

Abstract
The main purpose of this study was to determine whether enhancement of repair capacity would attenuate mitochondrial DNA oxidative damage and result in greater cell survival under stressful conditions. The repair of oxidative damage is initiated by DNA glycosylases, which catalyze the excision of oxidized bases, such as 8-hydroxydeoxyguanosine (8-oxodG). Drosophila DNA glycosylases, dOgg1 and RpS3, were ectopically expressed within the mitochondrial matrix in Drosophila S2 cells, causing a severalfold decrease in the levels of 8-oxodG in mitochondrial DNA. Unexpectedly, cells did not show increased resistance to oxidative stress, but instead became more susceptible to treatment with hydrogen peroxide or paraquat. Even in the absence of oxidative challenge, cells expressing RpS3 or dOgg1 in mitochondria exhibited increased apoptosis relative to controls, as determined by flow-cytometric analysis of Annexin V and DNA degradation measured by the Comet assay. Another notable finding was that ectopic expression of either dOgg1 or RpS3 in mitochondria increased cell survival after exposure to the nitric oxide donor SNAP. These results suggest that ectopic expression of one of the constituents of the DNA repair system in mitochondria may cause a perturbation in the base excision repair pathway and lower, rather than enhance, survivability.
Keywords: Free radicals
It is widely accepted that cellular DNA is subject to considerable structural damage caused by a variety of endogenous and exogenous agents, which if not repaired or erroneously repaired may result in gene mutations, chromosomal aberrations, or modification of gene regulation, among others. Mitochondrial DNA is believed to be particularly susceptible to oxidative damage for several reasons, including its exposure to mitochondrially generated reactive oxygen species, absence of association with histones, and limited DNA repair [1]. Alterations such as fragmentation, rearrangements, deletions, and point mutations in mtDNA have been quite extensively documented. Accrual of such lesions beyond a certain threshold has been hypothesized to lead to functional attenuation and threaten cell survival. A combination of such pathogenic events has also been hypothesized to be causal in the aging process [2]. Indeed, the level of 8-hydroxydeoxyguanosine (8-oxodG), formed by the oxidative modification of deoxyguanine at the C-8 position, is ∼3- to 16-fold higher in mitochondrial than in nuclear DNA and has also been found to increase exponentially with age [3–6].
In metazoans, DNA damage is repaired via three distinct pathways: cell cycle arrest (often referred to as DNA damage checkpoint), transcriptional induction, and DNA base excision (BER) [7–9]. Oxidized bases, such as 8-oxodG, are thought to be repaired primarily by the BER pathway [10], which is initiated by the hydrolysis of N-glycosyl bonds via DNA glycosylases, resulting in the release of bases and appearance of apurinic/apyrimidinic (AP) sites. The latter are cleaved by AP lyase or AP endonuclease, and the gap is closed by the combined action of DNA polymerase and ligase [11]. In Drosophila, there are two enzymes, dOgg1 and RpS3 [12,13], which possess glycosylase/AP lyase activity for the removal of 8-oxodG. The latter enzyme has been shown to restore resistance to H2O2 and MMS toxicity in a DNA repair-deficient Escherichia coli mutant [14,15]. Heterologous expression of Drosophila RpS3 has been previously demonstrated to enhance the removal of 8-oxodG in human cells [16,17].
The main purpose of the present study was to determine whether oxidative damage to mitochondrial DNA can be attenuated by the ectopic expression of DNA glycosylase/AP lyase within the mitochondrial matrix. Specifically, stable Drosophila S2 transfectant cell lines, expressing dOgg1 or RpS3 proteins in mitochondria, were generated and tested for DNA damage and cell viability under normal and stressful conditions.
Materials and methods
Generation of Drosophila S2 cells expressing dOgg1 and RpS3 in the mitochondria
EST clone LD19945 containing a cDNA corresponding to the Drosophila dOgg1 gene in a pBluescript vector and EST clone LD 47488 containing a cDNA corresponding to the RpS3 gene in a pOT2 vector were obtained from Research Genomics (Huntsville, AL, USA). The 22 amino-terminal codons of the Drosophila ornithine aminotransferase (OAT) gene, including a putative mitochondrial presequence, was attached to the N-termini of the coding regions of the dOgg1 and RpS3 genes, replacing the start codons, using a two-step splicing by overlapping extension (SOE) PCR amplification approach. In the first set of reactions, PCR products containing OAT and dOgg1 or RpS3 fusion sequences were generated. Primers for the generation of the OAT-derived PCR product were 5′-gatattggtaccatcATGTTCTCCAAGCTTTCCAC (F-OAT), 5′-gtcggaaAGCCGCTTTTTGGGCC (R-SOE-dOgg), and 5′-cgcattAGCCGCTTTTTGGGCC (R-SOE-RpS3). Primers for the generation of the dOgg1 and RpS3 products were 5′-GCGGCTaaggctgttttacaggatcg (F-SOE-dOgg), 5′-tagatatctagactttttagg (R-dOgg), 5′-GCGGCTaatgcgaaccttccgatttccaag (F-SOE-RpS3), and 5′-gtaaaactatctagacaaaactttcgcc (R-RpS3). The start codon is indicated by italic letters and KpnI and XbaI restriction sites are in bold. The part of the sequence corresponding to the OAT gene is indicated by capital letters. PCR products were gel purified and used in a second set of reactions as shown in Fig. 1.
