

Abstract
Although the tryptophan-degrading enzyme, indoleamine 2,3-dioxygenase (IDO), is a pivotal mediator of inflammation-induced depression, its mechanism of regulation has not yet been investigated in this context. Here, we demonstrate an essential role for interferon (IFN)γ and tumor necrosis factor (TNF)α in the induction of IDO and depressive-like behaviors in response to chronic immune activation. Wild-type (WT) control mice and IFNγR−− mice were inoculated with an attenuated form of Mycobacterium bovis, bacille Calmette-Guérin (BCG). Infection with BCG induced an acute episode of sickness that was similar in WT and IFNγR−− mice. Increased immobility during the forced swim and tail suspension tests occurred in WT mice 7 d after BCG inoculation but was entirely absent in IFNγR−− mice. In WT mice, these indices of depressive-like behavior were associated with chronic upregulation of IFNγ, interleukin(IL)-1β, TNFα, and IDO. Proinflammatory cytokine expression was elevated in BCG-infected IFNγR−− mice as well, but upregulation of lung and brain IDO mRNA was completely abolished. This was accompanied by an attenuation of BCG-induced TNFα mRNA and the lack of an increase in plasma kynurenine/tryptophan ratio in the BCG-inoculated IFNγR−− mice compared with WT controls. Pretreatment of mice with the TNFα antagonist, etanercept, partially blunted BCG-induced IDO activation and depressive-like behavior. In accordance with these in vivo data, IFNγ and TNFα synergized to induce IDO in primary microglia. Together, these data demonstrate that IFNγ, with TNFα, is necessary for induction of IDO and depressive-like behavior in mice after BCG infection.
Introduction
Chronic inflammation is often associated with depression (Evans et al., 2005; Adler et al., 2008; Dantzer et al., 2008a,b). A quasi-experimental model of this relationship is provided by cytokine immunotherapy (Maes et al., 2001; Capuron et al., 2002, 2003). Patients treated with interferon (IFN)α for cancer or hepatitis C develop transient flu-like symptoms that often culminate in major depressive disorders (Capuron et al., 2002). IFNα treatment in patients elicits systemic immune activation, and a pivotal factor for the development of inflammation-associated depression appears to be the tryptphan-degrading enzyme indoleamine 2,3-dioxygenase (IDO) (Raison et al., 2006). Activation of this ubiquitous enzyme by proinflammatory cytokines leads to the generation of several neuroactive metabolites including quinolic acid (QA), an NMDA receptor agonist that has been implicated in neurodegenerative disorders (Guillemin et al., 2005b; Spalletta et al., 2006). In animal studies, inhibition of IDO abrogates depressive-like behaviors induced by acute (O'Connor et al., 2008b) or chronic inflammation (O'Connor et al., 2009).
The exact nature of the proinflammatory cytokines responsible for upregulating IDO is still elusive within the context of inflammation-associated depression. IFNγ is the predominant cytokine implicated in the induction of IDO (Schroecksnadel et al., 2003). Two IFN-stimulated response elements (ISREs) and IFNγ-activated site (GAS) sequences are present in the 5′-promoter region of the IDO gene (Hassanain et al., 1993; Chon et al., 1995; Konan and Taylor, 1996). Clinically, individuals with an allelic variant (T allele) of the IFNγ gene resulting in high IFNγ production also have an elevated plasma kynurenine/tryptophan ratio, indicative of increased IDO activity (Raitala et al., 2005). However, subsequent studies have identified IFN-independent pathways, including TNFα and Toll-like receptor-4, capable of inducing IDO activity (Fujigaki et al., 2006), so multiple DNA regulatory elements are likely to be important in eliciting an appropriate response to complex immunological signals.
Here, we tested the hypothesis that both IFNγ and TNFα are the main IDO-inducing proinflammatory cytokines responsible for bacille Calmette-Guérin (BCG)-induced depressive-like behaviors. We first determined the role of IFNγ in depressive-like behavior induced by inoculation of BCG. Infection of mice with BCG is a well validated and classical model of chronic immune activation. Mice chronically infected with BCG display the two successive phases of sickness and depressive-like behaviors that are typically observed in patients undergoing immunotherapy (Moreau et al., 2005, 2008; O'Connor et al., 2009). For this reason, infection of mice with BCG represents a suitable model to study the depressive-like behavioral implications of chronic inflammation. BCG also chronically induces both IDO and proinflammatory cytokines such as IFNγ and TNFα (Moreau et al., 2008; O'Connor et al., 2009). After establishing an appropriate dose of BCG in C57BL/6J mice to induce depressive-like behavior, we show here that IFNγ receptor deficient mice (IFNγR−−) exhibited a typical acute sickness response to BCG, but were completely protected from the subsequent development of depressive-like behavior. Moreover, the robust BCG-induced increase in IDO mRNA expression and enzymatic activity that occurred in WT mice was absent in IFNγR−− mice. This was associated with an attenuated upregulation of central TNFα mRNA, and when BCG-infected mice were pretreated with etanercept, the soluble TNF receptor that prevents endogenous TNFα from binding to its biologically active receptor, the induction of IDO and depressive-like behavior were attenuated. We then confirmed in primary microglia, which are known to be key cellular mediators of neuroimmune responses, that IFNγ markedly induced IDO expression and primed microglia for additional cytokine signals, like TNFα. Together, these results demonstrate the critical role of IFNγ and TNFα in IDO upregulation and depressive-like behavior and indicate that the IFNγ and TNFα receptor pathways provide specific targets for therapeutic intervention in inflammation-associated depression.
Materials and Methods
Animals and treatments
All animal care and use were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council) and approved by the Institutional Animal Care and Use Committee. Experiments were performed on 8–12-week-old male C57BL/6J wild-type controls, B6.129S7-Ifngr1tm1Agt/J (stock #000664 and #003288) obtained from The Jackson Laboratory or Crl:CD1 (ICR) mice obtained from Charles River Laboratories. Mice were individually housed in standard shoebox cages, with wood shavings litter, in a temperature- (23°C) and humidity- (45–55%) controlled environment with a 12/12 h modified dark-light cycle (light on 10:00 P.M.–10:00 A.M.). Food and water were available ad libitum. Mice were handled individually every day for 7 d before the experiments.
On the day of BCG inoculation, fresh solutions were prepared by dispersing lyophilized BCG cultures (SanofiPasteur) in sterile endotoxin-free isotonic saline and injected intraperitoneally at the indicated doses. For the TNF-inhibition study, starting 1 d before BCG inoculation, mice were injected once daily subcutaneously for the duration of the experiment with either sterile physiological saline or 2.5 mg/kg etanercept (Amgen) dissolved in sterile endotoxin-free isotonic saline. This dose was chosen on the basis of previous studies demonstrating its ability to fully block TNFα activity (Popivanova et al., 2008).
