

Abstract
Cu ions have been suggested to enhance the assembly and pathogenic potential of the Alzheimer's disease amyloid-β (Aβ) peptide. To explore this relationship in vivo, toxic-milk (txJ) mice with a mutant ATPase7b transporter favoring elevated Cu levels were analyzed in combination with the transgenic (Tg) CRND8 amyloid precursor protein mice exhibiting robust Aβ deposition. Unexpectedly, TgCRND8 mice homozygous for the recessive txJ mutation examined at 6 months of age exhibited a reduced number of amyloid plaques and diminished plasma Aβ levels. In addition, homozygosity for txJ increased survival of young TgCRND8 mice and lowered endogenous CNS Aβ at times before detectable increases in Cu in the CNS. These data suggest that the beneficial effect of the txJ mutation on CNS Aβ burden may proceed by a previously undescribed mechanism, likely involving increased clearance of peripheral pools of Aβ peptide.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by extracellular deposition of amyloid-β (Aβ) as senile plaques and intracellular accumulation of hyperphosphorylated tau as neurofibrillary tangles (1). Aβ is generated by secretase-mediated endoproteolysis of the amyloid precursor protein (APP), and familial AD mutations skew APP processing to favor production of pro-amyloidogenic forms of the peptide or net Aβ production and thus drive disease pathogenesis. Although there is growing interest in defining pathogenic subvarieties of Aβ [e.g., secreted oligomeric assemblies such as Aβ-derived diffusible ligands (2) and intracellular forms (3)], modulators of APP and Aβ biology in sporadic disease have remained more elusive. One area of particular interest concerns the role of transition metals.
Cu and Zn ions are abundant in the normal brain (4–6), and direct measurements of metal levels have indicated altered homeostasis in AD (7–10). Interestingly, APP has a selective, high-affinity Cu-binding site in the extracellular (ecto-) domain that is capable of reducing Cu(II) to Cu(I) (11), and a recent structural analysis of this domain has revealed some similarity to previously identified Cu chaperone proteins (12). In addition to the ectodomain Cu-binding site of APP, Aβ peptide also contains binding sites for Zn and Cu (13–15). Aβ–metal interaction may drive both fibril formation and free radical production (13, 16–18), findings potentially relevant to AD pathogenesis in vivo, given that metal chelators can resolubilize Aβ aggregates from postmortem AD brain (19). On the other hand, studies of APP processing in cultured cells have revealed stimulation of the α-secretase pathway for APP processing by extracellular Cu ions (20). As this pathway cleaves the Aβ domain of APP into two fragments, it has a potential to be anti-amyloidogenic. Prompted by these divergent observations, we devised a genetic experiment to investigate how Cu might modulate Aβ-dependent pathologies in vivo by using the transgenic (Tg) CRND8 line of TgAPP mice (21, 22) in conjunction with a mutant allele of the CuATPase7b copper transporter.
In Wilson disease, mutations in the human CuATPase7B gene result in reduced ability to efflux Cu into the bile (23). The Wilson disease CuATPase7B protein (Wnd) resides in the trans-Golgi network (24) and relocates to cytoplasmic vesicles upon Cu loading. Cu builds up within the cytoplasm in CuATPase7B mutant cells because of an inability to load secretory vesicles and may eventually be released into extracellular compartments because of cell lysis (25, 26). In this regard, it is known that Cu is increased in the cerebrospinal fluid (CSF) of Wilson disease patients (27, 28). Similar homeostatic perturbations have been inferred for mouse toxic-milk (txJ) mutations of the CuATPase7b transporter (29, 30), and age-dependent elevations of CNS Cu (and Zn) are observed in tx mice harboring this mutation (this article). Using txJ mice and TgCRND8/txJ mice to explore APP–Cu and Aβ–Cu interactions, our analyses reveal unexpected effects of homozygosity for the txJ mutation.
Materials and Methods
Mice. All animal husbandry procedures were performed in accord with Canadian Council on Animal Care guidelines. TgCRND8 mice (22) were maintained on a C3H/HeJ × C57BL/6J genetic background. C3HeB/FeJ-Atp7btxJ (txJ) mice, homozygous for the txJ mutation and C3HeB/FeJ stock were obtained from The Jackson Laboratory. txJ mice harbor an autosomal recessive mutation in the gene encoding CuATPase7b (31). The txJ mutation is a second allele of the original tx mutation described in DL mice (32). TgCRND8 [(C3H/HeJ × C57BL/6J) × C3H/HeJ] females were paired with males homozygous for the txJ mutation. All female offspring confirmed to be APP-positive by hybridization of tail DNA with a human APP cDNA probe (22, 33, 34) were backcrossed with homozygous txJ males to produce experimental groups. txJ genotyping was established by sequencing of a PCR-amplified portion of Cu Atp7b exon 8 (31).
Metal Analysis. Analyses were performed in a dedicated trace element laboratory by using procedures published previously (35, 36). All txJ, C3HeB/FeJ, and TgCRND8/txJ mice were injected with sodium pentobarbitol then transcardially perfused with 0.1 M PBS made with trace metal-free double-distilled H2O. Brains were then removed and bisected in the midsagittal plane, and the olfactory bulb and cerebellum were removed. Liver samples were collected from the anterior lobe. Samples were stored in polypropylene tubes at –80°C until freeze-dried (FLEXI-DRY Microprocessor Manifold lyophilizer, FTS Systems, Stone Ridge, NY) to a stable dry weight. Tissue was then rehydrated in 500 μl of double-distilled H2O overnight and digested in 1 ml of ultrapure nitric acid (Ultrex II, J.T. Baker) at 90°C. Digests were diluted and placed within the autosampler of an atomic absorption spectrophotometer (Varian graphite furnace GTA 100-Varian Spectra 880) for Cu analysis or a flame atomic absorption spectrophotometer for Zn analysis.
CSF was collected from a subset of mice before removal of the brain. For this, a 23-gauge needle, attached to capillary tubing and a syringe, was inserted into the cisterna magna. CSF was aspirated and stored in Eppendorf tubes at –80°C. Duplicate samples (1 or 2 μl, depending on the volume collected) were diluted in 200 μl of 0.5% nitric acid overnight and placed within the autosampler of the atomic absorption spectrophotometer for Cu analysis.
