

Abstract
The purpose of this study was to evaluate the ability of novel semiselective matrix metalloproteinase inhibitors (MMPI) to protect myocardial structure–function in the setting of ischemia–reperfusion injury. For this purpose, an isolated rat model of myocardial stunning and infarction was used. Isolated hearts were subjected to 20–30 minutes of global no-flow ischemia and 30-minute reperfusion. Myocardial performance was assessed as the product of the heart rate and left ventricular developed pressure (rate–pressure product, RPP). Coronary flow rates, ventricular weights, indicators of muscle (troponin I), and fibrillar collagen damage (collagen opalation) were measured. Four MMPI were tested: 2 non-hydroxamate, semiselective inhibitors (PY-2 and 1,2-HOPO-2) and 2 broad-spectrum inhibitors (PD166793 and CGS27023A). The non-hydroxamate, semiselective inhibitors were shown to be nontoxic in cocultures of cardiac cells. Results indicate that semiselective inhibitors (in particular 1,2-HOPO-2) yield improved cardiac performance (~23% higher RPP vs. controls) and coronary flow rates (~22%), reducing muscle (~25%) and fibrillar collagen damage (~60%). Evidence suggests the involvement of matrix metalloproteinase-2 in these actions. Interestingly, broad-spectrum inhibitors only show modest improvement (~8% higher RPP vs. controls) without affecting the other measured parameters. In conclusion, semiselective MMPI can act as cardioprotectors in isolated perfused rat hearts. Protection is observed in all structural components of the myocardium translating into improved contractile function. Based on these findings, non-hydroxamate, semiselective MMPI warrant further studies as to their ability to protect ischemic myocardium in the in vivo setting.
Keywords: stunning, infarction, extracellular matrix, collagen, proteases
INTRODUCTION
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that have been shown to play a significant role in a number of physiological and pathophysiological processes. MMPs are best known for their ability to proteolyse components of the extracellular matrix such as collagen, fibronectin, and laminin. However, MMPs have recently been reported to also target sarcomeric and cytoskeletal proteins found inside cardiac cells such as troponin I, myosin light chain-1, and α-actinin.1–6 MMPs can also degrade components of the vascular compartment.3 MMPs have been well characterized for their participation in the pathophysiology of chronic diseases such as heart failure and cancer.3,7–12 However, less is known about their involvement in the mediation of acute injury such as seen with ischemia–reperfusion. MMP subtypes that may be involved in mediating damage to cardiac tissues remains to be determined. MMPs have also been postulated to play “protective” roles in that they may be necessary to facilitate a positive outcome after injury.7,9,11,13,14 The myocardium can be subdivided into vascular, pacemaker/electrical, muscle, and extracellular matrix compartments, and the disruption of any of these compartments can lead to a physiological imbalance.15,16 However, MMP subtypes implicated in damage to these individual compartments in the setting of ischemia–reperfusion injury are not well understood.
Many pharmaceutical companies developed programs to synthesize inhibitors of these enzymes.11,13,17 Multiple compounds with nanomolar inhibition profiles were developed.12,18 Some of these MMP inhibitors (MMPI) possess broad-spectrum inhibitory profiles, whereas others have more semiselective profiles for the inhibition of MMPs (ie, a compound that has an inhibitory profile between that of a broad-spectrum and a truly selective inhibitor).12,13,17,18 As the means to inhibit MMPs, most compounds relied on the use of either a hydroxamate or a carboxylate metal binding group to chelate the catalytic zinc(II) ion.11,17,19,20 MMPI failed clinical trials as they were hampered by toxicity and poor oral bioavailability.12,17,20,21 Nonetheless, hydroxamate- and carboxylate-based MMPI are still used for experimental purposes.22
Our laboratories have developed a new family of MMPI based on non-hydroxamate zinc-binding groups (ZBGs).23 These ZBGs have been previously shown to yield low toxicity profiles when tested in cultured cardiac cells.23 The ZBGs were also used to develop full-length MMPI with semiselective profiles.20 The availability of new inhibitors based on these ZBGs would allow us to inquire into their ability to act as cardioprotectors and compare them with known, potent, broad-spectrum hydroxamate/carboxylate-based inhibitors. The utilization of compounds with unique inhibitory profiles may also allow us to better identify the role that MMP subtypes have in mediating damage to myocardial compartments and thus impairing cardiac performance.