Primary glia isolation and culture
Primary mixed glial cultures were established from brains of <2-d-old C57BL/6J mice. After removal of the meninges, brains were mechanically minced and dissociated with 0.25% trypsin/0.5 mm EDTA. After inactivation of trypsin with fetal calf serum (FCS), tissue suspension was passed through a 70 μm nylon cell strainer (BD Falcon). Dissociated cells were suspended in DMEM glucose with 4 mm glutamine (HyClone) supplemented with 10% heat-inactivated FCS (HyClone), 100 U/ml penicillin, and 100 μg/ml streptomycin and plated in tissue culture flasks. After 15–20 d, microglia were separated from confluent astrocytes by shaking the flasks for 1 h at 37°C on an orbital shaker at 150 rpm. Isolated microglia were collected and cultured in L929 cell conditioned medium for 7–10 d before treatment. The purity of microglia was >90% CD11b+ verified by flow cytometry. Cells were treated with 1 ng/ml recombinant murine IFNγ (PeproTech; Catalog 315–05) or 10 ng/ml recombinant murine TNFα (Invitrogen; Catalog PRC3014). These doses were selected on the basis of previous dose–response experiments (data not shown).
Behavioral experiments
All behavioral experiments were performed during the first 4 h of the dark phase of the light cycle.
Locomotor activity.
The effects of BCG on locomotor activity (LMA) were assessed in mice individually placed into a clean, novel cage identical to the home cage, but devoid of bedding or litter. The cage was divided into four virtual quadrants, and LMA was measured by counting the number of quadrant entries and rearings over a 5 min period. Counting was done by a trained observer who was blind to the treatments.
Forced swim test.
The forced swim test (FST), a standardized test of depressive-like behavior in which depression is inferred from increased duration of immobility, was conducted as described previously (O'Connor et al., 2008b, 2009). The duration of immobility was determined during the test using the mobility function of the “Observer Basic” software (Noldus). Program analysis settings were: sampling rate = 3/s; detection method = subtraction with low threshold of 20 and high threshold of 255 and minimum detectable object size of 200 pixels; image filtering = 2 pixel erosion and dilation; mobility threshold of 20% with 3 interval averaging.
Tail suspension test.
The tail suspension test (TST), a standardized test of depressive-like behavior in which depression is inferred from increased duration of immobility, was conducted as previously described using the Mouse Tail Suspension Package (MED-TSS-MS, Med Associates) (O'Connor et al., 2008b, 2009). Program analysis settings were integration = on; resolution = 0.1 s; gain = 4; start trigger = 20.
RNA extraction and reverse transcription
Total RNA from lung and whole brain samples was extracted in TRIzol reagent. All reverse transcriptase (RT) reactions were performed in a Stratagene Robocycler Gradient 96, using an Ambion (cat # 1710) reverse transcriptase kit according to manufacturer instructions, using 125 ng of total RNA and random decamer primers for each reaction. All RNA samples were reverse transcribed simultaneously to minimize interassay variation associated with the reverse transcription reaction.
Real-time RT-PCR
Real-time RT-PCR was performed on an Applied Biosystems Prism 7900 using Taqman gene expression assays for TNFα (cat # Mm00443258_m1), IL-1β (cat # Mm00434228_m1), IFNγ (cat # Mm00801778_m1), IDO (cat # Mm00492586_m1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cat # Mm999999_g1), purchased from Applied Biosystems. Reactions were performed in duplicate according to manufacturer instructions using 125 ng of cDNA template for each reaction. Relative quantitative measurement of target gene levels was performed using the ΔΔCt method, where Ct is the threshold concentration. GAPDH was used as the endogenous housekeeping control gene. Transgenic mice were positively identified by PCR using cDNA from the brain tissue of wild-type or IFNγR−− mice to confirm the absence or presence of the neomycin gene. If the neomycin gene was present, a 492 bp amplification product was generated. The forward and reverse primer sequences were 5′-AGGAT CTCCTGTCATCTCACCTTGCTCCTG-3′ and 5′-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-3′, respectively.
HPLC
Plasma tryptophan and kynurenine were analyzed by HPLC using an ESA Coulochem II detector with a 5041 Enhanced Analytical cell containing a glassy carbon electrode (+600 mV). Mobile phase (pH = 4.6) consisted of 75 mm NaH2PO4, 25 μm EDTA (disodium salt), and 100 μl/L triethylamine in acetonitrile:water (6:94 v:v). Brain monoamines were analyzed at +320 mV. The monoamine mobile phase, pH = 3.0, consisted of 75 mm NaH2PO4, 25 μm EDTA (disodium salt), 1.7 mm octanesulfonic acid, and 100 μl/L triethylamine in acetonitrile:water (7:93 v:v). The chromatograms were integrated and quantified using Dynamax MacIntegrator II software (Rainin Instruments).
Plasma (50 μl) was mixed with a solution of 10% sulfosalicylic acid solution (10 μl) and allowed to precipitate proteins on ice for at least 30 min. After the precipitation, samples were centrifuged at 12,000 × g for 10 min at 4°C. The supernatant was extracted and loaded into a Costar Spin-X centrifuge tube filter (0.22 μm nylon part 8169 Corning) and centrifuged at 12,000 × g for 6 min at 4°C. For the current study, plasma extracts were diluted at 1:50 following the extraction steps described above.
Mouse brains were first weighed and then homogenized in 500 μl of a 0.1 N HClO4/10 μm ascorbate solution using a 1.5 ml centrifuge tube and disposable pestle. Upon complete homogenization, the brain samples were centrifuged at 12,000 × g for 5 min at 4°C. After homogenization, the supernatant was extracted and loaded into a Costar Spin-X tube filter and centrifuged at 12,000 × g for 5 min at 4°C. For the current study, the sample extracts were then diluted at 1:10 (v:v) in 0.02 N HClO4 and analyzed for kynurenine, tryptophan and 1-methyl-tryptophan. The brain extracts which were analyzed for 5-hydroxyindoleacetic acid (5-HIAA) and serotonin were diluted at 1:100 (w:v) and injected into the HPLC system. Data from plasma samples are expressed as μmol/L, while data from brain samples are expressed as pmol/mg tissue.
A standard curve was generated on each day from concentrated (2 μm) tryptophan, 1-methyl-tryptophan, and kynurenine standards made up in 0.02 N HClO4 and held at 4°C until a 20 μl volume was injected into the system. Standards were made using a serial dilution technique that made the standards to levels that would encompass expected levels in the plasma samples. The standard curve was created using the system software and samples were not analyzed unless a linear standard curve with r2 >0.995 was achieved.
Statistical analysis
Data (mean ± SEM) were analyzed using a one-way (treatment), two-way (pretreatment or genotype × treatment), or three-way (pretreatment or genotype × treatment × time) ANOVA with repeated measurement on the time factor where appropriate, followed by a post hoc pairwise multiple comparison procedure using the Fisher's least significant difference, if the interaction was significant.