Immunohistochemistry. Formalin fixed brains were paraffin-embedded and sectioned sagitally. Sections were histologically stained by hematoxylin and eosin, Bielschowsky's silver stain, thioflavin S, and Congo red (22). Cu was visualized by using a rhodamine stain (37). All sections for immunohistochemistry were pretreated with 3% H2O2 and nonimmune serum. The following antibodies were used: rabbit anti-cow glial fibrillary acidic protein (Roche Molecular Biochemicals; diluted 1:1,000), mouse anti-microtubule-associated protein 2 (Sigma; diluted 1:20), and mouse anti-synaptophysin (DAKO; diluted 1:500). Amyloid was visualized by using the monoclonal antibody 6F/3D (DAKO; diluted 1:400) following a 5-min formic acid treatment. In all cases primary antibodies were left to react overnight at 4°C. Sections were developed according to the manufacturer's instructions for StreptABC complex/HRP-conjugated “Duet” anti-mouse/rabbit antibody kit (DAKO). Diaminobenzidine was used as a chromagen. Sections were counterstained with hematoxylin or luxol fast blue and mounted with resin.
Amyloid Plaque-Load Determination. Amyloid plaque burden was estimated by directly measuring plaque area and numbers on seven 5-μm-thick saggital sections that were evenly spaced (≈100 μm apart) throughout the region of interest. Each section from the “systematic-uniform-random” set (38) was immunostained by using DAKO 6F/3D antibody as described above and lightly counterstained with luxol fast blue to better visualize regional borders. To reconstruct the hippocampal or cortical region from each section in the set, multipanel digital images (final magnification was ×100) were captured by using a Zeiss Axioskop 2 plus microscope fitted with a motorized stage (MAC 5000, Ludl Electronic Products, Hawthorne, NY) and a Photometrics Coolsnap digital camera (Tucson, AZ). Openlab imaging software (Improvision, Lexington, MA) was then used to measure the regional area, as well as to convert micrographs to binary images for plaque number and plaque area quantifications. The threshold for pixel detection was the same for each image. Brain regions were based on the Paxinos and Franklin mouse brain atlas (39). The data from the seven sections were summed (and divided by the regional area in the case of plaque burden) to derive representative values for each animal. Mean data were then analyzed for each genotype.
Protein Analysis and Aβ ELISA. Ten percent (wt/vol) sucrose homogenates were prepared from brain hemispheres dissected to remove the olfactory bulb and cerebellum as described (21, 40). One aliquot of each brain homogenate was directly used for SDS/PAGE, whereas a second was extracted in an equal volume of 0.4% diethylamine and spun at 135,000 × g for 1 h, and the supernatant was neutralized (40). This diethylamine extraction protocol quantitatively recovers nondeposited Aβ and secreted APP fragments (sAPP) in the supernatant, leaving membrane-associated APP in the pellet (40, 41). In human Aβ-depositing mice, Aβ was quantitated after extraction in formic acid (21). APP holoprotein and APP metabolites were resolved by SDS/PAGE on 4–20% Tris-glycine gradient gels (NOVEX, San Diego) and transferred to nitrocellulose membrane (Millipore) before Western blot analysis with antibody C1/6.1 [holo-APP and APP-CTF (42)] or antibody 22C11 (diethylamine-soluble sAPP, Chemicon, diluted 1:1000). Proteins were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences), with all samples normalized by protein concentration. Aβ sandwich ELISAs were performed as described (21, 40). ELISA plates also containing synthetic Aβ standards and were developed by using a color reaction (TMB Microwell Peroxidase Substrate System, Kirkegaard & Perry Laboratories). Each Aβ measurement represented the mean of two or more readings.
Statistical Analysis. A factorial model ANOVA was used. Post hoc comparisons were carried out by using Fisher's least significant difference (LSD) test or, whenever preplanned, multiple t test comparisons were performed. Comparisons between the means of two independent groups were carried out by using a two-tailed t test, adjusting degrees of freedom in cases of unequal variances. The critical α level was set to 0.05 for all statistical analyses. Statistical analyses were performed by using statview 5.0 (Abacus Concepts, Berkeley, CA). All reported values represent means ± SEM.
Results
Effect of APP on CNS Cu. Based on prior studies of Cu physiology and transport, we were interested in assessing the magnitude of the effects of APP overexpression on CNS Cu (43). By using atomic adsorption spectroscopy, a decrease in net Cu was detected in the brains of aged TgCRND8 mice (Fig. 1, P < 0.05; ANOVA). TgAPP mice between the ages of 3 and 5 months (early stages of amyloid deposition; ref. 22) had a small, nonsignificant reduction in net brain Cu compared with controls (P > 0.05; t test), whereas 9- to 10-month-old mice (advanced amyloid deposition) had a significant reduction in net brain Cu as compared with controls (P < 0.01; t test). In broad agreement with similar analyses for Tg2576 TgAPP mice (43), no difference in brain Zn was seen in TgCRND8 mice at either age investigated (Fig. 1).
Fig. 1.

Cu levels in TgCRND8 brain. At 3–5 months the reduction in brain Cu was nonsignificant. By 9–10 months of age the difference was significant (P < 0.05; t test; n = 11 non-Tg and 7 TgCRND8 at 3–5 months; n = 7 non-Tg and 10 TgCRND8 at 9–10 months). No significant difference in brain Zn (Right) was found. Asterisks in this and subsequent figures indicate where pairwise t test comparisons or Fisher's LSD post hoc test were significant. **, P < 0.01.
Phenotypic Analysis of txJ Mice. Two alleles of tx with similar properties are in common use, namely the “DL” and “J” alleles (29–31). Because the viability of mice overexpressing APP transgenes depends on strain backgrounds, our studies used the J allele, because this was isolated on a C3H genetic background potentially compatible with propagation of the TgCRND8 transgene array (22). Analyses were carried out on stock mice carrying the txJ allele to confirm the elevation of brain Cu and the absence of confounding CNS pathologies and to assess potential alterations in α-secretase processing (20).