Fig. 1.
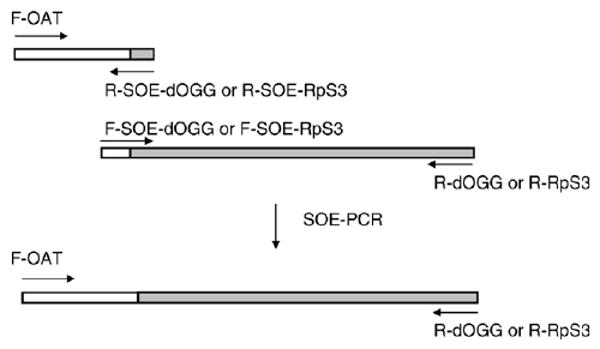
Generation of OAT-dOgg1 and OAT-RpS3 constructs. OAT sequence is shown as open bars, and dOgg1 or RpS3 sequences are indicated as gray bars. The resulting PCR products were digested with KpnI and XbaI endonucleases and ligated into corresponding sites of the pMT/V5-His vector (Invitrogen), allowing an inducible expression of the cloned genes, in frame with the V5 epitope. Subsequently, the fused constructs were shuttled into the pIB/V5-His vector (Invitrogen), permitting constitutive expression.
Stably transfected cell lines were generated according to standard procedures. Briefly, S2 Drosophila Schneider cells were maintained in complete DES Expression medium (Invitrogen) supplemented with 10% FBS and 50 μg/ml penicillin/streptomycin (Cellgro). Cells were transfected with 19 μg of plasmid DNA using the Calcium Phosphate Transfection Kit followed by selection of stable transfectant cell lines according to the manufacturer's manual (Invitrogen). After selection, cells were maintained in a DES medium containing 30 μg/ml blasticidin. All cell lines were transferred to fresh medium every 3–5 days at 1:3–1:5 dilution retaining one-third of the conditioned medium. Localization of recombinant OAT-dOgg1 and OAT-RpS3 proteins in mitochondria was assessed by immunoblot analysis of isolated cell fractions.
Experimentally induced stress and cell viability
For viability assays, overnight cell cultures that reached 1 × 106 cells/ml density were exposed to 20 mM hydrogen peroxide (Sigma), 10 mM paraquat (Sigma), or 1 mM S-nitroso-N-acetylpenicillamine (SNAP) (Molecular Probes) and incubated at room temperature for different durations. Cell viability was determined by staining with trypan blue (Invitrogen) and assessed as a percentage of live cells to the total number of cells. A minimum of 100 cells were counted in each experiment.
Preparation of mitochondrial fractions and immunoblot analysis
Mitochondria were isolated from 2 × 107 cells using the Mitochondria Isolation Kit (Pierce). Protein lysates for immunoblot analysis were extracted from mitochondrial fractions by T-PER Tissue Protein Extraction Reagent (Pierce) containing protease inhibitors (Roche). Protein concentrations were determined by the Bio-Rad Protein Assay reagent (Bio-Rad) and 10 μg of the protein extracts was then resolved by 10% SDS–PAGE followed by transfer to PVDF membrane (Millipore). Immunoblots were probed with anti-V5-HRP antibodies (Invitrogen) or with antibodies raised against mitochondrial peroxiredoxin or cytosolic SOD, as described previously [18], and visualized using the ECL+ Western blotting detection system (Amersham) according to the manufacturer's instructions.
Preparation and analysis of mitochondrial DNA
Mitochondrial DNA was extracted from cells using the mtDNA Extractor CT Kit (Wako) and dissolved in 100 μl of TE buffer. DNA hydrolysates for HPLC analysis were prepared as described by Hamilton et al. [19]. Briefly, DNA samples (50–100 μg) were dissolved in 100 μl of 20 mM sodium acetate buffer and digested with 20 μg of nuclease P1 (Boehringer Mannheim) for 11 min at 65°C. After digestion, DNA samples were treated with alkaline phosphatase (Boehringer Mannheim) at 37°C for 1 h. For the evaluation of mitochondrial DNA gaps, 5–10 μg of isolated DNA was incubated for 20 min at 65°C, samples were cooled to room temperature, and NaOH was added to a final concentration of 0.1 N, followed by incubation for 15 min at 37°C to cleave DNA at the sites of nucleotide gaps. Preparations were loaded onto a 1% agarose gel and analyzed by densitometric scanning using the digital imaging analysis system with AlphaEase Stand Alone Software (Alpha Innotech Corp., San Leandro, CA, USA).
Apoptosis and flow cytometry
DNA laddering was evaluated by agarose-gel electrophoresis as described [18] previously. For flow-cytometric analysis, 1 × 106 cells were plated and allowed to grow overnight, followed by exposure to H2O2, paraquat, or SNAP for specific lengths of time. Cells were pelleted and washed twice with PBS and the number of cells undergoing apoptosis was determined using the Annexin V–FITC Apoptosis kit (BD Pharmingen). Flow-cytometry analysis was performed on a FACSCalibur apparatus (Becton–Dickinson), using FlowJo software.