Results
BCG dose-dependently precipitates the development of depressive-like behaviors in C57BL/6J mice
To confirm that BCG inoculation induces the development of depressive-like behaviors in the inbred C57BL/6J mouse strain in a manner similar to that previously demonstrated in outbred CD-1 mice (Moreau et al., 2008), mice were treated with either non-pyrogenic physiological saline or a dispersed BCG suspension containing 106, 107, or 108 colony forming units (cfu). After treatment, mice were weighed daily and monitored for behavioral signs of sickness and depressive-like behavior. Consistent with our previous findings in CD-1 mice, BCG induced a transient, dose-dependent reduction in body weight (dose × time, F(3,112) = 9.06, p < 0.001) that returned to baseline by 3 (106 and 107 cfu) or 7 (108 cfu) d after BCG inoculation (Fig. 1A). After body weight had returned to pretreatment levels, mice were tested for locomotor deficits (a measure of sickness behavior). Horizontal locomotor activity remained modestly, yet significantly reduced (p = 0.024) only in mice inoculated with 108 cfu BCG compared with saline controls (Fig. 1B, left panel). However, there were no significant differences in locomotor activity in the mice inoculated with lower doses of BCG when compared with control mice. Also, there was no significant reduction in the number of rearings exhibited during the test by BCG-inoculated mice compared with saline-treated control mice (Fig. 1B, right panel).
Figure 1.
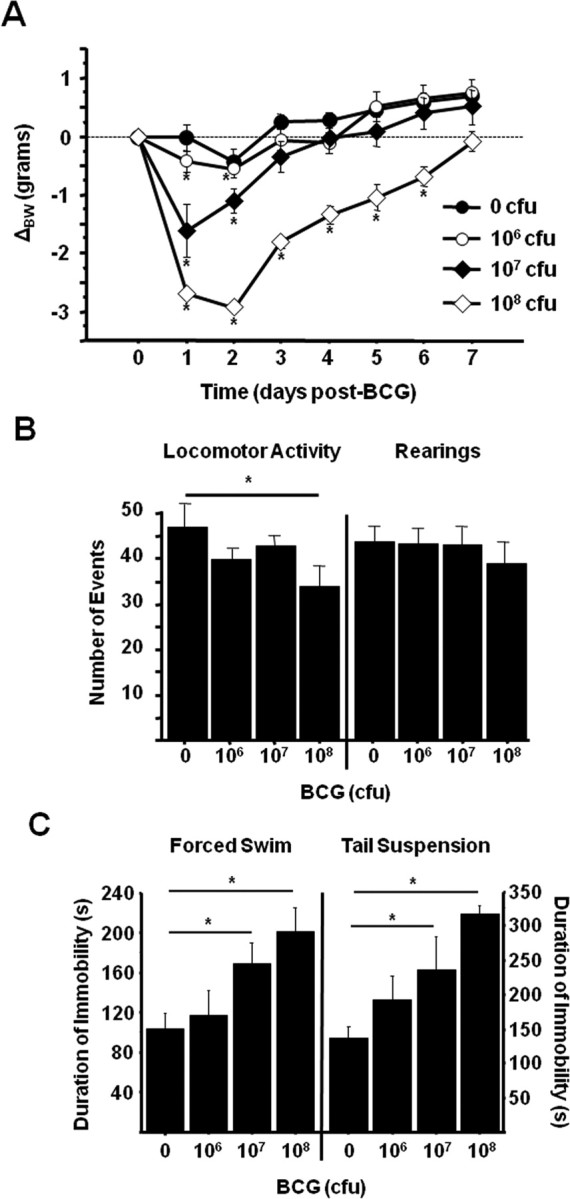
BCG dose-dependently induces depressive-like behavior in C57BL/6J mice after recovery from sickness behavior. Increasing doses of BCG were inoculated to mice via a single intraperitoneal injection. A, Mice were weighed daily as an indication of the acute sickness response. B, Seven days after BCG inoculation, horizontal locomotor activity (left) and rearings (right) were measured in a novel cage to ensure sickness-related motor deficits had returned to baseline levels. C, The duration of immobility during the forced swim test (left) and tail suspension test (right) was then recorded to assess the persistence of depressive-like behavior. Data represent means ± SEM (n = 5 mice per group). Error bars indicate statistical differences versus either same treatment baseline (A) or saline control (B, C). *p < 0.05.
Even in the absence of any overt signs of sickness behavior, BCG induced a clear dose-dependent increase in depressive-like behaviors 7 d after inoculation (treatment effect; p < 0.001 for FST and p = 0.006 for TST). When subjected to the FST, mice inoculated with 107 cfu BCG exhibited a significant increase (p = 0.017) in the duration of immobility (Fig. 1C, left panel). Four hours later, mice were subjected to the TST. Again, mice inoculated with 107 cfu of BCG exhibited a significant increase (p = 0.037) in the duration of immobility compared with control mice (Fig. 1C, right panel). Together, these findings validate the BCG model of chronic inflammation induced depressive-like behavior in the C57BL/6J inbred mouse strain and establish 107 cfu BCG/mouse as an effective dose with minimal impact on motor activity.
IFNγ receptor deletion specifically prevents the development of BCG-induced depressive-like behaviors
To determine whether BCG-induced depressive-like behaviors are dependent on IFNγ activity in vivo, WT or IFNγR−− mice were treated with either non-pyrogenic physiological saline or 107 cfu of BCG in a 2 × 2 factorial arrangement. After treatment, mice were weighed daily and monitored for behavioral signs of sickness. Figure 2, A and B, show that BCG induced a typical transient acute sickness response in WT and IFNγ−− mice that was completely resolved by 7 d after inoculation. There was a significant BCG × time interaction for body weight (F(1,126) = 15.92, p < 0.001) (Fig. 2A) and locomotor activity (F(1,72) = 11.11, p < 0.001) (Fig. 2B); however, there were no differences resulting from genotype (p > 0.5) for either dependent variable.
Figure 2.