In isolated perfused heart preparations, myocardial stunning can be generated after 20–25 minutes of global no-flow ischemia (GNFI) followed by reperfusion.3,24,25 If the period of ischemia is extended to ≥30 minutes, irreversible functional impairment occurs as cells undergo necrosis (ie, infarction).3,25–30 The exact mechanism responsible for myocardial stunning is yet to be fully elucidated, and the role of MMPs and their subtypes has not been systematically examined. With these issues in mind, using an isolated rat heart model of ischemia–reperfusion-induced injury and a select group of broad-spectrum and semiselective MMPI, we pursued the following goals: (1) to assess cardiac damage caused by MMPs during ischemia–reperfusion injury, (2) to identify the role exerted by MMP subtypes in causing cardiac damage, and (3) to contrast the protective effects exerted by novel semiselective versus broad-spectrum MMPI.
MATERIALS AND METHODS
Reagents
The chemical structures of the MMPI used in this study are shown in Figure 1. Our studies included 2 semiselective MMPI with different ZBGs: PY-2, a hydroxypyrone; and 1,2-HOPO-2, a hydroxypyridinone.20 Two broad-spectrum MMPI utilizing different ZBGs: CGS27023A, a hydroxamate18; and PD166793, a carboxylate.12 A negative control was also employed: PICO-2 (an MMPI-like hydroxypyridine molecule devoid of a functional ZBG, rendering it inactive).20 PD166793 was purchased from Tocris Bioscience (Ellisville, MO). CGS27023A was synthesized in our laboratory and its purity (>99%) verified by nuclear magnetic resonance and combustion analysis. Minocycline was obtained from Sigma–Aldrich (St Louis, MO). Recombinant human MMP-1, -2, -3, -7, -8, -9, -12, and -13 catalytic domains were obtained from BIOMOL International (Plymouth Meeting, PA).
FIGURE 1.

Structures of MMPI. Structures of the different compounds used in our studies. PY-2 and 1,2-HOPO-2 represent novel semiselective MMPI. PY-2 is a hydroxypyrone and 1,2-HOPO-2 is a hydroxypyridinone. PICO-2, the negative control, is a hydroxypyridine molecule bearing a nonfunctional ZBG. CGS27023A is a hydroxamate-based broad-spectrum MMPI, and PD166793 is a carboxylate-based broad-spectrum MMPI.
Recombinant MMP Assays
To determine IC50 values for the compounds, the MMP inhibitory activities of recombinant human MMP-1, -2, -3, -7, -8, -9, -12, and -13 catalytic domains were measured utilizing a 96-well microplate fluorescent assay kit (BIOMOL International), following the procedure provided with the kit. Fluorescence was measured using a BioTek FLx 800 microplate reader (BioTek, Winooski, VT) and white 96-well plates (Nalgene Nunc, Rochester, NY). Compounds were dissolved in dimethyl sulfoxide (DMSO), and the final concentration of DMSO was kept at 0.005% in assay buffer [50 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid, 10 mM CaCl2, 0.05% Brij-35, pH 7.5]. MMPs were incubated individually with varying concentrations of different inhibitors for 1 hour at 37°C, followed by addition of substrate to initiate the assay. Reactions were agitated by shaking for 1 second after each fluorescence measurement. Generated fluorescence (λex = 335 nm, λem = 405 nm) was measured at 60-second intervals for 30 minutes. Substrate cleavage rates were determined from the linear regions of kinetic curves. IC50 was determined from graphs of MMP activity versus log [MMPI]. The small molecule chelator o-phenanthroline was used to confirm that substrate cleavage was attributable to MMP activity.