Cu levels in txJ mice. As shown in Fig. 2a, brain Cu levels are increased versus isogenic controls as txJ homozygote mice aged (P < 0.0001; ANOVA). By 4 months of age, txJ/J mice had ≈1.2-fold more brain Cu than control C3H mice (P < 0.05; t test). This difference persisted in 6-month-old mice (P < 0.01; t test). At 6 months of age, CSF Cu was elevated 3-fold in mice homozygous for the txJ mutation compared with control mice [0.491 ± 0.035 μg/l (n = 4) versus 0.164 ± 0.024 μg/l (n = 4), P < 0.01; t test]. Brain Zn was also altered in txJ/J mice (Fig. 2a, P < 0.01; ANOVA), although the percent change was of smaller magnitude than that seen for Cu. Cu levels were also determined in the liver, an accepted site of expression and physiological action of the tx mutation and the Wnd protein (29). Whereas Cu levels in the liver of txJ homozygotes were comparable to levels in controls at 1 month of age, by 2 months of age Cu levels of txJ/J mice surpassed the values seen in controls by >50-fold [18 ± 1.1 μg/g (n = 6) versus 950.86 ± 52.3 μg/g (n = 6), P < 0.0001; t test]. Zn was ≈2.5× greater in txJ homozygotes versus control mice at 2 months of age [128 ± 2 μg/g (n = 6) versus 337 ± 10 μg/g (n = 6), P < 0.0001; t test], remaining at this level as the mice aged (not shown).
Fig. 2.

Brain Cu levels and APP processing in txJ mice. (a) txJ/J mice (hatched bars) had significantly elevated brain Cu levels as they aged compared with isogenic C3H controls (open bars, Left; ANOVA effect of genotype is F1,23 = 22.2, P < 0.0001; effect of age is F2,23 = 43.5, P < 0.0001, and genotype × age interaction is F2,48 = 10.0, P < 0.01). Brain Zn levels (Right) again varied less than Cu levels but were different between txJ/J and C3H mice with age (ANOVA effect of genotype is F1,20 = 14.8, P < 0.01, effect of age is F2,20 = 15.9, P < 0.0001, and genotype × age interaction is F2,20 = 14.5, P < 0.01). At 2 months txJ/J mice had less brain Zn than C3H mice (P < 0.05; t test), whereas at 6 months txJ/J mice had more Zn than C3H mice [P < 0.05; t test; n = 6 C3H and 6 txJ at 2 months; n = 5 C3H and 5 txJ (except Zn, n = 2 txJ) at 4 months; n = 4 C3H and 4 txJ at 6 months]. (b) Western blot analysis using both a C-terminal-specific APP antibody (C1/6.1; Upper) and an N-terminal-specific APP antibody (22C11; Lower) revealed no differences in the processing of endogenous APP between 2-month-old C3H and txJ/J mice. (c) Similarly, no difference was observed in mouse holo-APP or sAPP in control mice and txJ homozygous mice at 6 months of age (comparison of non-Tg mice that were txJ heterozygotes and homozygotes as a result of the TgCRND8/txJ crosses).
Pathology. Brain sections from mice between 1 and 6 months of age were stained with a variety of histochemical and immunohistochemical markers. Hematoxylin staining of coronal and sagittal sections failed to distinguish txJ/J mice from txJ/+ heterozygotes or WT mice at all ages examined. Immunohistochemical staining for an axonal protein (anti-NF-200), a dendritic marker (anti-microtubule-associated protein 2), and gliosis (anti-glial fibrillary acidic protein) failed to reveal differences between txJ mice and age-matched control mice. On the other hand, liver pathologies were evident in aged txJ homozygotes, in agreement with studies of txDL mice (refs. 44–46 and data not shown).
α-Secretase processing of endogenous APP. To determine whether increased CNS Cu increased α-secretase processing of APP, brain homogenates and the supernatant of diethylamine extracts were analyzed by Western blotting for APP holoprotein and sAPP, respectively. These experiments included txJ/J and control animals at 2 and 6 months of age and assessed the status of products of the endogenous mouse APP gene, using the 22C11 antibody directed against the APP ectodomain and the C1/6.1 antibody directed against the carboxyl terminus of APP. No effect of the txJ mutation was observed at either time point (Fig. 2 b and c). Control experiments performed on animals at 1 year of age (and with higher levels of CNS Cu) confirmed our ability to detect elevation of sAPP with this methodology (data not shown).
Phenotypic Analysis of TgCRND8/txJ Mice. TgCRND8 mice were crossed with txJ mice to establish Aβ-depositing mice with genetically altered brain Cu metabolism. The TgCRND8/txJ breeding scheme produced offspring of all four predicted genotypes, namely, (i) TgCRND8+/– × txJ/J (for the sake of simplicity these mice will be referred to as APP+/txJ/J), (ii) TgCRND8+/– × txJ/+ (APP+/txJ/+), (iii) TgCRND8–/– × txJ/J (APP–/txJ/J), and (iv) TgCRND8–/– × txJ/+ (APP–/txJ/+).
Survival. Because the APP695 transgene in TgCRND8 mice is associated with postnatal lethality (22, 47), the colony was monitored for survival. Analysis of the genotypes of the mice resulting from the cross (at age 1 month) indicated that the observed genotypic distribution departed significantly from the expected ratios (Fig. 3a, P < 0.02, 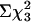 ). Further analysis confirmed that this was due to the underrepresentation of APP+/txJ/+ mice, by ≈50% (Fig. 3a, P < 0.01,
). Further analysis confirmed that this was due to the underrepresentation of APP+/txJ/+ mice, by ≈50% (Fig. 3a, P < 0.01, 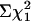 ), suggesting that homozygosity for the txJ mutation may protect against toxic effects of the mutant human APP695 transgene. To confirm that this observation was not an idiosyncrasy of the TgCRND8/txJ crosses, we also examined a cohort of mice from the parent TgCRND8 colony. When considering pups born to pairs similar to those in our experimental crosses (i.e., TgAPP-positive C3H/C57BL6 mothers mated to C57BL6 WT males), we found that APP-positive mice were also underrepresented, comprising 37% of mice surviving to the age of weaning (55/148 survivors;
), suggesting that homozygosity for the txJ mutation may protect against toxic effects of the mutant human APP695 transgene. To confirm that this observation was not an idiosyncrasy of the TgCRND8/txJ crosses, we also examined a cohort of mice from the parent TgCRND8 colony. When considering pups born to pairs similar to those in our experimental crosses (i.e., TgAPP-positive C3H/C57BL6 mothers mated to C57BL6 WT males), we found that APP-positive mice were also underrepresented, comprising 37% of mice surviving to the age of weaning (55/148 survivors;  ). Between 1 and 6 months of age, there was no evidence for heightened mortality in APP+/txJ/J mice (15/23 survivors, 65%) versus a similar figure for the parental TgCRND8 line (22).