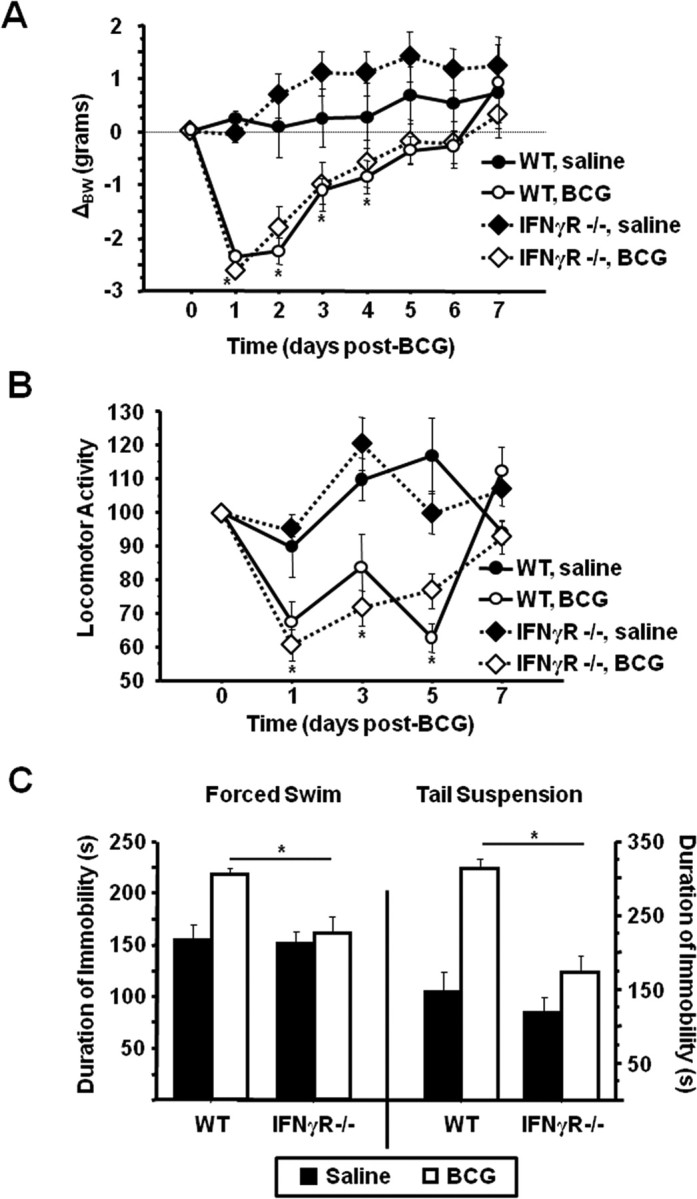
IFNγR−− mice exhibit normal sickness behavior but do not develop depressive-like behaviors in response to BCG. WT or IFNγR−− mice received an intraperitoneal injection of either saline or 107 cfu of BCG. A, B, Body weight was recorded daily (A), and horizontal locomotor activity in a novel cage (B) was measured every other day for 7 d after inoculation until behavioral indications of the acute sickness response were no longer apparent. C, One week after BCG inoculation, the duration of immobility in the forced swim test (left) and the tail suspension test (right) was recorded. Data represent means ± SEM (n = 3–9 mice per group). Error bars indicate statistical differences between groups. *p < 0.05.
As we have already established (Fig. 1C, left panel), BCG induced a significant increase in the duration of immobility during the FST 7 d after inoculation. However, this increase was completely absent in the IFNγR−−mice (genotype × BCG F(1,18) = 4.61, p = 0.042) (Fig. 2C, left panel). Four hours later, BCG-treated mice also displayed an increased duration of immobility during the TST that was absent in mice lacking functional IFNγ receptors (genotype × BCG F(1,18) = 7.74, p = 0.01) (Fig. 2C, right panel). Together, these results demonstrate that whereas IFNγ activity is not required for BCG-induced acute sickness responses, it is fully necessary for the subsequent development of depressive-like behaviors in response to BCG.
BCG-induced IDO expression and activity are dependent on IFNγ receptor function
IDO plays a pivotal role in mediating the development of depressive-like behaviors in response to both acute and chronic peripheral immune activation (Henry et al., 2008; Moreau et al., 2008; O'Connor et al., 2008b, 2009). To determine if the absence of BCG-induced depressive-like behaviors in IFNγR−− mice was the result of impaired IDO upregulation, steady-state mRNA expression of IDO was measured by real-time RT-PCR in both the lung and brain tissues of mice immediately after behavioral testing at 7 d after BCG inoculation. IDO mRNA was significantly upregulated in both the lungs (treatment effect; p < 0.001) (Fig. 3A) and brain (treatment effect; p < 0.001) (Fig. 3B) of BCG-inoculated WT mice. However, this BCG-induced upregulation of IDO mRNA was completely inhibited in IFNγR−− mice in both tissues, perfectly paralleling the failure of these mice to develop depressive-like behaviors in response to BCG (lung: genotype × BCG F(1,18) = 98.2, p < 0.001) and brain: genotype × BCG F(1,18) = 17.9, p < 0.001) (Fig. 3A,B).
Figure 3.

IFNγ receptors are necessary for BCG-induced IDO expression and activity. Immediately after behavioral testing at 7 d after BCG, mice were killed for blood collection, followed by perfusion with ice-cold heparin-containing PBS and tissue collection. A, B, Steady-state expression of IDO mRNA transcripts in both the lungs (A) and brain (B) were measured by real-time RT-PCR. C, E, To determine the relationship between relative mRNA expression and enzymatic activity, lungs (C) and brain (E) tissue from a separate group of BCG-inoculated mice were used. D, F, Plasma (D) and brain (F) concentrations of kynurenine and tryptophan were measured by HPLC with electrochemical detection. The ratio of kynurenine/tryptophan was determined as an indication of in vivo IDO activity. Data represent means ± SEM (n = 3–9 mice per group). Error bars indicate statistical differences between groups. *p < 0.05. Average Ct values for BCG treated WT mice were as follows: lung = 26.5 ± 0.7 and brain = 36.1 ± 0.6.
To demonstrate that the inability of BCG to induce IDO mRNA in IFNγR−− mice was of pathophysiological relevance, the correlation between relative mRNA expression level of IDO and ex vivo IDO enzymatic activity was tested in both lung and brain tissues of BCG-inoculated WT mice, and the kynurenine/tryptophan ratios were measured in the plasma and brains of separate groups of WT and IFNγR−− mice treated with either saline or BCG. There was a significant positive correlation between the relative mRNA expression of IDO and enzyme activity in lung tissue collected 7 d after BCG inoculation (r = 0.84, p < 0.001) (Fig. 3C). Concordant with the increase in lung IDO activity, BCG induced a significant increase in the plasma kynurenine/tryptophan ratio in WT mice; however, as with depressive-like behavior and IDO mRNA expression, the kynurenine/tryptophan ratio in the plasma of BCG-inoculated IFNγR−− mice was substantially attenuated (genotype × BCG F(1,18) = 7.05, p = 0.015) (Fig. 3D).
Although IDO enzymatic activity in brain tissue was markedly lower than in the lung, there was again a significant positive correlation between IDO expression and activity 7 d after BCG (r = 0.74, p < 0.001) (Fig. 3E). While the kynurenine/tryptophan ratio in whole-brain tissue was not significantly elevated in response to BCG, it was significantly reduced in the IFNγR−− mice (genotype, p = 0.002) (Fig. 3F). Together, these results indicate that IFNγ receptor function is necessary for BCG-induced activation of IDO and the subsequent development of depressive-like behavior.
Increased IDO metabolism does not reduce central 5-HT concentration or turnover
We have previously shown that increased IDO activity after acute immune activation with LPS has no significant impact on serotonin availability (O'Connor et al., 2008b). To determine whether the BCG-induced shift in tryptophan metabolism along the kynurenine pathway affected serotonin availability, brain levels of 5-HT and the major 5-HT metabolite 5-HIAA were measured in WT and IFNγR−− mice 7 d after inoculation, when depressive-like behaviors and IDO activation were fully apparent. Consistent with our previous findings in LPS-treated mice, brain concentrations of 5-HT and 5-HIAA were not significantly different as a result of BCG treatment or IFNγR deletion (p > 0.05) (Table 1). Similarly, there were no changes in the brain 5-HIAA:5-HT ratio (p > 0.05) (Table 1). These data indicate that BCG-induced depressive-like behaviors are unlikely to result from diminished 5-HT availability.
Table 1.
