

Abstract
Natural killer (NK) cells form a structure at their interface with a susceptible target cell called the activating NK cell immunologic synapse (NKIS). The mature activating NKIS contains a central and peripheral supramolecular activation cluster (SMAC), and includes polarized surface receptors, filamentous actin (F-actin) and perforin. Evaluation of the NKIS in human NK cells revealed CD2, CD11a, CD11b and F-actin in the peripheral SMAC (pSMAC) with perforin in the central SMAC. The accumulation of F-actin and surface receptors was rapid and depended on Wiskott–Aldrich syndrome protein-driven actin polymerization. The accumulation at and arrangement of these molecules in the pSMAC was not affected by microtubule depolymerization. The polarization of perforin, however was slower and required intact actin, Wiskott–Aldrich syndrome protein, and microtubule function. Thus the process of CD2, CD11a, CD11b, and F-actin accumulation in the pSMAC and perforin accumulation in the central SMAC of the NKIS are sequential processes with distinct cytoskeletal requirements.
Natural killer (NK) cells are lymphocytes that rely on activation or inhibition through germ-line-encoded receptors. They possess a variety of effector functions, including cytokine production and cytotoxicity and play important roles in defense against infectious disease and control of cancer cells (reviewed in refs. 1 and 2). The cytolytic capability of NK cells is enabled after ligation of specific cell-surface receptors, which leads to activation of a cascade of intracelluar signals resulting in Ca2+ flux, polarization of granules, and subsequent release of lytic molecules. Prominent in the lytic machinery is perforin, which forms pores in the target cell membrane, allowing the entry of other cytolytic granule contents. Although certain NK cell receptors alone can result in perforin release when ligated in sufficient density, NK cell activation in vivo most likely represents a coordinated effort of various receptors with activating potential.
Two families of receptors that are important in promoting function in NK and other cells are the Ig superfamily and integrins. An example of the former is CD2, and examples of the latter are CD11a (LFA-1) and CD11b (Mac1). All three can participate both in adhesion between the NK cell and its target cell and in stimulation. Blocking antibodies have been used to demonstrate adhesion (3, 4), which is additionally illuminated by leukocyte adhesion deficiency, in which β2-integrin is defective and surface expression of CD11a and CD11b is absent (3). Regarding NK cell activation signaling, ligation of CD2 induces kinase function and extension of lipid raft domains (5), whereas ligation of CD11a (6) and CD11b (7) induce phosphorylation-dependent NK cell activation. CD11b is of particular interest, because NK cells are the only lymphocytes that uniformly express this subunit in high density. The interaction of these receptors with their ligands on target cells is likely to be important in accessing NK cell function.
To form an interface between an NK cell and susceptible target cell, cell-surface receptors participate in cognate interactions that initiate formation of a specific time-dependent structure called the activating NK cell immunologic synapse (NKIS). A number of molecules have been found to accumulate at the activating NKIS (reviewed in ref. 8), and, although the majority are strictly intracellular, they include CD2 (5) and CD11a (9) on the cell surface. Immortalized or cultured NK cells accumulate CD11a in the peripheral supramolecular activation cluster (pSMAC) (9), but the three-dimensional arrangement of CD2 or CD11b in the activating NKIS and relative localization of cell-surface receptors to perforin is not known. Although studies in murine cytotoxic T cells (CTLs) have shown perforin is in the central SMAC (cSMAC), the mechanism by which it arrives at that location and its kinetics have not been specifically demonstrated (10). As the microtubule organizing center (MTOC) actively polarizes toward the IS in murine CTLs (11), cytolytic granules presumably move along the microtubules.
In T cells, the formation of the SMAC has been shown to rely in part on actin polymerization. Some IS-related molecules can cluster in the presence of inhibitors of actin polymerization and others cannot (12, 13). T cells from patients with Wiskott–Aldrich syndrome (WAS) have decreased actin polarization and lipid raft microdomain clustering at the IS (14), due to a mutation in the gene coding for the WAS protein (WASp), which promotes branch formation in actin filaments. Intact WASp is also required in T cells for the clustering of CD2, which is linked to actin and WASp through specific adaptor proteins (15). In contrast, the relation between β2-integrins and WASp is probably more indirect as integrin ligation can activate WASp, but the major link between actin and β2-integrins is through talin and filamin (reviewed in ref. 16), and in T cells, WASp is not needed for integrin clustering (17). A requirement for WASp in the polarization and localization of granules, however, has not been demonstrated in T cells.