Cardiac Cell Preparations and Cell Viability Assays
Neonatal rat ventricular fibroblasts and myocytes were prepared as previously described.31 Briefly, cells were prepared from hearts of 1- to 2-day old Sprague-Dawley rats (Harlan, Indianapolis, IN). After collagenase digestions (4 times), myocyte and nonmyocyte cells were isolated by Percoll density gradient. Equal numbers of fibroblasts and myocytes were cocultured in 96-well cell culture plates at 37°C in 5% CO2 in Dulbecco Modified Eagle Medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum, 100 U/mL penicillin G, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Invitrogen). When reaching 90% confluence, cells were serum deprived for 24 hours in Dulbecco Modified Eagle Medium, 100 U/mL penicillin G, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B, and treated according to the experimental design.
To assess MMPI cytotoxicity, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, MTT (Sigma–Aldrich) cell viability assays were performed. Tetrazolium salts such as MTT are metabolized by mitochondrial dehydrogenases to form a blue formazan dye, providing a colorimetric signal in response to viable mitochondria. Cardiac fibroblast/myocyte cocultures were incubated with variable concentrations of MMPI (0.01–1000 μM) for 24 hours. After incubation, the cell medium was removed, and cells were rinsed 3 times with fresh serum-free medium to remove excess MMPI, which could interfere with the MTT reaction. Cells were lysed and combined with MTT in medium for 2 hours. After solubilization of the MTT formazan crystals, absorbance at 570 nm was measured using a microplate reader Bio-Tek μ-Quant microplate spectrophotometer (Bio-Tek, Winooski, VT), and the percentage viability relative to untreated cardiac fibroblast/myocyte cocultures was calculated from triplicates. To further assess cell viability, the morphology of cells was examined before assaying by MTT.
Determination of Antioxidant Activity for MMPI
Antioxidant activity for the MMPI was measured using an in vitro Antioxidant Assay Kit (Cayman Chemical, Ann Arbor, MI) following the kit instructions. The assay relies on the ability of antioxidants in the sample to inhibit the oxidation of 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate] (ABTS) by metmyoglobin in comparison with Trolox, a water-soluble tocopherol analog. Minocycline (Sigma–Aldrich) was also used as a representative of a known antioxidant. The amount of oxidized ABTS generated was monitored by reading the absorbance at 750 nm. All compounds were initially dissolved in DMSO and subsequently in water. Same concentrations as indicated for Trolox were used for the different compounds. Experiments were performed in duplicate using a microplate reader.
Langendorff Heart Perfusion and Ischemia–Reperfusion Protocols
The protocols were approved by the University of California, San Diego Institutional Animal Care and Use Committee, and conform to the published National Institutes of Health guidelines for animal research. To determine the capacity of the MMPI to induce organ toxicity and/or cardio-protection, we used our ex vivo model of isolated stunned rat hearts.20 MMPI were solubilized in DMSO at 0.1 M. Initial dose–response studies were performed to identify optimal dosage to be used. Male Sprague-Dawley rats (250–300 g) (Harlan) were used for the experiments. Hearts were rapidly excised from ketamine-anesthetized animals and briefly rinsed by immersion in ice-cold Krebs–Henseleit buffer. Hearts were retrogradely perfused via the aorta at a constant pressure of 60 mm Hg with nonrecirculating Krebs–Henseleit buffer at 37°C. The composition of the buffer (mM) was NaCl (118), KCl (4.7), KH2PO4 (1.2), MgSO4 (1.2), CaCl2 (2.5–3.0), NaHCO3 (25), glucose (11), and EDTA (0.5). The buffer was continuously gassed with 95% O2/5% CO2 (pH 7.4). Spontaneously beating hearts were used in all experiments. A water-filled cling-wrap balloon connected to a pressure transducer was inserted into the left ventricle through an incision in the left atrium and through the mitral valve, and the volume was adjusted to achieve an end diastolic pressure of 8–12 mm Hg. A water-jacketed glass chamber was positioned around the heart to maintain its temperature at 37°C. Stock solutions of the various MMPI were initially solubilized in DMSO and subsequently diluted at the desired MMPI concentrations with Krebs–Henseleit buffer. The MMPI were provided to the isolated beating hearts during both the stabilization and reperfusion periods (ie, throughout the experiment). The final concentration of DMSO used in Krebs–Henseleit buffer was 0.005%. Control hearts were perfused with Krebs–Henseleit buffer containing 0.005% DMSO. The time interval between thoracotomy and attachment of the heart to the perfusion system for initiation of the stabilization period was <1 minute.