Comet assay
Comet assay was performed using a Comet Assay Kit (Trevigen). Cells at the density of 1 × 106 cells/ml were grown overnight followed by exposure to exogenous stressors. Aliquots of 105 cells were washed twice with PBS and mixed with 0.5% low-melting-point agarose (Fisher) in PBS at 37°C followed by immobilization on the Comet slides in duplicate. Gels were solidified for 5–10 min at 4°C followed by 30 min immersion in a 4°C prechilled lysis buffer. DNA was denatured for 30 min in alkaline buffer at room temperature. Slides were washed twice for 5 min with 1× TBE followed by electrophoresis in a horizontal apparatus for 10 min at 1 V/cm. Gels were fixed for 5 min in methanol, then for 5 min in ethanol, and allowed to dry overnight. All subsequent steps were performed in the dark or under dim light. For visualization, slides were stained with SYBR green dye (Molecular Probes) and pictures were captured with a FITC filter using a fluorescence microscope (Nikon) and MetaMorph software. The images of 50 randomly chosen individual cells per slide were captured and visually classified into four categories according to nucleus integrity and tail migration (score 1, intact nucleus, no DNA in the comet tail; score 2, some DNA in the comet tail; score 3, almost equal amounts of DNA in the nucleus and in the comet tail; score 4, all DNA in the comet tail).
Statistical analysis
All experiments were performed at least three times unless otherwise indicated. The results are means ± SD determined using STATISTICA or Microsoft Excel software.
Results
Expression and subcellular localization of recombinant mt-dOgg1 and mt-RPS3
Subcellular fractions of the stably transfected cell lines expressing either recombinant dOgg1 or recombinant RpS3 proteins, containing the OAT leader peptide [20] targeted to the mitochondrial matrix, were subjected to immunoblot analysis (Fig. 2).
Fig. 2.
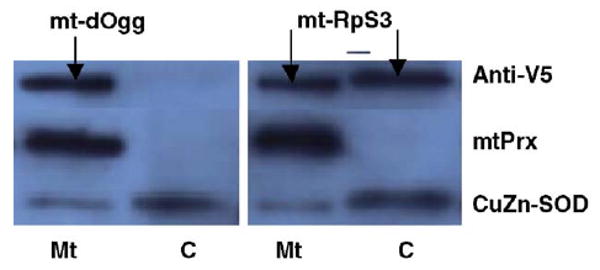
Immunoblot analysis of cytoplasmic (C) and mitochondrial (Mt) fractions isolated from cells expressing recombinant mt-dOgg1 and mt-RpS3 proteins. Cytoplasmic and mitochondrial fractions (5 μg) were resolved by SDS–PAGE and subjected to immunoblot analysis. Signals were obtained with anti-V5 antibodies and antibodies raised against mitochondrial peroxiredoxin (mtPrx) and cytosolic CuZn-SOD proteins. These last two antibodies were used to assess the specificity of the fractions.
The mt-dOgg1 was found to be exclusively located in the mitochondrial fraction, whereas the mt-RpS3 was found in both cytoplasmic and mitochondrial fractions. Results of immunocytological analysis, using FITC-labeled anti-V5 antibodies and MitoTracker red (Invitrogen), were identical to those obtained from the immunoblots, demonstrating the presence of mt-dOgg1 and mt-RpS3 recombinant proteins in the perinuclear region (data not shown). To investigate the possibility that endogenous dOgg1 and RpS3 may have been down-regulated by negative feedback, quantitative RT-PCR analysis was performed using primers that allowed differentiation between endogenous and recombinant mRNA transcripts. No differences were found in the levels of endogenous dOgg1 and RpS3 between the cells expressing the OAT-containing genes and the control cells; the levels of recombinant dOgg1 and RpS3 mRNAs were approximately 1.5- and 3-fold higher than the levels of the endogenous genes (data not shown).
HPLC analysis of mitochondrial DNA
To determine the effects of dOgg1 or RpS3 overexpression on mtDNA oxidation, the levels of 8-oxodG were measured in DNA preparations isolated from mitochondrial fractions using HPLC analysis. As shown in Table 1, a 1.4- to 6.7-fold decrease in the 8-oxodG levels was observed in samples from cells expressing dOgg1 or RpS3 in mitochondria, compared to control. Similar effects were observed in transfectant cells exposed to oxidants, although the levels of oxidized DNA were higher compared to untreated cells. Nevertheless, there were severalfold differences in the levels of 8-oxodG between the transfectants and the control cells, which was particularly evident after exposure to SNAP.
Table 1.