). Between 1 and 6 months of age, there was no evidence for heightened mortality in APP+/txJ/J mice (15/23 survivors, 65%) versus a similar figure for the parental TgCRND8 line (22).
Fig. 3.

Phenotypic analysis of TgCRND8/txJ mice. (a) Summary of the genotypes of the mice resulting from the TgCRND8 and txJ crosses. Note that the observed genotypic distribution significantly varied from the expected ratios (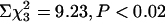 ). Binary analysis confirmed that this deviation was due to the underrepresentation of APP+/txJ/+mice (*,
). Binary analysis confirmed that this deviation was due to the underrepresentation of APP+/txJ/+mice (*, 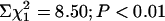 ). (b) Assessment of Cu levels at ages 6–7 months (Upper) confirmed significant differences between the genotypes (ANOVA is F3,12 = 12.6; P < 0.01). Post hoc analysis revealed a modest depletion of brain Cu in APP+/txJ/+ (downward hatched bar; n = 2) compared with APP–/txJ/+ (open bars; P > 0.05; LSD, n = 5) and an increase of ≈1.5× in APP–/txJ/J (upward hatched bars; n = 5) and APP+/txJ/J (crosshatched bars; n = 4) brain Cu over txJ/+ mice (P < 0.01, LSD). Cu levels in the APP+/txJ/J mice were not distinguishable from those of the APP–/txJ/J littermates (P > 0.05; LSD). The Lower histogram summarizes brain Zn levels in these mice. A small increase in APP+/txJ/J mice (n = 4) versus APP+/txJ/+ (n = 2), APP–/txJ/J (n = 5), and APP–/txJ/+ mice (n = 5) was noted (*, P < 0.05, LSD). (c) Western blotting of APP+/txJ/+ and APP+/txJ/J brains by using either a C-terminal-specific antibody (Upper) or an N-terminal-specific antibody (Lower) failed to reveal any overt difference in processing of transgene-encoded human APP. (d)Aβ-specific ELISAs performed on a formic acid extract of total brain demonstrated a lowering of both Aβ40 and Aβ42 levels in APP+/txJ/J brain (crosshatched bars; n = 3) compared with APP+/txJ/+ brain (downward-hatched bars; n = 5; P >0.05; t test). (e) Plasma Aβ levels were also determined in TgCRND8/txJ mice by using this ELISA. Both Aβ40 and Aβ42 were lower in APP+/txJ/J (n = 4) mice compared with APP+/txJ/+ (n = 3) mice (Aβ40, P < 0.05; Aβ42, P = 0.05).
). (b) Assessment of Cu levels at ages 6–7 months (Upper) confirmed significant differences between the genotypes (ANOVA is F3,12 = 12.6; P < 0.01). Post hoc analysis revealed a modest depletion of brain Cu in APP+/txJ/+ (downward hatched bar; n = 2) compared with APP–/txJ/+ (open bars; P > 0.05; LSD, n = 5) and an increase of ≈1.5× in APP–/txJ/J (upward hatched bars; n = 5) and APP+/txJ/J (crosshatched bars; n = 4) brain Cu over txJ/+ mice (P < 0.01, LSD). Cu levels in the APP+/txJ/J mice were not distinguishable from those of the APP–/txJ/J littermates (P > 0.05; LSD). The Lower histogram summarizes brain Zn levels in these mice. A small increase in APP+/txJ/J mice (n = 4) versus APP+/txJ/+ (n = 2), APP–/txJ/J (n = 5), and APP–/txJ/+ mice (n = 5) was noted (*, P < 0.05, LSD). (c) Western blotting of APP+/txJ/+ and APP+/txJ/J brains by using either a C-terminal-specific antibody (Upper) or an N-terminal-specific antibody (Lower) failed to reveal any overt difference in processing of transgene-encoded human APP. (d)Aβ-specific ELISAs performed on a formic acid extract of total brain demonstrated a lowering of both Aβ40 and Aβ42 levels in APP+/txJ/J brain (crosshatched bars; n = 3) compared with APP+/txJ/+ brain (downward-hatched bars; n = 5; P >0.05; t test). (e) Plasma Aβ levels were also determined in TgCRND8/txJ mice by using this ELISA. Both Aβ40 and Aβ42 were lower in APP+/txJ/J (n = 4) mice compared with APP+/txJ/+ (n = 3) mice (Aβ40, P < 0.05; Aβ42, P = 0.05).
Cu levels in TgCRND8/txJ mice. A cohort of TgCRND8/txJ mice at 6–7 months of age was subjected to detailed analysis. Cu levels at this time were in agreement with previous analyses (Fig. 3b, P < 0.0001; ANOVA). A modest depletion of brain Cu in APP+/txJ/+ compared with APP–/txJ/+ (P > 0.05; LSD) confirmed our observations of TgCRND8 mice versus controls in the parent colony (Fig. 1). txJ homozygotes had an ≈1.5× increase in brain Cu over APP–/txJ/+ mice (Fig. 3b, P < 0.01; LSD), also in agreement with earlier observations from the txJ colony (Fig. 2a). Cu levels in the APP+/txJ/J mice were not distinguishable from those of the APP–/txJ/J littermates, indicating that the APP-transgene did not offset the elevated Cu levels associated with the txJ mutation. Zn levels in this cohort of mice were broadly consistent with the data presented in Fig. 2, although a small elevation was seen in APP+/txJ/J mice (Fig. 3b; P < 0.05; LSD).