After 10 minutes of aerobic perfusion to achieve steady-state conditions, hearts were subjected to 20–25 minutes of GNFI, or in select cases 30-minute GNFI, by clamping of the aortic inflow line. This was followed by 30 minutes of aerobic reperfusion as the clamp was removed. Using 20- to 25-minute GNFI, hearts developed stunning conditions during reperfusion, and with 30-minute GNFI, hearts underwent infarction. In our hands, stunning is defined by the recovery of contractile function in hearts to ~50% of that observed during the stabilization period; when recovery of contractile function reached ≤30%, it was considered equivalent to infarction. Infarction was verified by staining of fresh equatorial slices from perfused hearts with 1% wt/vol 2,3,5-triphenyltetrazolium chloride (Sigma–Aldrich) in phosphate buffer, pH 7.4, for 20 minutes at 37°C. Contrasting white (necrotic)/red (viable) staining was only discernible in infarcted hearts.
Analysis of Cardiac Work and Vascular Function
Heart rate and left ventricular pressure were monitored on a polygraph and recorded on a computer system for subsequent analysis. Left ventricular developed pressure was calculated as the difference between left ventricular peak systolic and end diastolic pressures of the left ventricular pressure trace. The work index, rate–pressure product (RPP), was calculated as follows: (heart rate) × (left ventricular developed pressures). To assess vascular performance, coronary flow rate (mL/min) was measured by sampling perfusate with a graduated cylinder during reperfusion at 5-minute intervals, and compared with the coronary flow rate during baseline perfusion, 5 minutes before ischemia. At the end of perfusion, hearts were collected, the remaining atria and aorta were trimmed, and excess buffer was blotted. Ventricles were rapidly frozen in dry ice/methanol bath and weighed to determine wet weight. Ventricles were desiccated at 80°C for 24 hours and reweighed to determine dry weights. Edema was calculated as the wet to dry weight ratio.25,32
Assessment of Muscle Damage
Cardiac troponin I (cTnI) release was measured in coronary effluent as a molecular marker for sarcomeric (intracellular) damage to cardiomyocytes. Five-minute reperfusion buffer was analyzed for cTnI release using a rat cTnI enzyme-linked immunosorbent assay kit (Life Diagnostics Inc, West Chester, PA). As a biochemical marker for cell damage, creatine kinase activity was also measured in coronary effluent. For each heart, 0.5 mL of coronary effluent was collected every 5 minutes throughout the experiment. Creatine kinase activity was determined using a colorimetric assay creatine kinase reagent (Pointe Scientific Inc, Canton, MI) and expressed as the Vmax of the kinetic reaction in mean optical density per minute (mOD/min). Values were compared at specific time points (0- to 30-minute reperfusion) or as total creatine kinase released (area under the curve).