In NK cells actin and microtubule polymerization are required for cytotoxic function (18), and filamentous actin (F-actin) (19), as well as the MTOC (20), accumulates at the activating NKIS. Important actin-associated molecules are also found in the SMAC, including WASp and talin (9, 21). WASp function, in particular, is an important contributor to F-actin polarization in NK cells, and both actin polymerization and WASp function are required for effective localization of perforin-containing granules to the activating NKIS (21). The relation between the actin- and microtubule-directed events in activating NKIS formation, however, is unclear. In this study, the requirement of the actin cytoskeleton, WASp activity, and microtubule function in the formation and structure of the activating NKIS were evaluated.
Methods
Cells. Leukocyte-enriched or fresh peripheral blood was obtained from volunteer donors in compliance with institutional guidelines and used to prepare ex vivo NK cells by negative selection with Rosette-Sep NK (StemCell Technologies, Vancouver). Enriched NK cell populations were >85% CD56+/CD3– with <0.5% CD3+ cells. Ex vivo NK cells from a patient with a G252A point mutation in WASP without detectable expression of WASp (21) were also used. The NK tumor cell YTS expressing the murine ecotropic receptor (YTS-Eco) (22) was infected with ecotropic retroviruses packaged in the Plat-E 293-Eco cell line (23). MHC class I-negative K562 erythroleukemia, or 721.221 B-lymphoblastoid cell lines served as target cells.
YTS Transduction. The pBABE retroviral vector containing a puromycin resistance gene (22) was modified by introduction of GFP (Clontech), flanked by a 5′ EcoRI/AgeI and 3′ BamHI site. CD2 was cloned from human T cells, generating an EcoRI site 5′ and an AgeI site 3′ to CD2, omitting the stop codon, and then ligated into modified pBABE to obtain the fusion CD2/GFP protein. Vectors encoding CD2/GFP or GFP alone were packaged in Plat-E 293-Eco cells and harvested virus used to infect YTS-Eco cells (22). After puromycin selection, GFP+ cells were isolated by fluorescence-activated cell sorting (Moflo; Cytomation, Fort Collins, CO) and surface expression of CD2 was uniformly positive, but was negative in untransduced YTS-Eco cells or those transduced with GFP alone (YTS-GFP) (data not shown).
Antibodies and Fluorescent Probes. Antibodies with the following specificities were obtained from BD Biosciences (San Diego): CD2 (RPA-2.10), CD11a (HI111), CD56 (B159), IgG1k control (MOPC-21), perforin (δG9), and IgG2b control (27–35). Anti-CD11b (LM2.1) was a gift of L. Klickstein (Brigham and Women's Hospital, Boston). Alexa Fluor 568-conjugated goat anti-mouse and Alexa Fluor 647-conjugated phalloidin (Molecular Probes) were used to detect bound antibody or F-actin, respectively (Alexa Fluor 647-conjugated streptavidin was used as a phalloidin control).
Cell Conjugation and Analysis. To evaluate the NKIS, ex vivo NK cells were conjugated to K562 cells, or YTS cells to 721.221 cells at a 2:1 ratio for 15 min at 37°C in suspension, followed by adherence to poly-l-lysine-coated glass slides (Sigma) for 15 min at 37°C. In time course experiments, the conjugation and adherence times were 2.5, 5, 10, 15, or 20 min each (for a zero time, iced cell suspensions were mixed and adhered to a slide for 2.5 min). After adherence, cells were fixed with 4% formaldehyde, incubated with antibody against a cell-surface marker, followed by Alexa Fluor 568-conjugated goat anti-mouse, and were then permeabilized and stained intracellularly as described (21). Antibodies were used in the range of 1–20 μg/ml.
