprotein (good cholesterol) and lower levels of low-density lipoprotein (bad cholesterol) than age-matched controls, and they demonstrate significantly lower risks of heart disease and stroke compared with age-matched controls (45, 46). In addition, a “longevity syndrome” was described among families with extremely low levels of low-density lipoprotein particles (47). Although reasonable to believe that the impact of MTP on human longevity is through its impact on lipid profiles, the association studies above suggest that this gene may also affect susceptibility to insulin resistance and obesity.
MTP and ApoE. There are many parallels between the associations of MTP and ApoE. Both genes are risk factors implicated in cardiovascular disease and longevity, the latter being also being associated with Alzheimer's disease (AD). In light of this, it would be intriguing to explore the genetic epidemiology of MTP with respect to diseases of aging, such as AD. Using the well-matched set of 250 LLI and controls, we confirmed in our LLI and control samples that the ApoE ε2 allele is protective (12% vs. 7%), the ε3 allele is neutral (83% vs. 80%), and the ε4 allele is detrimental (5% vs. 13%) with respect to lifespan extension (P = 0.0001, Table 10, which is published as supporting information on the PNAS web site). No interaction between the MTP and ApoE alleles with respect to lifespan was detected, although the sample size may have been inadequate.
Implications. Our study lends credence to the claim that centenarians and near-centenarians offer an appealing model for studying human longevity and disease resistance (45). We have confirmed the hypothesis that a population that has escaped or delayed the lethal pathologies of old age is likely to be helpful in studies to detect genetic factors that impact the diseases of aging (48). By systematically examining a large stretch of the genome for genes associated with a phenotype, we have provided credibility to the use of public gene maps and SNP databases to perform studies of the full genome. As our study mapped 0.5% of the genome, it would have required a 200 times larger study to scan the entire genome. Because MTP can explain only a small fraction of the total genetic variance in human longevity and there may be dozens of genes with a similar association, much more expansive studies in the near future will likely yield a wealth of insight into the genetic basis of longevity, aging, and disease resistance.
Supplementary Material
Acknowledgments
We thank the many individuals involved in recruiting subjects for this study and the laboratory and bioinformatic staff that carried out the genotyping and analyses. We thank the study participants and their families for generously agreeing to participate in our study. We are most appreciative of the assistance provided by the American Association of Retired Persons in the subject recruitment efforts led by T.T.P. We thank Carl Weissman for his operational expertise during the experimental stages of this work. Last, we are grateful for the assistance of the Rutgers University Cell and DNA Repository. Funding for this study was largely contributed by Elixir Pharmaceuticals. This work was supported by grants from the National Institute on Aging (R01-AG1875), the Ellison Medical Foundation, the Alzheimer's Association's Templeton Discovery Award, the Institute for the Study of Aging, the Howard Hughes Medical Institute, and the French Ministry of Research. L.M.K. is an Investigator with the Howard Hughes Medical Institute.
Abbreviations: LLI, long-lived individuals; MTP, microsomal transfer protein; SNP, single-nucleotide polymorphism; HWE, Hardy–Weinberg equilibrium.
References