Assessments of Collagen Damage
To assess the integrity of the myocardial extracellular matrix, we measured collagen degradation using the fluorescent labeling reagent o-phthaldialdehyde. Left ventricle samples from perfused hearts were used. Myocardial collagen was extracted using sequential ethanol/salt precipitations as described previously.33 Collagen samples isolated from rat heart ventricles (100 mg each) were used for o-phthaldialdehyde conjugation to exposed amino termini (“opalation”) as described previously.33 Collagen content was calculated under the assumption that the average hydroxyproline content of collagen is 10%, and thus 1 μmol of hydroxyproline is equivalent to 1 mg of collagen. Collagen content in each tissue sample was expressed as the percentage of the total dry weight of sample. Values are reported as arbitrary fluorescent units (AFU).
Quantification of MMPs in Coronary Effluents
To determine the release of MMPs from sham and stunned hearts, we examined the presence of MMP-2, -3, and -13 in coronary effluents. Perfusate (1 mL) collected within 1-minute reperfusion (stunned hearts) or at 35-minute baseline perfusion (sham hearts) was used directly to quantitate the MMP of interest using Quantikine Immunoassay kits (R&D Systems, Minneapolis, MN): Human/mouse/rat MMP-2 (total), mouse MMP-3 (total), and human Pro-MMP-13. MMP quantifications were performed according to the manufacturer’s instructions.
Statistical Analysis
Results are expressed as mean ± SEM. Comparisons between means were analyzed, as appropriate, by Student t test or 1-way or 2-way analysis of variance followed by Bonferroni t test. Values in Table 1 are expressed as percentages of the maximum value found within the same experimental set of hearts. A P value of <0.05 was considered statistically significant.
TABLE 1.
Effects of MMPI on Ventricular Wet Weights and Troponin Coronary Effluent Values as Measured by Enzyme-linked Immunosorbent Assay in Untreated (Control) Versus Treated Hearts
| Ventricular Weight (mg) |
Troponin I Release (ng/mL) |
|||
|---|---|---|---|---|
| MMPI | Control (n = 5–6) | Treatment (n = 5–10) | Control (n = 5–6) | Treatment (n = 5–10) |
| PY-2 | 80.6 ± 1.4 | 85.3 ± 2.6 | 75.15 ± 7.2 | 55.55 ± 7.2# |
| 1,2-HOPO-2 | 71.8 ± 7.1 | 58.7 ± 2.1* | 78.76 ± 7.6 | 58.24 ± 5.2* |
| CGS27023A | 91 ± 3.5 | 87.9 ± 2.2 | 40.12 ± 14 | 47.20 ± 7.1 |
| PD166793 | 93.5 ± 2.1 | 93.7 ± 1.3 | 61.86 ± 15.8 | 42.16 ± 11.1 |
| PICO-2 | 85.8 ± 3.8 | 82 ± 2.2 | 38.23 ± 16.6 | 27.18 ± 4.1 |
Troponin values were assessed in paired experiments between treated hearts and their respective controls.
P < 0.05,
P = 0.06 by t test.
RESULTS
MMPI IC50 Profiles
Table 2 shows the MMP inhibitory profiles determined for the different compounds used in the studies described below. Broad-spectrum MMPI display higher potency (ie, lower IC50 values) versus all 8 MMPs tested compared to IC50 values obtained from semiselective MMPI. The broad-spectrum MMPI CGS27023A demonstrated IC50 values of 0.5 μM or lower for all MMPs tested. The IC50 values for PD166793 were 3 μM (MMP-1, -7, -9) or better. Semi-selective MMPI displayed lower IC50 values for MMP-3, -8, and -12 versus MMP-2 and -13. As expected, the control compound PICO-2 failed to inhibit MMPs at high micromolar concentrations. The IC50 values for PICO-2 were all greater than 50 μM.
TABLE 2.