BCG does not induce changes in 5-HT
| WT | IFNγR−− | |||
|---|---|---|---|---|
| Saline | BCG | Saline | BCG | |
| 5-HT | 2.47 ± 0.11 | 2.51 ± 0.16 | 2.56 ± 0.011 | 2.49 ± 0.13 |
| 5-HIAA | 1.07 ± 0.07 | 1.19 ± 0.07 | 1.20 ± 0.06 | 1.15 ± 0.09 |
| 5-HIAA/5-HT | 435 ± 0.05 | 475 ± 0.02 | 474 ± 0.03 | 466 ± 0.04 |
Serotonin and its major metabolite 5-HIAA were measured by HPLC in brain tissue from WT or IFNγR−− mice treated with either saline or BCG. Data represent mean (micromoles per milligram of tissue) ± SEM; n = 3–9 mice per group.
IFNγR−− mice develop a robust proinflammatory cytokine and immune response to BCG, although TNFa is reduced
To confirm that the inability of BCG to induce IDO meditated depressive-like behaviors in IFNγR deficient mice was not merely the result of a non-specific defective proinflammatory cytokine response to BCG, lung and brain expression levels of the major IDO-inducing cytokines were determined in WT and IFNγR−− mice. BCG-induced a significant increase in steady-state mRNA expression of IFNγ in the lung (treatment effect; p = 0.01); however, unlike either IDO expression or depressive-like behavior, there was no attenuation of IFNγ mRNA in IFNγR−− mice (Fig. 4A). Similarly, there was a significant upregulation of IFNγ mRNA in brain tissue from BCG-inoculated mice (treatment effect; p = 0.048) (Fig. 4B), but there was no significant genotype × BCG interaction.
Figure 4.

IFNγR−− mice exhibit a typical proinflammatory cytokine response. A–F, Steady-state mRNA expression of IFNγ (A, B), IL-1β (C, D), and TNFα (E, F) were measured by real-time RT-PCR in both the lung and brain tissue 7 d after saline or BCG administration. G, As an indicator of BCG infection, splenomegaly was measured 7 d after BCG. Data represent means ± SEM (n = 3–9 mice per group). Error bars indicate statistical differences among groups. *p < 0.05. Average Ct values for BCG treated WT mice were as follows: IFNγ: lung = 31.1 ± 0.9, brain = 37.0 ± 1.0; IL-1β: lung = 26.2 ± 0.5 brain = 30.8 ± 0.8; TNFα: lung = 28.3 ± 0.6, brain = 32.7 ± 0.3.
Figure 4, C and D, indicate that another proinflammatory cytokine that plays a key role in inflammation-associated sickness behavior, IL-1β, was significantly increased in the lung and brain of all BCG-inoculated mice (p < 0.001), and there was again no genotype × BCG interaction (genotype × BCG, p = 0.08 in lung and p = 0.16 in brain). BCG also induced a significant upregulation of TNFα mRNA in lung tissue of WT and IFNγR−− mice compared with saline-treated controls. However, this increase was significantly less marked in IFNγR−− mice (genotype × BCG F(1,18) = 5.60, p = 0.026) (Fig. 4E). In brain tissue of BCG-inoculated mice, there was a modest, yet significant effect of genotype (p < 0.02) in reducing the steady state mRNA expression of TNFα (Fig. 4F). However, the main effect of BCG did not reach statistical significance (p = 0.06), and there was no genotype × BCG interaction. IL-6, SOCS-3, and TLR2 mRNA were also measured in lung and brain tissue 7 d after saline or BCG injection, but in all cases there were no significant main effects or interactions (data not shown).
Finally, as a correlate of BCG infection (Bourassa et al., 1985), splenomegaly was measured in WT and IFNγR−− mice 7 d after injection with saline or inoculation with BCG. Consistent with previous studies, BCG infection resulted in a marked increase in spleen weight (treatment effect; p < 0.001) that did not significantly differ between WT and IFNγR deficient mice (genotype × BCG, p > 0.8) (Fig. 4G). Together, these data indicate that although IFNγR−− mice exhibited a similar proinflammatory neuroimmune response to BCG during the first week after inoculation with the exception of TNFα, there was no upregulation of IDO nor any development of depressive-like behavior in the absence of functional IFNγ receptors.
In vivo neutralization of TNFα attenuates BCG-induced IDO expression and depressive-like behaviors
As already presented, inoculation of IFNγR−− mice with BCG resulted in a typical acute neuroimmune response (Fig. 2A,B); however, TNFα mRNA expression was found to be significantly lower in the brains of BCG-infected IFNγR deficient mice. TNFα is able to induce IDO independently of IFNγ and can also synergize with IFNγ to induce IDO (Robinson et al., 2003). To determine whether the blockade of IDO upregulation and depressive-like behaviors in IFNγR−− mice could be caused, at least in part, by a diminished TNFα upregulation, WT mice were chronically administered either nonpyrogenic physiological saline or soluble TNF receptor, etanercept, (2.5 mg/kg once daily) beginning 1 d before infection with BCG. After treatment, mice were weighed daily and monitored for behavioral signs of sickness. Figure 5, A and B, show that BCG induced a typical transient acute sickness response in WT and etanercept-treated mice. There was a significant BCG × time interaction for body weight (F(1,140) = 12.95, p < 0.001) (Fig. 5A) and locomotor activity (F(1,80) = 3.76, p = 0.008) (Fig. 5B). There was no difference caused by pretreatment (p = 0.85) for body weight, but pretreatment with etanercept abolished the transient reduction in locomotor activity induced by BCG (etanercept × BCG × time; F(1,80) = 2.95, p = 0.025) (Fig. 5B). There was a significant etanercept × BCG interaction on the duration of immobility during both the FST (F(1,19) = 6.75, p = 0.018) (Fig. 5C, left panel) and TST (F(1,19) = 6.46,p = 0.019) (Fig. 5C, right panel). Post hoc analysis revealed that while etanercept significantly reduced the duration of immobility in both the FST (p = 0.003) and TST (p = 0.044) for BCG-infected wild-type mice, BCG-infected mice pretreated with etanercept still exhibited increased immobility during both tests (FST; p = 0.024 and TST; p = 0.048).
Figure 5.

In vivo inhibition of TNFα activity attenuates depressive-like behavior and IDO upregulation in BCG-infected mice. C57BL/6 mice were administered etanercept (2.5 mg/kg once daily) beginning 1 d before injection with 107 cfu BCG. A, B, Body weight was recorded daily (A), and horizontal locomotor activity in a novel cage (B) was measured every other day for 7 d after inoculation, until behavioral indications of the acute sickness response were no longer apparent in all mice. C, One week after BCG inoculation, the duration of immobility in the forced swim test (left) and the tail suspension test (right) was recorded. D–F, After behavioral testing, IDO mRNA expression was measured by real-time RT-PCR in the lung (D) and brain (E), and the kynurenine/tryptophan ratio was measured in the plasma (F). Data represent means ± SEM (n = 3–9 mice per group). Error bars indicate statistical differences between groups. *p < 0.05.