APP processing, human Aβ, and plaque burden. TgCRND8 mice were examined to determine the effect of the txJ mutation on processing of human APP and Aβ species encoded by the APP695 transgene array. In agreement with analyses of mouse APP, Western blot analysis of samples from 6-month-old TgCRND8 mice showed that the txJ/J mutation did not alter levels of either human APP holoprotein or human sAPP (Fig. 3c). Aβ-specific ELISAs on formic acid-extracted total brain from APP+/txJ/+ and APP+/txJ/J mice (Fig. 3d) revealed a tendency for reductions in Aβ40 and Aβ42, although the differences between experimental groups did not reach significance. A similar tendency was noted in analyses of soluble forms of CNS Aβ within diethylamine fractions (data not shown). Interestingly, the txJ mutation resulted in reduction of plasma Aβ in APP+/txJ/J versus APP+/txJ/+ mice (Fig. 3e). Both Aβ40 and Aβ42 were lower in the plasma of APP+/txJ/J compared with APP+/txJ/+ mice at 6 months of age (Aβ40, P < 0.05; Aβ42, P = 0.05; t test).
As net levels of Aβ in the CNS may fail to register regional or redistribution effects, the 6F/3D monoclonal antibody was used to visualize and quantify dense-cored plaques in a series of systematically spaced sections of formalin-fixed tissue (Fig. 4 a and b). Although no difference in plaque morphology was found between APP+/txJ+/+ and APP+/txJ/+ mice (thioflavin S and Congo red stains, not shown), a striking outcome of this analysis was a 45% reduction in the area occupied by plaques in the cortex and hippocampus in APP+/txJ/J mice (Fig. 4c, P < 0.05; t test) due to a reduced number of plaques (Fig. 4c, P < 0.05; t test).
Fig. 4.

Plaque burden in APP+/txJ/+ versus APP+/txJ/J mice. (a) Photomicrographs representing a 6-month-old APP+/txJ/+ demonstrate the significant deposition of Aβ plaques in the hippocampus (Left) and cortex (Center). (b)A reduction in the Aβ plaque load can be seen in the hippocampus (Left) and cortex (Center) of APP+/txJ/J mice when compared with APP+/txJ/+ (above). Higher magnification of Aβ plaques (Right) revealed that Aβ plaques were indistinguishable between TgCRND8 heterozygous for the txJ mutation (a)or homozygous for the txJ mutation (b). [Scale bars = 350 μm (Left and Center) and 100 μm (Right).] (c) Quantitative image analysis using a systematic uniform random series of brain sections from APP+/txJ/+ (downward-hatched bars; n = 5) and APP+/txJ/J mice (crosshatched bars; n = 5) confirmed a reduction in the area occupied by plaques in the hippocampus and cortex of APP+/txJ/J mice (Left, P < 0.05; t test) and a reduction in the number of plaques in the hippocampus and cortex of APP+/txJ/J mice (Center, P < 0.05; t test). However, no significant difference in plaque size was noted between the groups (Right, P > 0.05; t test).
Effect of txJ on Endogenous Mouse Aβ. Although human Aβ40 and Aβ42 are prone to fibrillogenesis, three substitutions present in the N terminus of the peptide attenuate the analogous process in the case of murine Aβ. We reasoned that if the txJ mutation has its primary effect on fibril assembly, then an impact on steady-state levels of soluble forms of Aβ may not be apparent. Conversely, if the txJ mutation has a primary effect on the synthesis or turnover of soluble Aβ, then this property should be apparent in a setting where measurements of net Aβ levels are not complicated by (i) peptide aggregation and (ii) the stoichiometric excess of insoluble forms of Aβ, as present in the CNS of aged TgCRND8 mice (22). Aβ levels in txJ/J and control mice were therefore assessed by using an ELISA specific for murine forms of Aβ40 and Aβ42 (Fig. 5). Compared with control mice, txJ/J mice had lower CNS Aβ40 (P < 0.01; t test) and Aβ42 at 2 months of age (P < 0.05; t test). Differences present at 6 months of age did not reach significance. In sum, these data support an effect of the txJ mutation on levels of endogenous Aβ in the CNS of young mice.
Fig. 5.

Effect of txJ on endogenous mouse brain Aβ. (a) ELISAs specific for the murine forms of Aβ40 and Aβ42 demonstrated a significant reduction in Aβ40 (P < 0.01; t test) and Aβ42 (P < 0.05; t test) in 2-month-old txJ/J mice (hatched bars; n = 6 Aβ40; n = 5 Aβ42) compared with isogenic age-matched C3H controls (open bars; n = 5Aβ40; n = 6Aβ42). (b) Similar analysis of 6-month-old non-TgAPP mice from the TgCRND8/txJ crosses revealed nonsignificant reductions in mouse Aβ40 and Aβ42 in homozygous txJ mice. (APP–/txJ/+: open bars, n = 3 Aβ40; n = 3 Aβ42. APP–/txJ/J: n = 6 Aβ40; txJ, n = 6 Aβ42.)
Discussion
Cu and Aβ. Although previous in vitro studies of Aβ peptides and studies of postmortem tissue have implied a pro-pathogenic effect of Cu on Aβ assembly and toxicity (ref. 48 and references therein), our in vivo studies define an inverse relationship between Cu levels in the CNS and amyloid burden. First, CNS Cu levels in brains of TgCRND8 mice were lower than in non-Tg controls (Fig. 1), even though the Tg mice exhibit a substantial burden of dense-cored plaques and Aβ levels. Moreover, reductions of a similar magnitude were present in the independently derived Tg2576 and TgAPP23 lines of amyloid-depositing TgAPP mice (43, 49). Second, Aβ species measured by a variety of assays were lower in txJ/J mice than in age-matched controls. This was manifested by an ≈45% reduction in dense-cored plaques composed of human Aβ in APP+/txJ/J mice, by a tendency for reduction of human Aβ peptides in the CNS and reduced levels in the plasma, and by reduction of endogenous mouse Aβ40 and Aβ42 in the CNS of young txJ/J mice (Figs. 3, 4, 5). This reduction of amyloid plaque burden in txJ homozygotes was of the same magnitude as that seen with Aβ42-directed immunization (ref. 21; now a well established intervention in mouse models of CNS amyloidosis), as measured in the same TgCRND8 line of TgAPP mice. Furthermore, reduced Aβ levels were noted in experiments where Cu was increased in TgAPP23 mice by dietary intervention (49), speaking to the general nature of this effect.