IC50 Values (μM) Determined for MMPI
| Semiselective |
Broad-Spectrum |
|||
|---|---|---|---|---|
| MMP | PY-2 | 1,2-HOPO-2 | CGS27023A | PD166793 |
| 1 | >50 | >50 | <0.05 | 3 |
| 2 | 4.4 | 0.92 | <0.05 | <0.05 |
| 3 | 0.077 | 0.56 | <0.05 | <0.05 |
| 7 | >50 | >50 | 0.5 | 3 |
| 8 | 0.248 | 0.086 | <0.05 | <0.05 |
| 9 | 32.3 | 27.1 | <0.05 | 3 |
| 12 | 0.085 | 0.018 | <0.05 | <0.05 |
| 13 | 6.6 | 4.1 | <0.05 | <0.05 |
MMPI and Cytotoxicity
Cytotoxicity MTT assay results for MMPI are shown in Figure 2. At concentrations ≤10 μM, the MMPI tested did not compromise cardiac cell viability in vitro. Microscopic inspection revealed considerable precipitation of the compounds at concentrations of 100 μM or greater where cell viability (ie, morphology) was compromised (data not shown).
FIGURE 2.
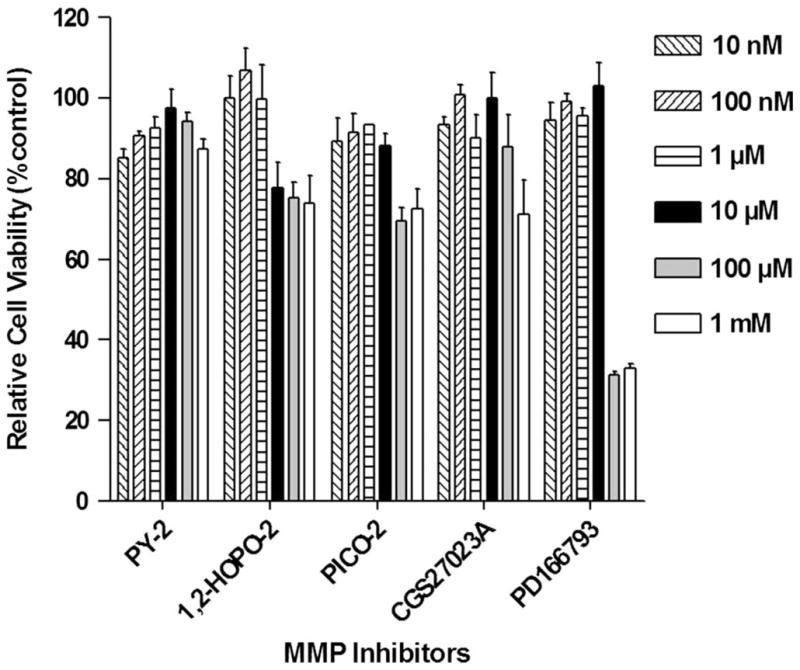
Cytotoxicity of MMPI. 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTT) cell viability assay results in cocultured cardiac fibroblasts and myocytes. Data indicate that all MMPI provided at concentrations ≤10 μM (black bar) sustain cell viability close to 100%.
MMPI Lack Antioxidant Potential
As shown in Figure 3, results indicate that none of the MMPI tested suppressed the oxidation of ABTS compared with the tocopherol analog Trolox or minocycline at equivalent concentrations (44, 135, 225 μM). These results rule out suppression of oxidative stress as a cardioprotective mode of action for these compounds.
FIGURE 3.

Antioxidant potential of MMPI. Oxidation of ABTS by metmyoglobin in the presence of MMPI compounds. In vitro assays indicate that MMPI lack antioxidant activity compared with that shown by the known antioxidants Trolox and minocycline at equivalent concentrations.
Effects of MMPI on Cardiac Hemodynamics
A group of control hearts (n = 51) was used to simulate ex vivo conditions of stunning, and an additional control group (n = 8) was used to simulate infarction. For each group of hearts, heart rate, left ventricular end diastolic pressure, and peak systolic pressure were compared. Heart rate was not affected by the different MMPI. Differences in the predisposition to arrhythmias or fibrillation were not observed. Interestingly, 5 μM 1,2-HOPO-2 was the only MMPI that did demonstrate a significant rise in PSP (data not shown).