Immediately after the last behavioral test, lung and brain tissue and plasma were collected for determination of IDO mRNA, proinflammatory cytokine mRNA expression, and the kynurenine/tryptophan ratio. BCG induced a significant increase in the steady-state mRNA expression of both brain and lung IFNγ (treatment effect; p = 0.012), IL-1β (treatment effect; p = 0.001), and TNFα (treatment effect; p = 0.039), but there was no effect of pretreatment with etanercept for any of these cytokines (etanercept × BCG; p > 0.7 for each measure) (data not shown). Conversely, similar to depressive-like behavior, etanercept pretreatment significantly attenuated IDO mRNA expression in both lung (etanercept × BCG; F(1,19) = 21.06,p < 0.001) (Fig. 5D) and brain (etanercept × BCG; F(1,19) = 7.06,p = 0.016) (Fig. 5E). Importantly, pretreatment with etanercept significantly reduced the plasma kynurenine/tryptophan ratio in BCG-infected mice (etanercept × BCG; F(1,19) = 4.42,p = 0.04) (Fig. 5F). Together, these results demonstrate that TNFα may play an important role in IDO upregulation and the development of depressive-like behaviors in response to BCG.
IFNγ and TNFα synergistically induce IDO expression in primary microglia
Inoculation of mice with BCG results in a marked upregulation of brain IDO which closely parallels increased IFNγ and TNFα expression (Moreau et al., 2005, 2008). To determine whether these two IDO-inducing proinflammatory cytokines interact to affect IDO upregulation in resident murine brain cells known to mediate neuroimmune responses, cultures of purified primary microglia were obtained from C57BL/6J mice, the same strain used in the in vivo studies. Primary microglia cultures were treated with either sterile PBS, 10 ng/ml recombinant murine TNFα, 1 ng/ml recombinant murine IFNγ or TNFα plus IFNγ for 6 h. At the end of the treatment period, steady-state mRNA expression of IDO was determined by real-time RT-PCR. TNFα alone did not induce IDO expression in primary microglia, while IFNγ alone induced a robust increase in IDO expression (p = 0.04) (Fig. 6). Interestingly, when primary microglia were treated with both IFNγ plus TNFα for the same period of time, upregulation of IDO was enhanced by slightly more than twofold when compared with IFNγ alone (p = 0.03) Together, these data support the idea that IFNγ which we have already shown to be elevated in the brain of BCG-treated mice is sufficient to induce IDO expression. Moreover, IFNγ, which is well known to prime peripheral macrophages to subsequent immune signals, may act in concert with TNFα in vivo to elicit the full neuroimmune response.
Figure 6.

IFNγ and TNFα synergistically induce IDO expression in primary microglia. Primary microglia cultures were treated with either PBS (Ctrl), 10 ng/ml recombinant murin TNFα, 1 ng/ml recombinant murine IFNγ, or TNFα plus IFNγ for 6 h. Steady-state expression of IDO mRNA transcripts were measured by real time RT-PCR. Data represent means ± SEM of three independent experiments. Error bars indicate statistical differences among groups. *p < 0.01. Average Ct values for IFNγ treated cells were 28.8 ± 0.4.
Discussion
The present results show that blockade of IFNγ receptor activation abrogates BCG-induced depressive-like behavior in mice by a mechanism that is at least partly mediated by TNFα and is dependent on the synergistic activating effect of IFNγ and TNFα on IDO.
Depressive-like behavior was measured by increased duration of immobility in the forced swim and tail suspension tests. Initially developed as behavioral screening tools for anti-depressant drug efficacy, these two tests are increasingly being used to measure a specific depressive-like behavior, behavioral despair, independently of any anti-depressant treatment (Dunn and Swiergiel, 2005; Banasr and Duman, 2008; Moreau et al., 2008; O'Connor et al., 2008b, 2009; Malisch et al., 2009). Although not assessed in the present series of experiments, sucrose preference would have likely provided similar data to those obtained in FST and TST because BCG has been shown previously to decrease sucrose preference independently of sickness (Moreau et al., 2008).
Infection of mice with BCG is a widely used and validated model of chronic cellular immunity that results in rapid and persistent dissemination of mycobacteria to all organs, especially the lungs, liver and spleen and, to a lesser extent, the CNS (Tsenova et al., 1999). BCG is recognized by the pathogen-associated molecular pattern receptors TLR2 and TLR4, and infection of mice with BCG results in production of proinflammatory cytokines, including IFNγ, TNFα, and IL-1β that remain elevated for several weeks (Moreau et al., 2005, 2008). IFNγ and TNFα are critical to the host's immunological defense against intracellular pathogens such as BCG. These cytokines maintain granuloma integrity and coordinate the cellular immune response (Kindler et al., 1989; Mittrucker et al., 2007). In fact, Kamijo et al. (1993) demonstrated that IFNγR−− mice had an increased sensitivity to BCG infection. Although BCG infection was not lethal in WT mice, all BCG-infected IFNγR deficient mice died by 9 weeks after inoculation. This observation is important because if IFNγR−− mice are more susceptible to BCG infection, one would predict BCG-induced neuroimmune responses and subsequent depressive-like behaviors to be exacerbated rather than inhibited, as we report in the present series of experiments. The dose of BCG used by Kamijo et al. (1993) was very similar to ours (2 × 107 vs 1 × 107 cfu), but pathological differences between the WT and IFNγR−− mice infected with BCG occurred later than 2 weeks after inoculation in the Kamijo et al. study. Our own experiments were conducted within the first 7 d after inoculation, when cytokine levels were not significantly different between WT and IFNγR−− mice, and the standard physiological correlate of BCG infection, splenomegaly, was not different. It is therefore unlikely that our results were biased by a differential sensitivity of IFNγR−− mice to BCG.
As anticipated, inoculation of mice with BCG caused an increase in IDO expression in WT mice, and this resulted in a significant increase in the plasma kynurenine/tryptophan levels. Moreover, because IDO upregulation did not occur in IFNγR−− mice, the plasma kynurenine/tryptophan levels were unchanged. Unlike our previous studies using acute immune activation with LPS, the ratio of kynurenine/tryptophan in the brains of BCG-inoculated mice (WT or IFNγR−−) was not higher than saline-treated control mice. This result was unexpected because IDO expression was significantly increased in the brains of BCG-inoculated WT mice, and peripheral kynurenine is readily transported into the brain via saturable transport mechanisms (Kita et al., 2002; Schwarcz and Pellicciari, 2002). We expected that a substantial increase in the peripheral kynurenine/tryptophan ratio would be mirrored by a parallel response in the brain. A possible explanation for this apparent disconnect might involve the upregulation of kynurenine pathway enzymes downstream of IDO. Glial cells can metabolize kynurenine to a number of intermediate metabolites (Kita et al., 2002; Schwarcz and Pellicciari, 2002). Astrocytes express high amounts of kynurenine aminotransferase, and degrade kynurenine to kynurenic acid. Microglia highly express the enzymes responsible for the generation of quinolinic acid, kynurenine monooxygenase (KMO) and kynureninase. Like IDO, KMO is also inducible by IFNγ (Alberati-Giani et al., 1996). However, the relative importance of specific kynurenine metabolites to inflammation-induced depressive behaviors is not known. As such, future studies aimed at characterizing differential kynurenine pathway metabolism within this context are necessary. Interestingly, we found that the baseline kynurenine/tryptophan ratio in the brain was significantly reduced in IFNγR knock-out mice (Fig. 4F) compared with WT control mice, while this was not the case in the plasma (Fig. 4D). This finding indicates that the IFNγ pathway may play an important role in regulating brain IDO expression even during periods of immunoquiescence. That the plasma kynurenine/tryptophan ratio did not follow this same pattern is likely because hepatic tryptophan dioxygenase (TDO) is the predominant tryptophan degrading enzyme at the periphery in the absence of inflammation (Schröcksnadel et al., 2006).