Comparison of 8-oxodG levels in mtDNA isolated from Drosophila cells expressing dOgg1 and RpS3 in mitochondria
| Cell line/treatment | 8-OxodG/105 dG ratio | |||
|---|---|---|---|---|
| Experiment 1 | Experiment 2 | Experiment 3 | Experiment 4 | |
| Untreated | ||||
| Control | 0.364 | 0.259 | 0.200 | 0.266 |
| RpS3 | 0.054 | 0.181 | 0.102 | 0.152 |
| dOgg1 | 0.140 | 0.117 | 0.118 | 0.113 |
| Hydrogen peroxide | ||||
| Control | 0.691 | 0.707 | ||
| RpS3 | 0.420 | 0.498 | ||
| dOgg1 | 0.305 | 0.354 | ||
| Paraquat | ||||
| Control | 0.560 | 0.501 | ||
| RpS3 | 0.345 | 0.401 | ||
| dOgg1 | 0.242 | 0.275 | ||
| SNAP | ||||
| Control | 0.675 | 0.543 | ||
| RpS3 | 0.269 | 0.297 | ||
| dOgg1 | 0.100 | 0.178 | ||
Samples were isolated from cells cultured under normal conditions and in the presence of 20 mM H2O2 for 6 h or 10 mM paraquat or 1 mM SNAP for 24 h. The differences between controls and cells expressing mt-RpS3 or mt-dOgg1 were statistically significant (p < 0.03).
On the basis of these results, it can be concluded that the recombinant dOgg1 and RpS3 glycosylases targeted to the mitochondrial matrix are functional and that the 8-oxodG excision in mt-dOgg1- and mt-RpS3-transfected S2 cells is more efficient than in control cells.
The effect of ectopic expression of dOgg1 and RpS3 in mitochondria on cell viability
To determine whether the ectopic expression of dOgg1 and RpS3 translates into enhanced cellular survival after oxidative stress, cells were subjected to 20 mM hydrogen peroxide, 10 mM paraquat (superoxide anion donor), and 1 mM SNAP (nitric oxide donor), followed by evaluation of cell viability by trypan blue exclusion (Fig. 3).
Fig. 3.
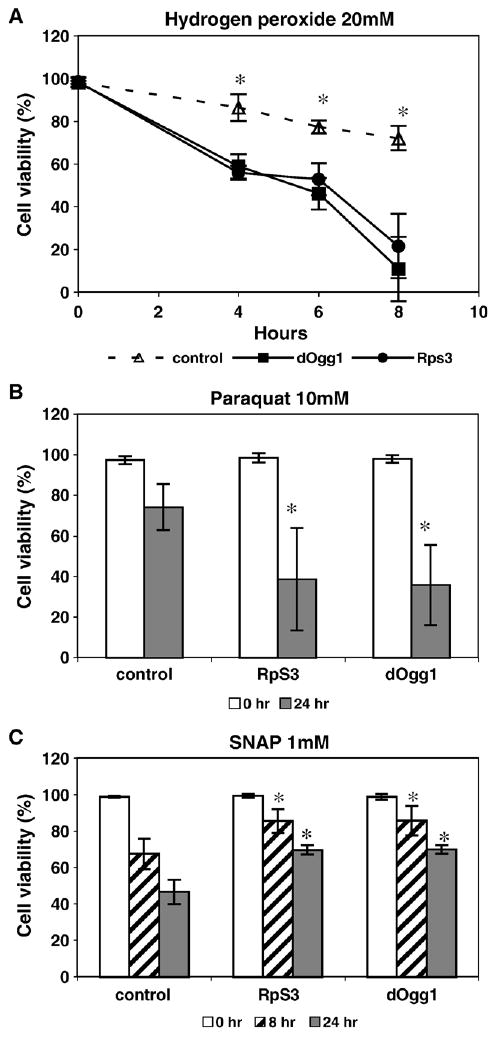
Viability of cells after H2O2, paraquat, and SNAP treatment. Cells were exposed to (A) 20 mM H2O2, (B) 10 mM paraquat, and (C) 1 mM SNAP and percentage cell survival was determined at various time intervals. The survival percentages are shown as the means ± SD of three separate experiments. The statistically significant differences (p < 0.05) are indicated by asterisks.
Under unchallenged (normal) conditions, cells ectopically expressing either dOgg1 or RpS3 enzyme were as viable as the controls (95–100%). However, compared to the control cells, the transfectant were more sensitive to H2O2 and paraquat, but had higher viability when treated with SNAP.
Incidence of apoptosis
To investigate further the effect of an increase in sensitivity to H2O2 and paraquat, apoptosis-associated DNA fragmentation was quantified in cells expressing RpS3 or dOgg1 in mitochondria. No DNA degradation was observed in samples isolated from the untreated control cells, but a typical internucleosomal fragmentation pattern was observed in untreated dOgg1 and RpS3 transfectants (Fig. 4). We also observed an increase in DNA fragmentation in mitochondrial preparations isolated from cells overexpressing dOgg1 or RpS3 compared to control; however, we have not seen substantial differences in DNA laddering between untreated cells or cells exposed to H2O2, paraquat, or SNAP.
Fig. 4.