Cell conjugates were visualized by using a Zeiss LSM 510 laser-scanning confocal microscope. An NK cell (small cell), or a YTS-CD2/GFP cell (GFP+ cell) in the conjugate was confirmed by the presence of perforin, after which the accumulation of fluorescent molecules at the NKIS was examined (in ≥50 conjugated NK cells per slide). In the majority, results were independently confirmed by two separate investigators who, when possible, were blinded as to the slides they were viewing. The percent of YTS-CD2/GFP cells conjugated to 721.221 cells was determined by evaluation of 100 YTS-CD2/GFP cells in randomly selected fields. For three-dimensional analysis, 20 images were acquired through the z axis at 0.5- to 1-μM increments which were integrated, projected, and rotated by using the lsm510 software package. At least 10 synapses per slide were evaluated in this manner and were then analyzed by using nih image for a sustained central depression in pixel intensity of ≥40%, which was assessed for perforin. Mean data were compiled from individual experiments and compared by using a Student's t test.
Cytoskeletal Inhibition. Actin or microtubule polymerization was interrupted by incubating cells in10 μM cytochalasin-D (Cyt-D) or 10 μM colchicine (Sigma), respectively, for 30 min at 37°C. Cyt-D contained 0.1% DMSO, which was used without Cyt-D as a vehicle control. Inhibitor-treated effector cells were maintained in the presence of the drug when combined with target cells, due to the reversible effects of colchicine. As target cells were not preincubated, the final concentration of drug during cell conjugation was 5 μM, which was capable of inhibiting NK cell cytolytic activity in 51Cr-release assay (data not shown and refs. 18 and 21).
Results
CD2, CD11a, CD11b, F-Actin, and Perforin Accumulate at the Activating NKIS. Accumulation of several NK cell-surface receptors in the activating NKIS relative to the previously studied WASp, F-actin, and perforin (21) has been examined here. In conjugates formed between ex vivo NK cells and K562 cells, CD2 was found to accumulate along with F-actin at the interface (Fig. 1). Actin polarization occurred in ≈80% of conjugates and CD2 polarization was also present in 89% of these (see summaries in Fig. 6, and Fig. 7, which is published as supporting information on the PNAS web site). Staining was specific for the NK cells, because K562 cells did not express CD2 (or CD11a and CD11b) as determined by fluorescence-activated cell sorting (data not shown). Perforin was also accumulated at the activating NKIS, but at about half the frequency of CD2 or F-actin (Figs. 1 and 6). The polarization of CD2, F-actin, and perforin in the same NK cell at the activating NKIS occurred in only 34% of conjugates (Fig. 7). This structure was termed a mature lytic synapse. Thus, many of the synapses appeared to be immature, because they lack perforin.
Fig. 1.

F-actin, along with CD2, CD11a, CD11b, and perforin, all polarize to the mature lytic NKIS. Ex vivo NK cells (small cells) are shown conjugated with K562 cells (large cells) in the top three rows, and a YTS-CD2/GFP cell (top cell) with a 721.221 cell (bottom cell) in the bottom row. Differential interference contrast images are in the left column and fluorescent images are in the right columns. F-actin is in the second column (blue). CD2, CD11a, or CD11b (red) and CD2/GFP (green) are in the third column. Perforin (green) in ex vivo NK cells and in YTS-CD2/GFP cells (red) is in the fourth column. A merged overlay of all fluorescent channels is in the fifth column.
Fig. 6.

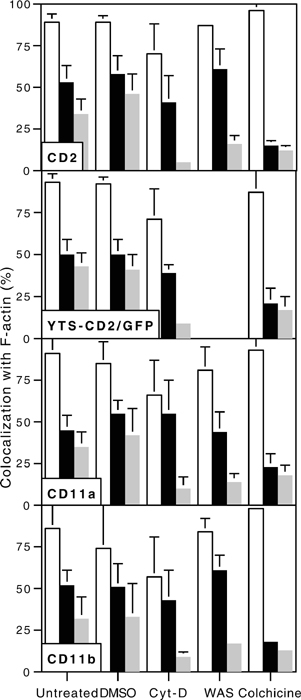
Cumulative summary of molecular polarization to the activating NKIS. Accumulation of F-actin (white bars), CD2, CD2/GFP, CD11a, or CD11b and perforin (gray bars) at the activating NKIS in ex vivo NK cells conjugated with K562 cells, or in YTS-CD2/GFP cells conjugated with 721.221 cells are shown. Cells were untreated, DMSO-treated, Cyt-D-treated, from a WAS patient, or colchicine-treated, as displayed on the x axis. Each cluster of bars represents the mean ± SD of three to six independent experiments, and the entire figure summarizes ≈4,000 individual conjugates. In all untreated samples, perforin was polarized less frequently than F-actin or the respective cell-surface receptor (P < 0.005). All molecular accumulations in Cyt-D-treated or WAS NK cells were less frequent than those found in their respective controls (P < 0.005), whereas in colchicine-treated cells only perforin polarization was decreased (P < 0.005).