MMPI conferred different levels of recovery and preservation of contractile function as indicated by the cardiac work index, RPP. PY-2 at 5 μM (Fig. 4A) yielded ~9% improvement (from 50% in controls to 59% in treated) in RPP versus controls at 30-minute reperfusion, whereas PY-2 at 10 μM (Fig. 4B) significantly increased improvement to ~19% under same conditions. The difference in RPP between PY-2 at 5 and 10 μM was significant (P = 0.045). 1,2-HOPO-2 at 5 μM (Fig. 5) yielded the highest recovery and preservation of contractile function among all inhibitors tested (~23% improvement in RPP versus controls at 30-minute reperfusion, P = 0.0001).
FIGURE 4.

Effects of 5 μM (A) and 10 μM (B) PY-2 on isolated, perfused rat hearts subjected to 20 minutes of GNFI and 30-minute reperfusion. RPP results indicate that under conditions of cardiac stunning, an increase in PY-2 from 5 to 10 μM enhances the recovery of contractile function versus the corresponding controls throughout reperfusion. ***P < 0.0001 by 2-way analysis of variance.
FIGURE 5.

Effect of 5 μM 1,2-HOPO-2 on isolated, perfused rat hearts subjected to 20 minutes of GNFI and 30-minute reperfusion. In stunned hearts, treatment with 1,2-HOPO-2 conferred an improved recovery of contractile function versus controls throughout reperfusion. ***P < 0.0001 by 2-way analysis of variance.
Broad-spectrum MMPI conferred a more modest but significant recovery in RPP. CGS27023A at 5 μM (Fig. 6A) and PD166793 at 5 μM (Fig. 6B) displayed comparable ~8% improvements in RPP versus controls at 30-minute reperfusion. This is notable in light of the much greater potency of these compounds against all MMPs tested (Table 2). As expected, 5 μM PICO-2 yielded no significant improvements in RPP versus controls (Fig. 7). Because 5 μM 1,2-HOPO-2 conferred higher levels of improved recovery, additional approaches were pursued to test this compound. In the isolated rat heart, the time of ischemia was increased to 30 minutes in order to simulate infarction conditions. In infarcted hearts, 5 μM 1,2-HOPO-2 (Fig. 8) yielded significantly improved recovery of ~17% in RPP versus controls (from 28% in controls to 45% in treated) at 30-minute reperfusion.
FIGURE 6.
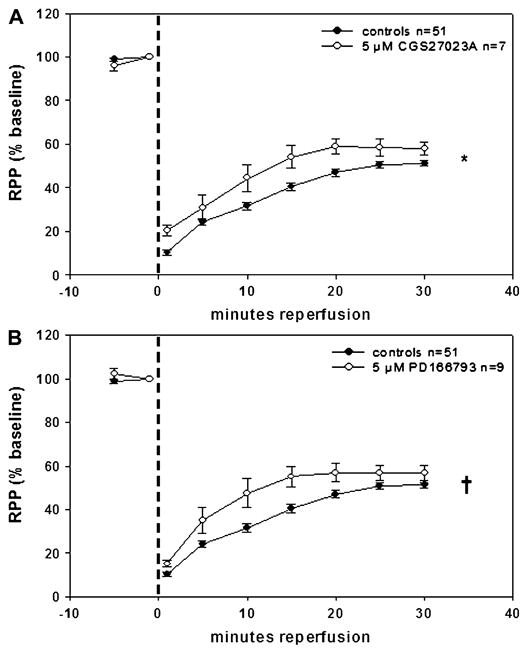
Effects of 5 μM CGS27023A (A) and 5 μM PD166793 (B) on isolated, perfused rat hearts subjected to 20 minutes of GNFI and 30-minute reperfusion. Broad-spectrum MMPI provided a modest but significant recovery in contractile function of stunned hearts versus controls during reperfusion. *P = 0.0484 and †P = 0.0464 by 2-way analysis of variance.