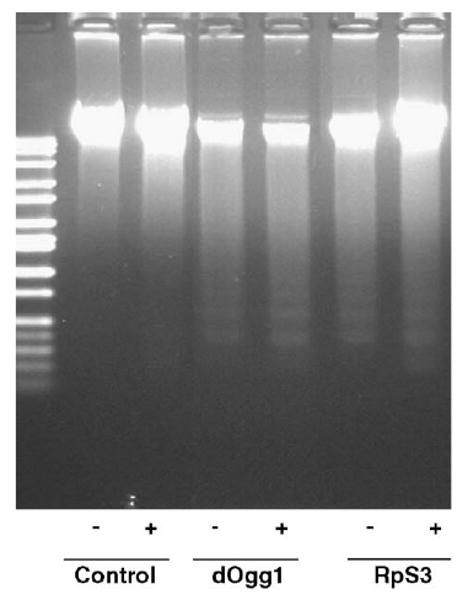
DNA fragmentation analysis of mt-dOgg1 and mt-RpS3 cell lines. DNA preparations were derived from control cells transfected with vector only and cells expressing dOgg1 or RpS3 in mitochondria. Cells were untreated (−) or treated (+) with 20 mM H2O2 for 6 h. DNA degradation after exposure to paraquat is not shown.
The possible cause of the increase in sensitivity of the transfected cells to H2O2 and paraquat was investigated by the identification of cells approaching apoptosis, using flow cytometry and staining with Annexin V–FITC and PI (Fig. 5).
Fig. 5.
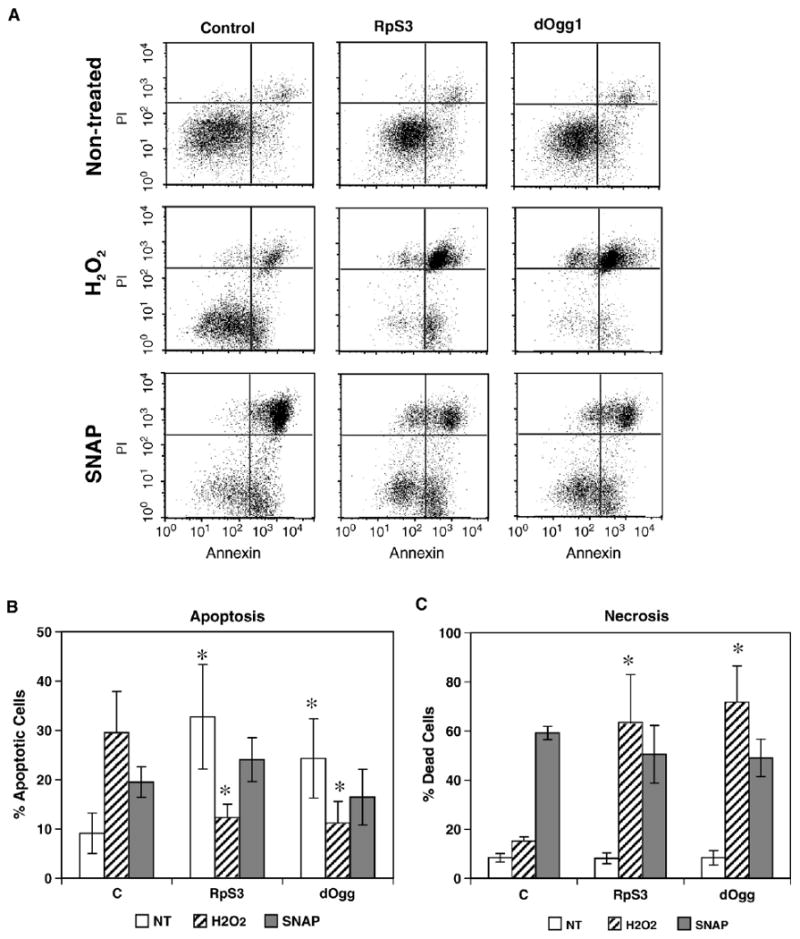
Flow-cytometric analysis of apoptosis and necrosis in the RpS3 and dOgg1 transfectant and control cell lines. (A) Dot plots of cells not treated and treated with H2O2 and SNAP. Cells that are undergoing apoptosis are Annexin V–FITC positive and PI negative (lower right quadrant). The dead cells produced through apoptosis are located in the right upper quadrant and are Annexin V–FITC- and PI-positive. (B) The numbers of apoptotic cells, both live and dead, are represented graphically. (C) The numbers of necrotic cells are represented graphically. The results are means ± SD of three to six independent experiments.
Flow cytometry data revealed that a relatively greater fraction of dOgg1 and RpS3 transfectant cells were shifted toward being Annexin positive compared to the controls. Consistent with the survival data shown in Fig. 3, exposure of cells to 20 mM H2O2 led to a dramatically higher percentage of dead cells among dOgg1 or RpS3 transfectants than among the controls. However, there were no significant differences between controls and experimentals in the number of apoptotic and dead cells, after treatment with SNAP (Fig. 5).