CD11a and CD11b also accumulated at the activating NKIS in ex vivo NK cells (Fig. 1), in a frequency statistically indistinguishable from those containing polarized CD2 (Fig. 6). The accumulation of perforin to form mature lytic synapses containing a β2-integrin was ≤35%, which was similar to that found in CD2 containing lytic synapses (Fig. 7).
To create a more flexible model in which to study lytic synapse formation, the CD2– YTS NK tumor cell line was transduced with a CD2/GFP construct using a retroviral system (22). YTS-CD2/GFP transductants expressed CD2 at the cell surface at levels similar to those found on other immortalized NK cells, including NK92, NKL, and NK3.3 (data not shown). When YTS-CD2/GFP cells were conjugated with the NK-susceptible target cell 721.221, CD2/GFP polarized to the activating NKIS (Fig. 1, 721.221 was used as YTS cells do not readily lyse K562 cells). YTS cells transduced with GFP alone demonstrate diffuse green fluorescence that does not polarize in conjugates with 721.221 cells (data not shown). The accumulation of F-actin, CD2, and perforin, and their colocalization at the activating NKIS in YTS-CD2/GFP cells was not significantly different from that found in ex vivo NK cells (Figs. 6 and 7). Thus, YTS-CD2/GFP transductants appeared to accurately recapitulate the molecular accumulations occurring at the activating NKIS in ex vivo NK cells.
CD2, CD11a, and CD11b Colocalize with F-Actin in the pSMAC, Whereas Perforin Is in the cSMAC of the Activating NKIS. To determine the three-dimensional arrangement of molecules in the mature NK cell lytic synapse, images collected through the z axis were integrated and rotated so that the entire synapse could be viewed. The majority of lytic synapses studied in ex vivo NK cells contained CD2, CD11a, or CD11b in the pSMAC, along with F-actin (Fig. 2). In contrast, perforin was consistently accumulated in the cSMAC, although not always at its geometric center. A similar accumulation of CD2/GFP, along with F-actin in the pSMAC, and perforin in the cSMAC, in the lytic synapse of YTS-CD2/GFP cells was also seen (Fig. 2). Thus, the structure of the mature lytic synapse in NK cells had a distinct pSMAC and cSMAC with CD2 and the two β2-integrins in the former and perforin in the latter.
Fig. 2.

F-actin with CD2, CD11a, or CD11b accumulate in the pSMAC and perforin in the cSMAC of the mature lytic NKIS. Representative lytic synapses were evaluated throughout their volume, and the z, x plane was reconstructed. The first three columns show lytic synapses in ex vivo NK cells conjugated with K562 cells. F-actin (blue, top row), perforin (green, second row), or CD2 in the first column, CD11a in the second column, or CD11b in the third column (red, third row) are shown. A synapse between a YTS-CD2/GFP cell and 721.221 cell is in the fourth column with F-actin (blue), perforin (red), and CD2/GFP (green) being shown. A merged overlay of all fluorescent channels is shown in the bottom row. (Scale bars, 5 μm). The mean frequency of synapses with an F-actin and CD2, CD11a, or CD11b in the pSMAC and perforin in the cSMAC are above the representative images (n = 3–5 experiments/donors ± SD).
Maximal F-Actin and CD2 Accumulation Occurs Before Perforin Accumulation at the Activating NKIS. As shown above, F-actin and cell-surface receptor aggregation at the activating NKIS were more prevalent than perforin polarization at a given time point, suggesting that perforin polarization was slower. Additionally perforin polarization may only occur in a subset of conjugated NK cells. To further evaluate the phenomenon, the polarization of F-actin, CD2/GFP, and perforin was assessed over time at the activating NKIS in YTS-CD2/GFP cells. Conjugates of YTS and 721.221 cells formed rapidly and peaked by 5 min (Fig. 3A). The accumulation of F-actin and CD2 also occurred at a similar rapidity, but the peak accumulation of perforin was found at 15 min of conjugation (the time used in preceding experiments), and was greater than that found at all other time points (P < 0.05; Fig. 3B). Unlike F-actin and CD2 accumulation, the percentage of conjugates with polarized perforin dropped after 15 min. Thus, the accumulation of perforin at the activating IS followed the aggregation of, and was at a rate distinct from, CD2 and F-actin.