Impact of the txJ Mutation on APP- and Aβ-Related Phenotypes. Analyses of txJ homozygous mice revealed beneficial effects on two phenotypic traits related to expression of the APP transgene, namely, survival and amyloid burden. Like other Tg mice overexpressing the APP695 form of APP (47, 50), TgCRND8 mice exhibit heightened mortality in the postnatal period, both before weaning and in the subsequent period up to 4 months of age (22). In our studies, genotypic ratios of TgCRND8 to non-Tg mice at weaning were improved in txJ homozygotes, and no deleterious effects of homozygosity for the txJ mutation upon genotypic ratios were observed at subsequent time points up to 6 months of age. Although the beneficial effects of Cu load may derive from normalization of deficits in CNS Cu and of Cu/Zn superoxide dismutase activity in aged mice (49, 51), we have noted inconsistencies between the susceptibility of inbred strains to the toxic effect of APP695 overexpression (47, 51) and baseline CNS Cu levels (data not presented). These inconsistencies suggest that additional mechanisms, mechanisms perhaps involving the metabolism of the Aβ peptide, may also have an impact on the survival of TgAPP695 mice.
Mechanism of Aβ Reduction in txJ Mice. The CuATPase7b transporter is a P-type ATPase associated with the trans-Golgi network. Although the ATPase7b transporter contains a Cu-selective domain (52), the txJ mutation in this protein is associated not only with an elevation of CNS Cu (up to 50%) but also with a 7% elevation in Zn. The question therefore arises as to whether elevated Zn levels contribute to lowering CNS Aβ. However, studies in vitro and of synaptic zinc in vivo strongly indicate a pro-amyloidogenic effect of this abundant metal (13, 15, 53), excluding this possibility.
How might elevated Cu modulate Aβ burden? Our data are compatible with mechanisms involving either reduced synthesis or, perhaps more likely, elevated clearance of Aβ. Although increases in α-secretase processing have been documented in Chinese hamster ovary cells exposed to heightened Cu (20), changes in the levels of sAPP were not clearly evident up to 6 months of age in the mice analyzed in this study. Although it is possible that the cumulative effect of a subtle increase in α-secretase processing in the CNS could contribute to a reduction in plaque burden, it seems more plausible that the main effect of txJ on Aβ proceeds by means of a fundamentally different mechanism. In this regard, an intriguing observation emerging from our analyses is that 2-month-old txJ/J mice, with no increases in sAPP or CNS Cu, have potent reductions in soluble forms of Aβ, namely, endogenous mouse Aβ40 and Aβ42. At this early time point the most striking property of txJ homozygote mice is the rise in peripheral levels of Cu and Zn. For example, in the liver, levels of Cu and Zn rise to 50× and 2.5× values in control animals, respectively.
Parenchymal, CSF, and plasma Aβ have been proposed to equilibrate, such that CNS-produced Aβ may make a strong contribution to net Aβ measured in the plasma (54, 55). Thus, the effect of the txJ mutation on CNS Aβ may be a consequence of changes in peripheral Aβ catabolism. Although plasma levels of endogenous mouse Aβ lay below the detection limit of our assay (data not presented), a significant reduction was noted for plasma levels of human Aβ40 and Aβ42 in APP+/txJ/J mice (Fig. 3e), establishing perturbed levels of Aβ in peripheral compartments. These data provide support for the hypothesis that effects of txJ on CNS amyloid burden may be a consequence of altered CNS/plasma equilibration driven by a strong reduction in pools of peripheral Aβ. Whether this effect on peripheral Aβ catabolism is mediated by an altered repertoire of metalloprotein or metalloprotease expression remains to be established.
Based on prior extrapolations from the effects of Cu and chelators on Aβ fibril assembly (19) and the ability of metal–Aβ complexes to generate free radical damage, the antibiotic and broad range Cu–Zn chelator clioquinol was administered to Tg2576 TgAPP mice (56). In 21-month-old mice, clioquinol treatment lowered the burden of the Aβ plaque surface area by ≈25%, lowered insoluble Aβ by 49%, and increased soluble forms of Aβ by ≈50%. Interestingly, these changes were accompanied by a 15% increase in soluble Cu (and Zn). Our studies, which increased Cu pools by way of the txJ mutation, also reveal an apparent “antipathogenic” effect on Aβ metabolism. However, because it is unclear whether changes of the CNS pools of Cu are relevant to this effect of txJ, our genetic studies raise questions about the anti-amyloidogenic mode of action (peripheral versus CNS) of Cu/Zn chelators. More broadly, besides improving our understanding of AD pathogenesis and risk factors, discerning the mechanism whereby txJ can modulate pools of Aβ may prove to be of practical use.
Acknowledgments
We thank Erwan Paitel, Paul Fraser, JoAnne McLaurin, and Rosemary Ahrens for discussions, and Marc Mercken (Johnson & Johnson Pharmaceutical Research and Development/Janssen Pharmaceutica, Beerse, Belgium) for anti-APP and anti-Aβ antibodies. This work was supported by the Alzheimer Society of Ontario (D.W.) and the National Institute on Aging (P.M.M. and R.A.N.). A.L.P. and D.W. were supported by Canadian Institutes for Health Research fellowships and Investigator Grants MFE41490 and MSC46763, respectively.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Aβ, amyloid-β; AD, Alzheimer's disease; APP, amyloid precursor protein; sAPP, secreted APP fragments; CSF, cerebrospinal fluid; LSD, least significant difference; Tg, transgenic.