Effects on DNA strand breaks
Single-cell gel electrophoresis (Comet assay) was used to compare DNA damage between control and experimental cells. In untreated controls, viability was found to be about 95–100%. The number of cells exhibiting DNA damage was higher in cell lines expressing mt-dOgg1 or mt-RpS3 compared to controls (Fig. 6A). When cells were treated with SNAP for 24 h, other than a marginal increase in the number of control cells at score 2 (cells showing a modest degree of DNA damage), there were no substantial differences between cells ectopically expressing DNA repair enzymes and the controls (Fig. 6C). Exposure of cells to paraquat resulted in a dramatic increase in the number of cells exhibiting the highest degree of DNA degradation (score 4) (Fig. 6B).
Fig. 6.
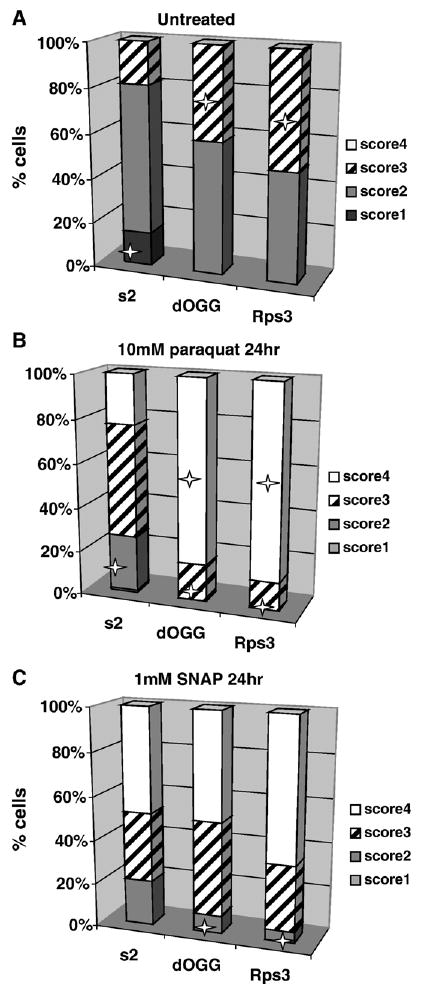
Evaluation of DNA damage in control cell line and cell lines expressing mt-RpS3 and mt-dOgg1 proteins by single-cell Comet assay. Frequency distribution of nuclei in four different classes of increasing total DNA damage is shown (see Materials and methods). The mean percentage values ± standard deviation of the mean of three separate experiments are shown. Statistically significant differences (p < 0.05) are identified by a star. (A) Untreated cells, (B) cells treated with 10 mM paraquat for 24 h, or (C) 1 mM SNAP for 24 h.
Discussion
The present study tested the idea that bolstering the mitochondrial DNA repair system by the import of ectopic repair enzymes into the mitochondrial matrix will lower the level of DNA oxidation, indicated by 8-oxodG, and thus enhance cellular survival under oxidative stress. Accordingly, glycosylases dOgg1 and RpS3, which are involved in the excision of oxidized bases such as 8-oxodG, the first step in the repair process, were ectopically expressed in mitochondria. The import was achieved by the insertion of the mitochondrial targeting sequence, OAT, into the glycosylases. This approach was previously used by us for the transport of Drosophila catalase into the mitochondrial matrix [20]. HPLC analysis established that overexpression of either dOgg1 or RpS3 resulted in significantly lower levels of 8-oxodG in the mitochondrial DNA of the transfected cell lines. Despite the initial expectation that such enhanced mitochondrial DNA repair capacity and the resultant decrease in the accumulation of 8-oxodG would provide greater protection against oxidative stress, the susceptibility of the stable RpS3 and dOgg1 transfectants to H2O2 and paraquat treatment was found to have been increased. However, cells ectopically expressing either dOgg1 or RpS3 in mitochondria showed greater resistance to nitrosative stress, exerted by the nitric oxide donor, SNAP.
These observations are apparently in disagreement with the earlier reports suggesting that ectopic expression of DNA repair enzymes in mitochondria alleviates oxidative stress-induced DNA damage and thus enhances the resistance of cells to exogenous stressors. For instance, the overexpression of mammalian Ogg1 in mitochondria protected against mtDNA damage induced by xanthine oxidase as well as cell death [21]. Similar results were obtained by targeting human 8-oxoguanine glycosylase to mitochondria [22]. In this case, there was not only a significant enhancement in the repair of menadione-induced oxidative lesions in mtDNA, but also a decrease in cytochrome c release and activation of caspase 9. Thus, cells overexpressing Ogg1 were reportedly better protected from menadione-induced apoptosis.
Our investigation of the possible causes of the increased cell death indicated that under unchallenged conditions, the number of cells undergoing apoptosis increased in cell lines expressing RpS3 or dOgg1 in mitochondria, compared to the control cells, as indicated by the characteristic DNA laddering effect (Fig. 4) and positive staining with Annexin V–FITC, a sensitive probe for the identification of apoptotic cells (Fig. 5). Furthermore, the Comet assay also showed an enhanced level of DNA strand breaks in the cell lines expressing the ectopic enzymes (Fig. 6). We also explored other potential factors, such as levels of oxidative stress that may contribute to the decrease in cell viability after ectopic expression of DNA repair enzymes in mitochondria. We did not find any significant increase in the levels of 8-oxo-dG in nuclear DNA preparations (data not shown), which may be an indicator of the shift to the pro-oxidative state in cells overexpressing DNA glycosylases.