Fig. 3.

CD2 and F-actin accumulate at the activating NKIS more readily than does perforin. (A) Conjugation of YTS-CD2/GFP cells with 721.221 over time (⋄) was calculated as a percentage of total YTS-CD2/GFP cells present. (B) The accumulation of F-actin (▴), CD2/GFP (▪), and perforin (•) at the IS of YTS-CD2/GFP cells conjugated with 721.221 cells was evaluated for each time point (except time 0 due to the insignificant number of conjugates found). Individual points are the mean of four to eight experiments ± SD.
Accumulation of CD2, CD11a, and CD11b at the Activating NKIS Requires Actin Polymerization and WASp Function. To determine whether the accumulation of cell-surface receptors relative to perforin at the activating NKIS depended on reorganization of the actin cytoskeleton, two methods of interfering with actin polymerization were used. First, actin polymerization was inhibited with Cyt-D. Preincubation of ex vivo NK cells with Cyt-D almost completely inhibited the polarization of CD2, CD11a, CD11b, or perforin, as well as F-actin (Fig. 4A top three rows). Similarly, Cyt-D also inhibited the reorientation of CD2 and perforin as well as F-actin in YTS transductants (Fig. 4A, bottom row). In all cases, the formation of the mature lytic synapse was decreased by >75% (P < 0.05; Fig. 7). Thus, actin polymerization is necessary for the polarization of CD2, CD11a, and CD11b to the pSMAC, as well as of perforin-containing granules to the cSMAC in the activating NKIS.
Fig. 4.

Polarization of cell-surface receptors and perforin to the activating NKIS depends on the actin cytoskeleton and WASp function. (A) Ex vivo NK cells treated with Cyt-D are shown conjugated to K562 cells (top three rows) and similarly treated YTS-CD2/GFP cells conjugated to 721.221 cells (bottom row). DMSO was used as a diluent for Cyt-D stocks, and this vehicle alone did not affect molecular polarizations (see Fig. 6). (B) Ex vivo NK cells from a patient with WAS are shown conjugated with K562 cells. The colors, specific molecules evaluated, and figure layout are the same as in Fig. 1.
As a second means of investigating the importance of actin polymerization for the formation of the activating NKIS, NK cells from a patient with WAS that have been previously shown to have defective cytolytic activity (21) were evaluated. Cell-surface expression of CD2, CD11a, and CD11b on CD56+/CD3– patient NK cells were found at levels comparable to those on NK cells from normal donors (data not shown). In the vast majority of conjugates, however, CD2, CD11a, CD11b, and perforin failed to polarize to the activating NKIS (Figs. 4B and 6). Thus, mature lytic synapses in patient NK cells were found less frequently, compared with those of healthy subjects (16% for those containing CD2, 18% for CD11a, and 13% for CD11b, all P < 0.05 compared with controls; Fig. 7). Thus, polarization of activating surface receptors to the pSMAC and perforin to the cSMAC depended on appropriate actin polymerization and to a large degree on WASp function.
Perforin Accumulation in the cSMAC, but Not CD2, CD11a, CD11b, and F-Actin Accumulation in the pSMAC, Requires Microtubule Function. To determine whether the formation of the mature lytic synapse also depended on microtubule function, colchicine was used as a depolymerizing agent. Because colchicine inhibits NK cell cytotoxicity (18), it should also interfere with the organization of the activating NKIS. Ex vivo NK cells that had been treated with colchicine demonstrated unusual cell morphologies, but normal accumulation of F-actin and cell-surface receptors at the activating NKIS (Figs. 5A, top three rows, and 6). A similar polarization of F-actin and CD2 were seen in at the activating IS in YTS-CD2/GFP cells (Fig. 5A, bottom row). In both systems, the colocalization of the surface receptor with F-actin at the activating IS was statistically indistinguishable from that seen without colchicine (Fig. 7). Despite the fact that CD2, CD11a, CD11b, and F-actin accumulation appeared unaffected by colchicine treatment, the polarization of perforin granules was severely impaired. The percentage of NK cell, target-cell conjugates with perforin present at the activating NKIS was reduced from ≈50% in untreated ex vivo NK cells, or YTS cells (Fig. 1) to <20% in those that had been colchicine-treated (P < 0.005; Figs. 5A and 6). Similarly the occurrence of mature lytic synapses was reduced by >50% (P < 0.05; Fig. 7). These reductions in perforin polarization or in the frequency of mature lytic synapses were similar to those found after inhibition of actin polymerization with Cyt-D. Thus, microtubule polymerization was required for the formation of the mature lytic synapse in NK cells, because it was needed for perforin localization, but not for surface receptor aggregation.