References
- 1.Braak, E., Griffing, K., Arai, K., Bohl, J., Bratzke, H. & Braak, H. (1999) Eur. Arch. Psychiatry Clin. Neurosci. 249, Suppl. 3, 14–22. [DOI] [PubMed] [Google Scholar]
- 2.Klein, W. L. (2002) Neurochem. Int. 41, 345–352. [DOI] [PubMed] [Google Scholar]
- 3.Glabe, C. (2001) J. Mol. Neurosci. 17, 137–145. [DOI] [PubMed] [Google Scholar]
- 4.Plantin, L. O., Lying-Tunell, U. & Kristensson, K. (1987) Biol. Trace Elem. Res. 13, 69–75. [DOI] [PubMed] [Google Scholar]
- 5.Atwood, C. S., Huang, X., Moir, R. D., Tanzi, R. E. & Bush, A. I. (1999) Met. Ions Biol. Syst. 36, 309–364. [PubMed] [Google Scholar]
- 6.Bush, A. I. (2000) Curr. Opin. Chem. Biol. 4, 184–191. [DOI] [PubMed] [Google Scholar]
- 7.Ehmann, W. D., Markesbery, W. R., Alauddin, M., Hossain, T. I. & Brubaker, E. H. (1986) Neurotoxicology 7, 195–206. [PubMed] [Google Scholar]
- 8.Samudralwar, D. L., Diprete, C. C., Ni, B. F., Ehmann, W. D. & Markesbery, W. R. (1995) J. Neurol. Sci. 130, 139–145. [DOI] [PubMed] [Google Scholar]
- 9.Thompson, C. M., Markesbery, W. R., Ehmann, W. D., Mao, Y. X. & Vance, D. E. (1988) Neurotoxicology 9, 1–7. [PubMed] [Google Scholar]
- 10.Cornett, C. R., Markesbery, W. R. & Ehmann, W. D. (1998) Neurotoxicology 19, 339–345. [PubMed] [Google Scholar]
- 11.Multhaup, G., Schlicksupp, A., Hesse, L., Beher, D., Ruppert, T., Masters, C., L. & Beyreuther, K. (1996) Science 271, 1406–1409. [DOI] [PubMed] [Google Scholar]
- 12.Barnham, K. J., McKinstry, W. J., Multhaup, G., Galatis, D., Morton, C. J., Curtain, C. C., Williamson, N. W., White, A. R., Hinds, M. G., Norton, R. S., et al. (2003) J. Biol. Chem. 278, 17401–17407. [DOI] [PubMed] [Google Scholar]
- 13.Bush, A. I., Multhaup, G., Moir, R. D., Williamson, T. G., Small, D. H., Rumble, B., Pollwein, P., Beyreuther, K. & Masters, C. L. (1993) J. Biol. Chem. 268, 16109–16112. [PubMed] [Google Scholar]
- 14.Atwood, C. S., Moir, R. D., Huang, X., Scarpa, R. C., Bacarra, N. M., Romano, D. M., Hartshorn, M. A., Tanzi, R. E. & Bush, A. I. (1998) J. Biol. Chem. 273, 12817–12826. [DOI] [PubMed] [Google Scholar]
- 15.Yang, D. S., McLaurin, J., Qin, K., Westaway, D. & Fraser, P. E. (2000) Eur. J. Biochem. 267, 6692–6698. [DOI] [PubMed] [Google Scholar]
- 16.Bush, A. I., Pettingell, W. H., Jr., de Paradis, M., Tanzi, R. E. & Wasco, W. (1994) J. Biol. Chem. 269, 26618–26621. [PubMed] [Google Scholar]
- 17.Huang, X., Atwood, C. S., Moir, R. D., Hartshorn, M. A., Vonsattel, J. P., Tanzi, R. E. & Bush, A. I. (1997) J. Biol. Chem. 272, 26464–26470. [DOI] [PubMed] [Google Scholar]
- 18.Atwood, C. S., Scarpa, R. C., Huang, X., Moir, R. D., Jones, W. D., Fairlie, D. P., Tanzi, R. E. & Bush, A. I. (2000) J. Neurochem. 75, 1219–1233. [DOI] [PubMed] [Google Scholar]
- 19.Cherny, R. A., Legg, J. T., McLean, C. A., Fairlie, D. P., Huang, X., Atwood, C. S., Beyreuther, K., Tanzi, R. E., Masters, C. L. & Bush, A. I. (1999) J. Biol. Chem. 274, 23223–23228. [DOI] [PubMed] [Google Scholar]
- 20.Borchardt, T., Camakaris, J., Cappai, R., Masters, C. L., Beyreuther, K. & Multhaup, G. (1999) Biochem. J. 344, Part 2, 461–467. [PMC free article] [PubMed] [Google Scholar]
- 21.Janus, C., Pearson, J., McLaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., Chishti, M. A., Horne, P., Heslin, D., French, J., et al. (2000) Nature 408, 979–982. [DOI] [PubMed] [Google Scholar]
- 22.Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., Strome, R., Zuker, N., Loukides, J., French, J., et al. (2001) J. Biol. Chem. 276, 21562–21570. [DOI] [PubMed] [Google Scholar]
- 23.Cox, D. W. & Roberts, E. A. (1998) in Sleisenger and Fordtran's Gastrointestinal and Liver Disease, eds. Feldman, M., Schlarschmidt, B. F. & Sleisenger, M. H. (Saunders, Philadelphia), pp. 1104–1111.