- 1.Gudmundsson, H., Gudbjartsson, D. F., Frigge, M., Gulcher, J. R. & Stefansson, K. (2000) Eur. J. Hum. Genet. 8, 743–749. [DOI] [PubMed] [Google Scholar]
- 2.Perls, T., Shea-Drinkwater, M., Bowen-Flynn, J., Ridge, S., Kang, S., Joyce, E., Daly, M., Brewster, S. J., Kunkel, L. M. & Puca, A. A. (2000) J. Am. Geriatr. Soc. 48, 1483–1485. [PubMed] [Google Scholar]
- 3.Herskind, A. M., McGue, M., Holm, N. V., Sorensen, T. I., Harvald, B. & Vaupel, J. W. (1996) Hum. Genet. 97, 319–323. [DOI] [PubMed] [Google Scholar]
- 4.McGue, M., Vaupel, J. W., Holm, N. & Harvald, B. (1993) J. Gerontol. 48, B237–B244. [DOI] [PubMed] [Google Scholar]
- 5.Perls, T., Kunkel, L. & Puca, A. (2002) Curr. Opin. Genet. Dev. 12, 362–369. [DOI] [PubMed] [Google Scholar]
- 6.Perls, T., Wilmoth, J., Levenson, R., Drinkwater, M., Cohen, M., Bogan, H., Joyce, E., Brewster, S. J., Kunkel, L. M. & Puca, A. A. (2002) Proc. Natl. Acad. Sci. USA 99, 8442–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Bockxmeer, F. M. (1994) Nat. Genet. 6, 4–5. [DOI] [PubMed] [Google Scholar]
- 8.Schachter, F., Faure-Delanef, L., Guenot, F., Rouger, H., Froguel, P., Lesueur-Ginot, L. & Cohen, D. (1994) Nat. Genet. 6, 29–32. [DOI] [PubMed] [Google Scholar]
- 9.Arking, D. E., Krebsova, A., Macek, M., Sr., Macek, M., Jr., Arking, A., Mian, I. S., Fried, L., Hamosh, A., Dey, S., McIntosh, I. & Dietz, H. C. (2002) Proc. Natl. Acad. Sci. USA 99, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kervinen, K., Savolainen, M. J., Salokannel, J., Hynninen, A., Heikkinen, J., Ehnholm, C., Koistinen, M. J. & Kesaniemi, Y. A. (1994) Atherosclerosis 105, 89–95. [DOI] [PubMed] [Google Scholar]
- 11.Schachter, F. (1998) Am. J. Hum. Genet. 62, 1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wachter, K. W. (1997) In Between Zeus and the Salmon: The Biodemography of Longevity (Natl. Acad. Press, Washington, DC). [PubMed]
- 13.Puca, A. A., Daly, M. J., Brewster, S. J., Matise, T. C., Barrett, J., Shea-Drinkwater, M., Kang, S., Joyce, E., Nicoli, J., Benson, E., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 10505–10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Excoffier, L. & Slatkin, M. (1995) Mol. Biol. Evol. 12, 921–927. [DOI] [PubMed] [Google Scholar]
- 15.Sokal, R. R. & Rohlf, F. J. (2000) Biometry (Freeman, New York).
- 16.Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J. & Lander, E. S. (2001) Nat. Genet. 29, 229–232. [DOI] [PubMed] [Google Scholar]
- 17.Rioux, J. D., Daly, M. J., Silverberg, M. S., Lindblad, K., Steinhart, H., Cohen, Z., Delmonte, T., Kocher, K., Miller, K., Guschwan, S., et al. (2001) Nat. Genet. 29, 223–228. [DOI] [PubMed] [Google Scholar]
- 18.Johnson, G. C., Esposito, L., Barratt, B. J., Smith, A. N., Heward, J., Di Genova, G., Ueda, H., Cordell, H. J., Eaves, I. A., Dudbridge, F., et al. (2001) Nat. Genet. 29, 233–237. [DOI] [PubMed] [Google Scholar]
- 19.De Benedictis, G., Falcone, E., Rose, G., Ruffolo, R., Spadafora, P., Baggio, G., Bertolini, S., Mari, D., Mattace, R., Monti, D., et al. (1997) Hum. Genet. 99, 312–318. [DOI] [PubMed] [Google Scholar]
- 20.Stephens, J. C. (1999) Mol. Diagn. 4, 309–317. [DOI] [PubMed] [Google Scholar]
- 21.Davidson, S. (2000) Nat. Biotechnol. 18, 1134–1135. [DOI] [PubMed] [Google Scholar]
- 22.Patil, N., Berno, A. J., Hinds, D. A., Barrett, W. A., Doshi, J. M., Hacker, C. R., Kautzer, C. R., Lee, D. H., Marjoribanks, C., McDonough, D. P., et al. (2001) Science 294, 1719–1723. [DOI] [PubMed] [Google Scholar]
- 23.Stephens, J. C., Schneider, J. A., Tanguay, D. A., Choi, J., Acharya, T., Stanley, S. E., Jiang, R., Messer, C. J., Chew, A., Han, J. H., et al. (2001) Science 293, 489–493. [DOI] [PubMed] [Google Scholar]
- 24.Hirschhorn, J. N., Lohmueller, K., Byrne, E. & Hirschhorn, K. (2002) Gen. Med. 4, 45–61. [DOI] [PubMed] [Google Scholar]