Fig. 5.

Perforin, but not cell-surface receptor or F-actin accumulation at the activating NKIS, depends on microtubule function. (A) Ex vivo NK cells or YTS-CD2/GFP cells treated with colchicine were conjugated with K562 or 721.221 cells, respectively. Layout, specific molecules evaluated, and colors displayed are the same as for Fig. 1. (B) Synapses from colchicine-treated ex vivo NK or YTS-CD2/GFP were analyzed throughout their volume and reconstructed in the z, x plane. The colors, specific molecules evaluated, and layout are the same as in Fig. 2. (Scale bars, 5 μm).
Because F-actin, CD2, CD11a, and CD11b all appeared to polarize to the activating NKIS in the presence of colchicine, whether or not the three-dimensional arrangements were altered in the absence of microtubule function and perforin granule polarization, was examined. Interestingly, the accumulation of CD2, CD11a, and CD11b in the pSMAC of ex vivo NK cells at the activating IS was unaffected by colchicine treatment (Fig. 5B). A similar preservation of CD2/GFP accumulation in YTS transductants was also found. As expected from the two-dimensional images, the cSMAC did not contain perforin. Importantly, the percentage of synapses in colchicine-treated NK cells with discrete CD2, CD11a, or CD11b accumulations in the pSMAC was not statistically different from that found in untreated cells (Fig. 5B). Granule polarization to the cSMAC required both actin and microtubule function, whereas receptor aggregation at the pSMAC required only the former.
Discussion
A variety of adhesion receptors with activating potential, including the β2-integrins, CD11a and CD11b, as well as the Ig superfamily member, CD2, all accumulate in the pSMAC of the activating NKIS. This clustering requires both actin polymerization and WASp function. Thus, the role of these receptors in cell signaling and NKIS formation should be preceded by signaling events promoting the activation of WASp and rearrangement of the cortical actin cytoskeleton. Alternatively the ligation of these receptors in small numbers before their clustering may be capable of generating signals required for further maturation.
Suprisingly, CD2, which can generate strong activating signals in NK cells (5) and promote WASp activation in T cells (15), was not found in the cSMAC. CD2 has been shown to associate with PLC-γ in NK cells (5) which, in turn has been localized at the cSMAC of the activating NKIS (9). In T cells, however, CD2 clusters centrally or in an intermediate ring between the cSMAC and pSMAC (24). In T cells, CD2 can activate WASp (14), but may also activate β-integrins (25). In NK cells, however, CD2 signaling has been reported not to activate β-integrins (26). These observations support a different role for, and localization of, CD2 in the IS of T and NK cells. The fine mapping of signals from these receptors relative to WASp in NK cells may provide insight into distinctions between T and NK cells.
Similar to CD2, CD11a and CD11b were both localized in the pSMAC. Although they can have distinct ligands, investigation of CD11a- vs. CD11b-deficient mice suggest CD11a has a more central role in leukocyte adhesion and extravasation from the circulation, whereas CD11b may function more prominently in regulation of these processes (27). Thus, independent study of these receptors in NK cells and in NKIS formation and function is warranted.
Formation of the pSMAC was extremely dependent on the actin cytoskeleton, and in particular on WASp activity. The dependence of the activating IS on WASp function may relate to the use of ex vivo NK cells that had not been activated with exogenous mitogens, cytokines, or feeder cells. The use of these stimulants may circumvent the regulatory control provided by WASp. Interestingly, functional activity (cytotoxicity) can be restored by culture of NK cells from the WAS patient studied here with IL-2 and feeder cells (unpublished results). β2-integrin, as well as CD2 clustering in ex vivo NK cells, depended on WASp function, which was in contrast to observations made in murine T cells (17). Dependence of synapse formation on the cytoskeleton may be more pronounced in NK cells than in T cells, as evidenced by recent experiments comparing cytoskeletal requirements for synapse formation in murine NKs and CTLs (28). Presumably, the greater specificity of T cells necessitates less opportunity for regulation.