An important objective of the present study was the identification of the main cytokines that are responsible for the development of depressive-like behavior in response to BCG. Based on its pivotal role in the induction of IDO, IFNγ was an obvious candidate. However, the inhibition of depressive-like behaviors in IFNγR deficient mice in the present study was accompanied by a reduced upregulation of TNFα mRNA in the brain. TNFα is a well known mediator of neuroimmune responses after immune activation (Kelley et al., 2003; O'Connor et al., 2008a), and there is some evidence to indicate TNFα may be involved in the pathophysiology of depression. Not only do patients suffering from major depression, who are otherwise medically healthy, often have elevated levels of circulating proinflammatory cytokines, including TNFα (Maes, 1995; Raison et al., 2006), but also mice lacking functional type 1 or type 2 TNFα receptors display an antidepressant phenotype (Simen et al., 2006). While the role of specific cytokines in mediating chronic inflammation-induced (comorbid) depression is not yet understood, administration of the TNFα inhibitor, etanercept, to patients with chronic psoriasis has been reported to alleviate symptoms of depression (Tyring et al., 2006). Pretreatment of mice with etanercept prevented the transient acute reduction in locomotor activity after BCG infection (Fig. 5B). However, this was not the case with the transient reduction in body weight (Fig. 5A) probably because IL-1β is the predominant proinflammatory cytokine responsible for modulating the anorexic response to immune activation (Layé et al., 2000). Inhibition of TNFα activity in vivo only partly inhibited the induction of depressive-like behaviors and IDO expression (Fig. 5C–F), indicating that the total inhibition of depressive-like behaviors observed in IFNγ−− mice cannot be attributed solely to TNFα.
IFNγ is a potent inducer of IDO in peripheral myeloid cells and cell lines, including dendritic cells (Jung et al., 2007), monocytes and fibroblasts (Fujigaki et al., 2006). However, IFNγ-independent IDO activation pathways have also been reported (Fujigaki et al., 2006; Jung et al., 2007). We have previously demonstrated that not only does IDO play a critical role in mediating LPS or BCG-induced depressive-like behaviors in mice, but also the upregulation of IDO in the brain parallels increased expression of IFNγ (Moreau et al., 2005, 2008; O'Connor et al., 2008b, 2009). Subsequent in vivo experiments have confirmed this positive correlation (André et al., 2008; Connor et al., 2008). However, LPS has been shown to induce IDO expression in primary mixed glial cultures without an increase in detectible IFNγ (Connor et al., 2008). The IFNγ-independent upregulation of IDO has first been described in the LPS model of immune activation, and it was not known whether it extends to other forms of immune activation. Although direct stimulation of human glial cells with IFNγ induces IDO (Guillemin et al., 2005a), TNFα can also induce the upregulation of IDO, although at relatively high doses. In fact, IFNγ and TNFα have been reported to synergistically induce IDO expression in the HeLa human endothelial cell line (Robinson et al., 2003). Consistent with these reports, we confirmed that IFNγ induced a robust increase in IDO expression in primary murine microglial cells, and while TNFα alone did not induce IDO, IFNγ plus TNFα treatment resulted in a robust synergistic effect on IDO upregulation. These results indicate that the necessary role of IFNγ on BCG induced IDO upregulation and depressive-like behaviors may be partly mediated through interactions with other cytokines, like TNFα.
In conclusion, the present study identifies IFNγ, together with TNFα, as the essential proinflammatory cytokine responsible for inducing IDO and depressive-like behaviors in the context of chronic inflammation induced by BCG. These findings are important because they indicate that targeting the cytokine pathways that lead to IDO activation specifically and effectively prevent the development of depressive behaviors. Thus, anti-inflammatory treatments may have therapeutic benefit in the treatment of inflammation associated depression.
Footnotes
This work was supported by National Institutes of Health Grants R01 MH 071349 and R01 MH 079829 to R.D. and R01 AG 029573 to K.W.K.
References
- Adler UC, Marques AH, Calil HM. Inflammatory aspects of depression. Inflamm Allergy Drug Targets. 2008;7:19–23. doi: 10.2174/187152808784165216. [DOI] [PubMed] [Google Scholar]
- Alberati-Giani D, Ricciardi-Castagnoli P, Köhler C, Cesura AM. Regulation of the kynurenine metabolic pathway by interferon-gamma in murine cloned macrophages and microglial cells. J Neurochem. 1996;66:996–1004. doi: 10.1046/j.1471-4159.1996.66030996.x. [DOI] [PubMed] [Google Scholar]
- André C, O'Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N. Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J Neuroimmunol. 2008;200:90–99. doi: 10.1016/j.jneuroim.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64:863–870. doi: 10.1016/j.biopsych.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa D, Forget A, Pelletier M, Skamene E, Turcotte R. Cellular immune response to Mycobacterium bovis (BCG) in genetically-susceptible and resistant congenic mouse strains. Clin Exp Immunol. 1985;62:31–38. [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH. Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology. 2002;26:643–652. doi: 10.1016/S0893-133X(01)00407-9. [DOI] [PubMed] [Google Scholar]
- Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry. 2003;54:906–914. doi: 10.1016/s0006-3223(03)00173-2. [DOI] [PubMed] [Google Scholar]
- Chon SY, Hassanain HH, Pine R, Gupta SL. Involvement of two regulatory elements in interferon-gamma-regulated expression of human indoleamine 2,3-dioxygenase gene. J Interferon Cytokine Res. 1995;15:517–526. doi: 10.1089/jir.1995.15.517. [DOI] [PubMed] [Google Scholar]
- Connor TJ, Starr N, O'Sullivan JB, Harkin A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: a role for IFN-gamma? Neurosci Lett. 2008;441:29–34. doi: 10.1016/j.neulet.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008a;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Capuron L, Irwin MR, Miller AH, Ollat H, Perry VH, Rousey S, Yirmiya R. Identification and treatment of symptoms associated with inflammation in medically ill patients. Psychoneuroendocrinology. 2008b;33:18–29. doi: 10.1016/j.psyneuen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH. Effects of interleukin-1 and endotoxin in the forced swim and tail suspension tests in mice. Pharmacol Biochem Behav. 2005;81:688–693. doi: 10.1016/j.pbb.2005.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DL, Charney DS, Lewis L, Golden RN, Gorman JM, Krishnan KR, Nemeroff CB, Bremner JD, Carney RM, Coyne JC, Delong MR, Frasure-Smith N, Glassman AH, Gold PW, Grant I, Gwyther L, Ironson G, Johnson RL, Kanner AM, Katon WJ, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry. 2005;58:175–189. doi: 10.1016/j.biopsych.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Fujigaki H, Saito K, Fujigaki S, Takemura M, Sudo K, Ishiguro H, Seishima M. The signal transducer and activator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopolysaccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. 2006;139:655–662. doi: 10.1093/jb/mvj072. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. 2005a;49:15–23. doi: 10.1002/glia.20090. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ, Noonan CE, Takikawa O, Cullen KM. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in Alzheimer's disease hippocampus. Neuropathol Appl Neurobiol. 2005b;31:395–404. doi: 10.1111/j.1365-2990.2005.00655.x. [DOI] [PubMed] [Google Scholar]