- 24.Hung, I. H., Suzuki, M., Yamaguchi, Y., Yuan, D. S., Klausner, R. D. & Gitlin, J. D. (1997) J. Biol. Chem. 272, 21461–21466. [DOI] [PubMed] [Google Scholar]
- 25.Forbes, J. R. & Cox, D. W. (2000) Hum. Mol. Genet. 9, 1927–1935. [DOI] [PubMed] [Google Scholar]
- 26.Voskoboinik, I., Greenough, M., La Fontaine, S., Mercer, J. F. & Camakaris, J. (2001) Biochem. Biophys. Res. Commun. 281, 966–970. [DOI] [PubMed] [Google Scholar]
- 27.Weisner, B., Hartard, C. & Dieu, C. (1987) J. Neurol. Sci. 79, 229–237. [DOI] [PubMed] [Google Scholar]
- 28.Kodama, H., Okabe, I., Yanagisawa, M., Nomiyama, H., Nomiyama, K., Nose, O. & Kamoshita, S. (1988) Pediatr. Neurol. 4, 35–37. [DOI] [PubMed] [Google Scholar]
- 29.Theophilos, M. B., Cox, D. W. & Mercer, J. F. (1996) Hum. Mol. Genet. 5, 1619–1624. [DOI] [PubMed] [Google Scholar]
- 30.La Fontaine, S., Theophilos, M. B., Firth, S. D., Gould, R., Parton, R. G. & Mercer, J. F. (2001) Hum. Mol. Genet. 10, 361–370. [DOI] [PubMed] [Google Scholar]
- 31.Coronado, V., Nanji, M. & Cox, D. W. (2001) Mamm. Genome 12, 793–795. [DOI] [PubMed] [Google Scholar]
- 32.Bronson, R. T., Sweet, H. O. & Davisson, M. T. (1995) Mouse Genome 93, 152–154. [Google Scholar]
- 33.Scott, M., Foster, D., Mirenda, C., Serban, D., Coufal, F., Wälchli, M., Torchia, M., Groth, D., Carlson, G., DeArmond, S. J., et al. (1989) Cell 59, 847–857. [DOI] [PubMed] [Google Scholar]
- 34.Citron, M., Westaway, D., Xia, W., Carlson, G. A., Diehl, T., Levesque, G., Johnson-Wood, K., Lee, M., Seubert, P., Davis, A., et al. (1997) Nat. Med. 3, 67–72. [DOI] [PubMed] [Google Scholar]
- 35.Lugowski, S., Smith, D. C. & Van Loon, J. C. (1990) Clin. Mater. 6, 91–104. [Google Scholar]
- 36.Lugowski, S. J., Smith, D. C., McHugh, A. D. & Van Loon, J. C. (1991) J. Biomed. Mater. Res. 25, 1443–1458. [DOI] [PubMed] [Google Scholar]
- 37.Lindquist, R. R. (1969) Arch. Pathol. 87, 370–379. [PubMed] [Google Scholar]
- 38.Mouton, P. R. (2000) The Stereologer Handbook (Systems Planning and Analysis, Alexandria, VA).
- 39.Paxinos, G. & Frankin, K. B. J. (2001) The Mouse Brain in Stereotaxic Coordinates (Academic, San Diego).
- 40.Rozmahel, R., Huang, J., Chen, F., Liang, Y., Nguyen, V., Ikeda, M., Levesque, G., Yu, G., Nishimura, M., Mathews, P., et al. (2002) Neurobiol. Aging 23, 187–194. [DOI] [PubMed] [Google Scholar]
- 41.Savage, M. J., Trusko, S. P., Howland, D. S., Pinsker, L. R., Mistretta, S., Reaume, A. G., Greenberg, B. D., Siman, R. & Scott, R. W. (1998) J. Neurosci. 18, 1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathews, P. M., Jiang, Y., Schmidt, S. D., Grbovic, O. M., Mercken, M. & Nixon, R. A. (2002) J. Biol. Chem. 277, 36415–36424. [DOI] [PubMed] [Google Scholar]
- 43.Maynard, C. J., Cappai, R., Volitakis, I., Cherny, R. A., White, A. R., Beyreuther, K., Masters, C. L., Bush, A. I. & Li, Q. X. (2002) J. Biol. Chem. 277, 44670–44676. [DOI] [PubMed] [Google Scholar]
- 44.Rauch, H. (1983) J. Hered. 74, 141–144. [DOI] [PubMed] [Google Scholar]
- 45.Howell, J. M. & Mercer, J. F. (1994) J. Comp. Pathol. 110, 37–47. [DOI] [PubMed] [Google Scholar]
- 46.Biempica, L., Rauch, H., Quintana, N. & Sternlieb, I. (1988) Lab. Invest. 59, 500–508. [PubMed] [Google Scholar]
- 47.Hsiao, K. K., Borchelt, D. R., Olson, K., Johannsdottir, R., Kitt, C., Yunis, W., Xu, S., Eckman, C., Younkin, S., Price, D., et al. (1995) Neuron 15, 1203–1218. [DOI] [PubMed] [Google Scholar]
- 48.Bush, A. I. (2002) Neurobiol. Aging 23, 1031–1038. [DOI] [PubMed] [Google Scholar]
- 49.Bayer, T., Schäfer, S., Simons, A., Kemmling, A., Kamer, T., Eckert, A., Schüssel, K., Eikenberg, O., Sturchler-Pierrat, C., Abramowski, D., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 14187–14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moechars, D., Dewachter, I., Lorent, K., Reverse, D., Baekelandt, V., Naidu, A., Tesseur, I., Spittaels, K., Haute, C. V., Checler, F., et al. (1999) J. Biol. Chem. 274, 6483–6492. [DOI] [PubMed] [Google Scholar]
- 51.Carlson, G. A., Borchelt, D. R., Dake, A., Turner, S., Danielson, V., Coffin, J. D., Eckman, C., Meiners, J., Nilsen, S. P., Younkin, S. G. & Hsiao, K. K. (1997) Hum. Mol. Genet. 6, 1951–1959. [DOI] [PubMed] [Google Scholar]
- 52.Lutsenko, S., Petrukhin, K., Cooper, M. J., Gilliam, C. T. & Kaplan, J. H. (1997) J. Biol. Chem. 272, 18939–18944. [DOI] [PubMed] [Google Scholar]
- 53.Lee, J. Y., Cole, T. B., Palmiter, R. D., Suh, S. W. & Koh, J. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeMattos, R. B., Bales, K. R., Cummins, D. J., Dodart, J. C., Paul, S. M. & Holtzman, D. M. (2001) Proc. Natl. Acad. Sci. USA 98, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeMattos, R. B., Bales, K. R., Parsadanian, M., O'Dell, M. A., Foss, E. M., Paul, S. M. & Holtzman, D. M. (2002) J. Neurochem. 81, 229–236. [DOI] [PubMed] [Google Scholar]
- 56.Cherny, R. A., Atwood, C. S., Xilinas, M. E., Gray, D. N., Jones, W. D., McLean, C. A., Barnham, K. J., Volitakis, I., Fraser, F. W., Kim, Y., et al. (2001) Neuron 30, 665–676. [DOI] [PubMed] [Google Scholar]