Conversely, the completely intact structure of the pSMAC in the absence of microtubule polymerization is notable. Thus, surface receptors move to the pSMAC of the NKIS laterally through the cell membrane, and are probably not brought to the surface from within in a microtubule-dependent manner. However, CD11b can be delivered in granules to the cell surface on cell activation (29). Clearly, that delivery mechanism is either not essential for pSMAC formation in NK cells or utilizes microtubule-independent transport. Additionally, microtubule-dependent membrane structures, such as actin-rich podosomes, as found in other cell types (30), are not required for formation of the pSMAC at the activating NKIS.
In contrast, perforin polarization to the cSMAC depended on microtubule function. In conjunction with the results demonstrating the dependence of perforin polarization on actin polymerization and WASp function, microtubule polymerization and perforin mobilization must follow actin reorganization, i.e., the activating NKIS is formed in a distinct sequence involving the actin cytoskeleton first and the microtubules second. It is not clear as to whether this mechanism can be generalized to CTLs, but the separation may be exaggerated in NK cells providing additional opportunity for regulation of activation through innate immune receptors having broader specificity.
Thus, two stages of formation of the activating NKIS have been described. At least two more stages not studied here must occur: polarization of the MTOC to the synaptic regions, and the active clustering of surface receptors. Actin dependence of these events or of the synaptic anchoring region of microtubules would explain the cyt-D-mediated inhibition of perforin polarization. Presumably, both of these events are also essential for mature lytic synapse formation and function.
Supplementary Material
Acknowledgments
We thank K. Krzewski, I. Chiu, and M. Fassett for assistance. This work was supported by National Institutes of Health Grants AI-50207 (to J.L.S.), HL-59561 (to R.S.G.), and AI-55602 (to J.S.O.).
Abbreviations: IS, immunologic synapse; NKIS, natural killer cell IS; SMAC, supramolecular activation cluster; c, central; p, peripheral; WAS, Wiskott–Aldrich Syndrome; WASp, WAS protein; F-actin, filamentous actin; Cyt-D, cytochalasin-D.
References
- 1.Orange, J. S. (2002) Microbes Infect. 4, 1545–1558. [DOI] [PubMed] [Google Scholar]
- 2.Miller, J. S. (2002) Cancer Invest. 20, 405–419. [DOI] [PubMed] [Google Scholar]
- 3.Kohl, S., Springer, T. A., Schmalstieg, F. C., Loo, L. S. & Anderson, D. C. (1984) J. Immunol. 133, 2972–2978. [PubMed] [Google Scholar]
- 4.Bolhuis, R. L., Roozemond, R. C. & van de Griend, R. J. (1986) J. Immunol. 136, 3939–3944. [PubMed] [Google Scholar]
- 5.Inoue, H., Miyaji, M., Kosugi, A., Nagafuku, M., Okazaki, T., Mimori, T., Amakawa, R., Fukuhara, S., Domae, N., Bloom, E. T. & Umehara, H. (2002) Eur. J. Immunol. 32, 2188–2198. [DOI] [PubMed] [Google Scholar]
- 6.Shibuya, K., Lanier, L. L., Phillips, J. H., Ochs, H. D., Shimizu, K., Nakayama, E., Nakauchi, H. & Shibuya, A. (1999) Immunity 11, 615–623. [DOI] [PubMed] [Google Scholar]
- 7.Vetvicka, V., Thornton, B. P. & Ross, G. D. (1996) J. Clin. Invest. 98, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyas, Y. M., Maniar, H. & Dupont, B. (2002) Immunol. Rev. 189, 161–178. [DOI] [PubMed] [Google Scholar]