The shift toward a proapoptotic state and eventually cell death became more pronounced in ectopic enzyme-expressing cells after treatment with H2O2 or paraquat. This was, however, not the case after SNAP treatment, in which no substantial differences were found in the number of cells undergoing apoptosis between expressing and control cells, and only marginal effects were observed using the Comet assay (Fig. 6). The cause of the shift toward an apoptotic state in the cells stably expressing mt-RPS3 or mt-dOgg1 is unclear. A similar observation has been made by Fishel et al. [23] in response to the import of N-methylpurine DNA glycosylase into mitochondria, which resulted in apoptosis of a significant number of cells even in the absence of any exogenous stressors. Because N-methylpurine DNA glycosylase lacks lyase activity, it was postulated that one possible mechanism of the decrease in cell viability could be the accumulation of AP sites that would lead to the increased probability of mutagenesis, disruption of DNA replication, or some other detrimental event. Unlike N-methylpurine DNA glycosylase, dOgg1 and RpS3 possess both glycosylase and AP lyase activity, which removes the 8-oxodG, thereby leaving a one-nucleotide gap in DNA. The accumulation of such nicked/gapped DNA could outstrip the resolving capacity of polymerases and/or ligases and this repair deficit could serve as a potent inducer of apoptosis. Indeed, we observed more damage to mitochondrial DNA in preparations isolated from cells expressing mt-dOgg1 and mt-RpS3 after treatment with paraquat, H2O2, or SNAP, relative to controls, as was assessed by gel electrophoresis of samples treated with alkaline solution (data not shown). Thus, our data are consistent with the hypothesis that the expression in mitochondria of one of the constituents of the DNA repair system may cause a perturbation in the BER pathway and lead to cell death. On this basis, it is possible to postulate that the positive effects observed in the mammalian study involving Ogg1 could be due to a greater resolving capacity of the mammalian repair system.
In the presence of oxidants, such as H2O2 or paraquat, cell death was more pronounced in cells expressing dOgg1 and RpS3 in the mitochondria (Fig. 3). Reactive oxygen species, such as the superoxide anion radical, hydrogen peroxide, and the hydroxyl radical, all can damage DNA bases and sugar residues and also cause strand breaks. It follows then that the exposure of cells to oxidants further exacerbated the deterioration of mtDNA status.
The other significant finding in this study is that targeting the expression of RpS3 and dOgg1 to mitochondria was associated with the attenuation of cytotoxicity caused by SNAP, an NO donor. NO and/or peroxynitrite, which is formed by the reaction of NO with the superoxide anion, can damage DNA by several different mechanisms. One of the common effects is the conversion of guanine to 8-nitroguanine and 8-oxoguanine [24,25]. It is known that NO not only causes oxidative DNA damage, but also directly inhibits the activity of DNA repair enzymes [26]. The protective effects of Rps3 and dOgg1 expression after exposure to SNAP might then be attributed to the alleviation of this inhibition. On the other hand, it is known that at certain concentrations NO may inhibit apoptosis at several steps, further allowing the DNA-damaged cell to survive (reviewed in Jaiswal et al. [27]). The beneficial effects of dOgg1 and RpS3 expression on cell survival under nitrosative stress may then represent the net balance between these factors.
In conclusion, ectopic expression of DNA glycosylases in mitochondria is able to reduce significantly steady-state levels of 8-oxodG in both normal and stressed cells, but does not confer greater resistance to oxidative stress. It would follow that targeting of additional DNA repair factors to the mitochondria may be required to promote a balanced DNA repair capacity and effectively enhance resistance, a possibility that is currently being explored.
Acknowledgments
We are grateful to Sharon Waldrop (Southern Methodist University, Dallas, TX, USA) for technical assistance on conducting flow cytometry analysis. This work was supported by NIH Grant AG7657.