- Hassanain HH, Chon SY, Gupta SL. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J Biol Chem. 1993;268:5077–5084. [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, Sheridan JF, Godbout JP. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5:15. doi: 10.1186/1742-2094-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung ID, Lee CM, Jeong YI, Lee JS, Park WS, Han J, Park YM. Differential regulation of indoleamine 2,3-dioxygenase by lipopolysaccharide and interferon gamma in murine bone marrow derived dendritic cells. FEBS Lett. 2007;581:1449–1456. doi: 10.1016/j.febslet.2007.02.073. [DOI] [PubMed] [Google Scholar]
- Kamijo R, Le J, Shapiro D, Havell EA, Huang S, Aguet M, Bosland M, Vilcek J. Mice that lack the interferon-gamma receptor have profoundly altered responses to infection with bacillus Calmette-Guerin and subsequent challenge with lipopolysaccharide. J Exp Med. 1993;178:1435–1440. doi: 10.1084/jem.178.4.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley KW, Bluthe RM, Dantzer R, Zhou JH, Shen WH, Johnson RW, Broussard SR. Cytokine-induced sickness behavior. Brain Behav Immun 17 Suppl. 2003;1:S112–S118. doi: 10.1016/s0889-1591(02)00077-6. [DOI] [PubMed] [Google Scholar]
- Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- Kita T, Morrison PF, Heyes MP, Markey SP. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the L-kynurenine and quinolinic acid pools in brain. J Neurochem. 2002;82:258–268. doi: 10.1046/j.1471-4159.2002.00955.x. [DOI] [PubMed] [Google Scholar]
- Konan KV, Taylor MW. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J Biol Chem. 1996;271:19140–19145. doi: 10.1074/jbc.271.32.19140. [DOI] [PubMed] [Google Scholar]
- Layé S, Gheusi G, Cremona S, Combe C, Kelley K, Dantzer R, Parnet P. Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am J Physiol Regul Integr Comp Physiol. 2000;279:R93–R98. doi: 10.1152/ajpregu.2000.279.1.R93. [DOI] [PubMed] [Google Scholar]
- Maes M. Evidence for an immune response in major depression: a review and hypothesis. Prog Neuropsychopharmacol Biol Psychiatry. 1995;19:11–38. doi: 10.1016/0278-5846(94)00101-m. [DOI] [PubMed] [Google Scholar]
- Maes M, Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, Almerighi C, Meltzer H. Treatment with interferon-alpha (IFN alpha) of hepatitis C patients induces lower serum dipeptidyl peptidase IV activity, which is related to IFN alpha-induced depressive and anxiety symptoms and immune activation. Mol Psychiatry. 2001;6:475–480. doi: 10.1038/sj.mp.4000872. [DOI] [PubMed] [Google Scholar]
- Malisch JL, Breuner CW, Kolb EM, Wada H, Hannon RM, Chappell MA, Middleton KM, Garland T., Jr Behavioral despair and home-cage activity in mice with chronically elevated baseline corticosterone concentrations. Behav Genet. 2009;39:192–201. doi: 10.1007/s10519-008-9246-8. [DOI] [PubMed] [Google Scholar]
- Mittrücker HW, Steinhoff U, Köhler A, Krause M, Lazar D, Mex P, Miekley D, Kaufmann SH. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci U S A. 2007;104:12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau M, Lestage J, Verrier D, Mormede C, Kelley KW, Dantzer R, Castanon N. Bacille Calmette-Guerin inoculation induces chronic activation of peripheral and brain indoleamine 2,3-dioxygenase in mice. J Infect Dis. 2005;192:537–544. doi: 10.1086/431603. [DOI] [PubMed] [Google Scholar]
- Moreau M, André C, O'Connor JC, Dumich SA, Woods JA, Kelley KW, Dantzer R, Lestage J, Castanon N. Inoculation of bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior. Brain Behav Immun. 2008;22:1087–1095. doi: 10.1016/j.bbi.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, McCusker RH, Strle K, Johnson RW, Dantzer R, Kelley KW. Regulation of IGF-I function by proinflammatory cytokines: at the interface of immunology and endocrinology. Cell Immunol. 2008a;252:91–110. doi: 10.1016/j.cellimm.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2008b doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JC, Lawson MA, Andre C, Briley EM, Szegedi SS, Lestage J, Castanon N, Herkenham M, Dantzer R, Kelley KW. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol. 2009;182:3202–3212. doi: 10.4049/jimmunol.0802722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitala A, Pertovaara M, Karjalainen J, Oja SS, Hurme M. Association of interferon-gamma +874(T/A) single nucleotide polymorphism with the rate of tryptophan catabolism in healthy individuals. Scand J Immunol. 2005;61:387–390. doi: 10.1111/j.1365-3083.2005.01586.x. [DOI] [PubMed] [Google Scholar]
- Robinson CM, Shirey KA, Carlin JM. Synergistic transcriptional activation of indoleamine dioxygenase by IFN-gamma and tumor necrosis factor-alpha. J Interferon Cytokine Res. 2003;23:413–421. doi: 10.1089/107999003322277829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröcksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin Chim Acta. 2006;364:82–90. doi: 10.1016/j.cca.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Schroecksnadel K, Kaser S, Ledochowski M, Neurauter G, Mur E, Herold M, Fuchs D. Increased degradation of tryptophan in blood of patients with rheumatoid arthritis. J Rheumatol. 2003;30:1935–1939. [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- Simen BB, Duman CH, Simen AA, Duman RS. TNFalpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol Psychiatry. 2006;59:775–785. doi: 10.1016/j.biopsych.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Spalletta G, Bossù P, Ciaramella A, Bria P, Caltagirone C, Robinson RG. The etiology of poststroke depression: a review of the literature and a new hypothesis involving inflammatory cytokines. Mol Psychiatry. 2006;11:984–991. doi: 10.1038/sj.mp.4001879. [DOI] [PubMed] [Google Scholar]
- Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumor necrosis factor alpha is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc Natl Acad Sci U S A. 1999;96:5657–5662. doi: 10.1073/pnas.96.10.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyring S, Gottlieb A, Papp K, Gordon K, Leonardi C, Wang A, Lalla D, Woolley M, Jahreis A, Zitnik R, Cella D, Krishnan R. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]