- 9.Vyas, Y. M., Mehta, K. M., Morgan, M., Maniar, H., Butros, L., Jung, S., Burkhardt, J. K. & Dupont, B. (2001) J. Immunol. 167, 4358–4367. [DOI] [PubMed] [Google Scholar]
- 10.Stinchcombe, J. C., Bossi, G., Booth, S. & Griffiths, G. M. (2001) Immunity 15, 751–761. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn, J. R. & Poenie, M. (2002) Immunity 16, 111–121. [DOI] [PubMed] [Google Scholar]
- 12.Wulfing, C., Sumen, C., Sjaastad, M. D., Wu, L. C., Dustin, M. L. & Davis, M. M. (2002) Nat. Immunol. 3, 42–47. [DOI] [PubMed] [Google Scholar]
- 13.Krummel, M. F., Sjaastad, M. D., Wulfing, C. & Davis, M. M. (2000) Science 289, 1349–1352. [DOI] [PubMed] [Google Scholar]
- 14.Dupre, L., Aiuti, A., Trifari, S., Martino, S., Saracco, P., Bordignon, C. & Roncarolo, M. G. (2002) Immunity 17, 157–166. [DOI] [PubMed] [Google Scholar]
- 15.Badour, K., Zhang, J., Shi, F., McGavin, M. K., Rampersad, V., Hardy, L. A., Field, D. & Siminovitch, K. A. (2003) Immunity 18, 141–154. [DOI] [PubMed] [Google Scholar]
- 16.Calderwood, D. A., Shattil, S. J. & Ginsberg, M. H. (2000) J. Biol. Chem. 275, 22607–22610. [DOI] [PubMed] [Google Scholar]
- 17.Krawczyk, C., Oliveira-dos-Santos, A., Sasaki, T., Griffiths, E., Ohashi, P. S., Snapper, S., Alt, F. & Penninger, J. M. (2002) Immunity 16, 331–343. [DOI] [PubMed] [Google Scholar]
- 18.Katz, P., Zaytoun, A. M. & Lee, J. H., Jr. (1982) J. Immunol. 129, 2816–2825. [PubMed] [Google Scholar]
- 19.Carpen, O., Virtanen, I., Lehto, V. P. & Saksela, E. (1983) J. Immunol. 131, 2695–2698. [PubMed] [Google Scholar]
- 20.Kupfer, A., Dennert, G. & Singer, S. J. (1983) Proc. Natl. Acad. Sci. USA 80, 7224–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orange, J. S., Ramesh, N., Remold-O'Donnell, E., Sasahara, Y., Koopman, L., Byrne, M., Bonilla, F. A., Rosen, F. S., Geha, R. S. & Strominger, J. L. (2002) Proc. Natl. Acad. Sci. USA 99, 11351–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen, G. B., Gandhi, R. T., Davis, D. M., Mandelboim, O., Chen, B. K., Strominger, J. L. & Baltimore, D. (1999) Immunity 10, 661–671. [DOI] [PubMed] [Google Scholar]
- 23.Morita, S., Kojima, T. & Kitamura, T. (2000) Gene Ther. 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- 24.van der Merwe, P. A., Davis, S. J., Shaw, A. S. & Dustin, M. L. (2000) Semin. Immunol. 12, 5–21. [DOI] [PubMed] [Google Scholar]
- 25.Kivens, W. J., Hunt, S. W., III, Mobley, J. L., Zell, T., Dell, C. L., Bierer, B. E. & Shimizu, Y. (1998) Mol. Cell. Biol. 18, 5291–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinowich, H., Lin, W. C., Herberman, R. B. & Whiteside, T. L. (1994) J. Immunol. 153, 3504–3513. [PubMed] [Google Scholar]
- 27.Ding, Z. M., Babensee, J. E., Simon, S. I., Lu, H., Perrard, J. L., Bullard, D. C., Dai, X. Y., Bromley, S. K., Dustin, M. L., Entman, M. L., et al. (1999) J. Immunol. 163, 5029–5038. [PubMed] [Google Scholar]
- 28.Wulfing, C., Purtic, B., Klem, J. & Schatzle, J. D. (2003) Proc. Natl. Acad. Sci. USA 100, 7767–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sengelov, H., Follin, P., Kjeldsen, L., Lollike, K., Dahlgren, C. & Borregaard, N. (1995) J. Immunol. 154, 4157–4165. [PubMed] [Google Scholar]
- 30.Linder, S., Hufner, K., Wintergerst, U. & Aepfelbacher, M. (2000) J. Cell Sci. 113, 4165–4176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}