Abbreviations
- 8-oxodG
8-hydroxydeoxyguanosine
- BER
DNA base excision
- AP
apurinic/apyrimidinic sites
- OAT
ornithine aminotransferase
- SNAP
S-nitroso-N-acetylpenicillamine
References
- 1.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25:502–850. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 2.Stevnsner T, Thorslund T, de Souza-Pinto NC, Bohr VA. Mitochondrial repair of 8-oxoguanine and changes with aging. Exp Gerontol. 2002;37:1189–1196. doi: 10.1016/s0531-5565(02)00142-0. [DOI] [PubMed] [Google Scholar]
- 3.Agarwal S, Sohal RS. DNA oxidative damage and life expectancy in houseflies. Proc Natl Acad Sci USA. 1994;91:12332–12335. doi: 10.1073/pnas.91.25.12332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 5.Bohr VA, Anson RM. Mitochondrial DNA repair pathways. J Bioenerg Biomembr. 1999;31:391–398. doi: 10.1023/a:1005484004167. [DOI] [PubMed] [Google Scholar]
- 6.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song YH. Drosophila melanogaster: a model for the study of DNA damage checkpoint response. Mol Cells. 2005;19:167–179. [PubMed] [Google Scholar]
- 8.Davis T, Meyers M, Patten CWV, Shandra N, Yang CR, et al. Transcriptional responses to DNA damage created by ionizing radiation. In: Nickologg JA, Hoekstra MF, editors. DNA damage and repair. Totowa, NJ: Humana Press; 1998. pp. 223–262. [Google Scholar]
- 9.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 10.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gros L, Saparbaev MK, Laval J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21:8905–8925. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- 12.Dherin C, Dizdaroglu M, Doerflinger H, Boiteux S, Radicella JP. Repair of oxidative DNA damage in Drosophila melanogaster: identification and characterization of dOgg1, a second DNA glycosylase activity for 8-hydroxyguanine and formamidopyrimidines. Nucleic Acids Res. 2000;28:4583–4892. doi: 10.1093/nar/28.23.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deutsch WA, Yacoub A, Jaruga P, Zastawny TH, Dizdaroglu M. Characterization and mechanism of action of Drosophila ribosomal protein S3 DNA glycosylase activity for the removal of oxidatively damaged DNA bases. J Biol Chem. 1997;272:32857–32860. doi: 10.1074/jbc.272.52.32857. [DOI] [PubMed] [Google Scholar]
- 14.Yacoub A, Augeri L, Kelley MR, Doetsch PW, Deutsch WA. A Drosophila ribosomal protein contains 8-oxoguanine and abasic site DNA repair activities. EMBO J. 1996;15:2306–2312. [PMC free article] [PubMed] [Google Scholar]
- 15.Kelley GG, Ondrako JM, Reks SE. Fuel and hormone regulation of phospholipase C beta 1 and delta 1 overexpressed in RINm5F pancreatic beta cells. Mol Cell Endocrinol. 2001;177:107–115. doi: 10.1016/s0303-7207(01)00453-1. [DOI] [PubMed] [Google Scholar]
- 16.Cappelli E, D'Osualdo A, Bogliolo M, Kelley MR, Frosina G. Drosophila S3 ribosomal protein accelerates repair of 8-oxoguanine performed by human and mouse cell extracts. Environ Mol Mutagen. 2003;42:50–58. doi: 10.1002/em.10166. [DOI] [PubMed] [Google Scholar]
- 17.Ropolo M, Geroldi A, Rossi O, Degan P, Zupo S, Poggi A, Frosina G. Expression of the Drosophila melanogaster S3 ribosomal/repair protein in T24 human bladder cells. Anticancer Res. 2004;24:3811–3818. [PubMed] [Google Scholar]
- 18.Radyuk SN, Sohal RS, Orr WC. Thioredoxin peroxidases can foster cytoprotection or cell death in response to different stressors: over- and under-expression of thioredoxin peroxidase in Drosophila cells. Biochem J. 2003;371:743–752. doi: 10.1042/BJ20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A. A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res. 2001;29:2117–2126. doi: 10.1093/nar/29.10.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwong LK, Mockett RJ, Bayne AC, Orr WC, Sohal RS. Decreased mitochondrial hydrogen peroxide release in transgenic Drosophila melanogaster expressing intramitochondrial catalase. Arch Biochem Biophys. 2000;383:303–308. doi: 10.1006/abbi.2000.2093. [DOI] [PubMed] [Google Scholar]
- 21.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am J Physiol Lung Cell Mol Physiol. 2002;283:L205–L210. doi: 10.1152/ajplung.00443.2001. [DOI] [PubMed] [Google Scholar]
- 22.Druzhyna NM, Hollensworth SB, Kelley MR, Wilson GL, Ledoux SP. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia. 2003;42:370–378. doi: 10.1002/glia.10230. [DOI] [PubMed] [Google Scholar]
- 23.Fishel ML, Seo YR, Smith ML, Kelley MR. Imbalancing the DNA base excision repair pathway in the mitochondria: targeting and overexpressing N-methylpurine DNA glycosylase in mitochondria leads to enhanced cell killing. Cancer Res. 2003;63:608–615. [PubMed] [Google Scholar]
- 24.Mannick EE, Bravo LE, Zarama G, Realpe JL, Zhang XJ, Ruiz B, Fontham ETH, Mera R, Miller MJS, Correa P. Inducible nitric oxide synthase, nitrotyrosine and apoptosis in Helicobacter pylori gastritis. Cancer Res. 1996;56:3238–3243. [PubMed] [Google Scholar]
- 25.Witherell HL, Hiatt RA, Replogle M, Personnet J. Helicobacter pylori infection and urinary excretion of 8-hydroxy-2-deoxyguanine, an oxidative DNA adduct. Cancer Epidemiol Biomarkers Prev. 1998;7:91–96. [PubMed] [Google Scholar]
- 26.Jaiswal M, LaRusso NF, Nishioka N, Nakabeppu Y, Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 27.Jaiswal M, LaRusso NF, Gores GJ. Nitric oxide in gastrointestinal epithelial cell carcinogenesis: linking inflammation to oncogenesis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G626–G634. doi: 10.1152/ajpgi.2001.281.3.G626. [DOI] [PubMed] [Google Scholar]