- 25.Reich, D. E. & Goldstein, D. B. (2001) Gen. Epidemiol. 20, 4–16. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen, D. M., Ehm, M. G. & Weir, B. S. (1998) Am. J. Hum. Genet. 63, 1531–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petot, G. J., Traore, F., Debanne, S. M., Lerner, A. J., Smyth, K. A. & Friedland, R. P. (2003) Metabolism 52, 279–281. [DOI] [PubMed] [Google Scholar]
- 28.Ribalta, J., Vallve, J. C., Girona, J. & Masana, L. (2003) Curr. Opin. Clin. Nutr. Metab. Care 6, 177–187. [DOI] [PubMed] [Google Scholar]
- 29.Wetterau, J. R., Lin, M. C. & Jamil, H. (1997) Biochim. Biophys. Acta 1345, 136–150. [DOI] [PubMed] [Google Scholar]
- 30.Shelness, G. S. & Sellers, J. A. (2001) Curr. Opin. Lipidol. 12, 151–157. [DOI] [PubMed] [Google Scholar]
- 31.Jamil, H., Chu, C. H., Dickson, J. K., Jr., Chen, Y., Yan, M., Biller, S. A., Gregg, R. E., Wetterau, J. R. & Gordon, D. A. (1998) J. Lipid Res. 39, 1448–1454. [PubMed] [Google Scholar]
- 32.Wu, X., Zhou, M., Huang, L. S., Wetterau, J. & Ginsberg, H. N. (1996) J. Biol. Chem. 271, 10277–10281. [DOI] [PubMed] [Google Scholar]
- 33.Berriot-Varoqueaux, N., Aggerbeck, L. P., Samson-Bouma, M. & Wetterau, J. R. (2000) Annu. Rev. Nutr. 20, 663–697. [DOI] [PubMed] [Google Scholar]
- 34.Wetterau, J. R., Gregg, R. E., Harrity, T. W., Arbeeny, C., Cap, M., Connolly, F., Chu, C. H., George, R. J., Gordon, D. A., Jamil, H., et al. (1998) Science 282, 751–754. [DOI] [PubMed] [Google Scholar]
- 35.Lin, M. C., Wang, E. J., Lee, C., Chin, K. T., Liu, D., Chiu, J. F. & Kung, H. F. (2002) J. Nutr. 132, 1165–1168. [DOI] [PubMed] [Google Scholar]
- 36.Lin, M. C., Li, J. J., Wang, E. J., Princler, G. L., Kauffman, F. C. & Kung, H. F. (1997) FASEB J. 11, 1145–1152. [DOI] [PubMed] [Google Scholar]
- 37.Wilcox, L. J., Borradaile, N. M., de Dreu, L. E. & Huff, M. W. (2001) J. Lipid Res. 42, 725–734. [PubMed] [Google Scholar]
- 38.Karpe, F., Lundahl, B., Ehrenborg, E., Eriksson, P. & Hamsten, A. (1998) Arterioscler. Thromb. Vasc. Biol. 18, 756–761. [DOI] [PubMed] [Google Scholar]
- 39.Couture, P., Otvos, J. D., Cupples, L. A., Wilson, P. W., Schaefer, E. J. & Ordovas, J. M. (2000) Atherosclerosis 148, 337–343. [DOI] [PubMed] [Google Scholar]
- 40.Juo, S. H., Han, Z., Smith, J. D., Colangelo, L. & Liu, K. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 1316–1322. [DOI] [PubMed] [Google Scholar]
- 41.Ledmyr, H., Karpe, F., Lundahl, B., McKinnon, M., Skoglund-Andersson, C. & Ehrenborg, E. (2002) J. Lipid Res. 43, 51–58. [PubMed] [Google Scholar]
- 42.St-Pierre, J., Lemieux, I., Miller-Felix, I., Prud'homme, D., Bergeron, J., Gaudet, D., Nadeau, A., Despres, J. P. & Vohl, M. C. (2002) Atherosclerosis 160, 317–324. [DOI] [PubMed] [Google Scholar]
- 43.Talmud, P. J., Palmen, J., Miller, G. & Humphries, S. E. (2000) Ann. Hum. Genet. 64, 269–276. [DOI] [PubMed] [Google Scholar]
- 44.Herrmann, S. M., Poirier, O., Nicaud, V., Evans, A., Ruidavets, J. B., Luc, G., Arveiler, D., Bao-Sheng, C. & Cambien, F. (1998) J. Lipid Res. 39, 2432–2435. [PubMed] [Google Scholar]
- 45.Barzilai, N., Gabriely, I., Gabriely, M., Iankowitz, N. & Sorkin, J. D. (2001) J. Am. Geriatr. Soc. 49, 76–79. [DOI] [PubMed] [Google Scholar]
- 46.Terry, D. F., Wilcox, M., McCormick, M. A., Lawler, E. & Perls, T. T. (2003) J. Gerontol. A Biol. Sci. Med. Sci. 58, M425–M431. [DOI] [PubMed] [Google Scholar]
- 47.Glueck, C. J., Gartside, P. S., Mellies, M. J. & Steiner, P. M. (1977) Trans. Assoc. Am. Physicians 90, 184–203. [PubMed] [Google Scholar]
- 48.Silverman, J. M., Smith, C. J., Marin, D. B., Birstein, S., Mare, M., Mohs, R. C. & Davis, K. L. (1999) Am. J. Hum. Genet. 64, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}