FIGURE 7.
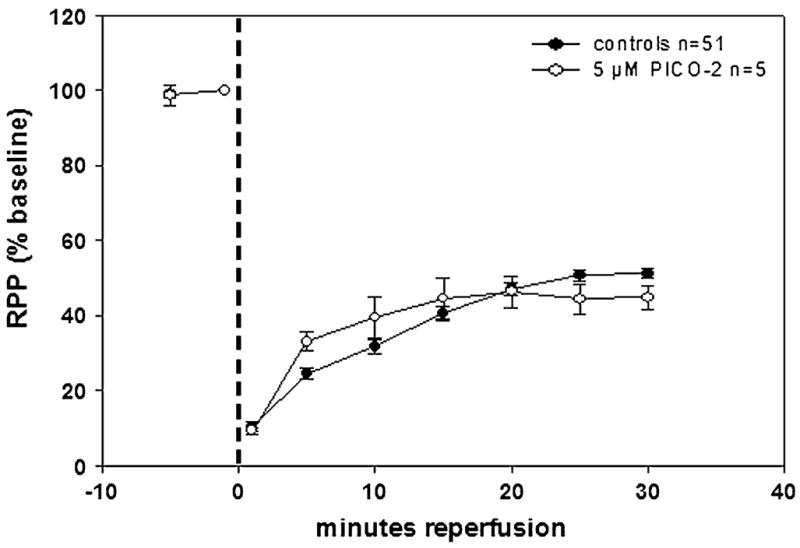
Effect of 5 μM PICO-2 on isolated, perfused rat hearts subjected to 20 minutes of GNFI and 30-minute reperfusion. The negative control compound, devoid of a functional ZBG, failed to improve contractility for stunned hearts during reperfusion.
FIGURE 8.

Effect of 5 μM 1,2-HOPO-2 on isolated, perfused rat hearts subjected to 30 minutes of GNFI and 30-minute reperfusion. Under a more severe type of ischemia–reperfusion injury (infarction), 5 μM 1,2-HOPO-2 still preserves contractile function during reperfusion. **P = 0.002 by 2-way analysis of variance.
Effects of MMPI on Vascular Function
When MMPI were provided at the same concentration (5 μM), perfusion of stunned hearts with the semiselective MMPI (PY-2 and 1,2-HOPO-2) yielded higher coronary flow rates versus controls (Figs. 9A, B). Perfusion with the broad-spectrum MMPI (CGS27023A and PD166793) did not improve coronary flow rates after ischemia (Figs. 9C, D). The negative control PICO-2 also failed to improve coronary flow rates (data not shown). 1,2-HOPO-2 at 5 μM when tested under conditions of infarction also yielded higher coronary flow rates versus controls (data not shown).
FIGURE 9.

Effect of MMPI on vascular function in stunned hearts. Semiselective MMPI (PY-2 and 1,2-HOPO-2) preserved and improved coronary flow rates during reperfusion versus controls. In contrast, the broad-spectrum MMPI (CGS27023A and PD166793) failed to improve coronary flow rates during reperfusion versus controls. **P < 0.01, #P = 0.06 by t test.
Only stunned hearts perfused with 5 μM 1,2-HOPO-2 (n = 5) displayed a significant reduction of ~15% in ventricular wet weight versus controls (n = 4) at the end of reperfusion (700 ± 34 vs. 820 ± 72 mg, P = 0.01). Furthermore, stunned hearts perfused with 5 μM 1,2-HOPO-2 were also analyzed for wet to dry weight ratios and compared with controls. Results indicate a significant reduction of ~10% in myocardial edema versus controls (4.9 ± 0.02 vs. 5.4 ± 0.21, P = 0.001).
Effects of MMPI on Muscle Damage
Hearts perfused with the semiselective MMPI 1,2-HOPO-2 displayed significant reductions in cTnI release versus controls after ischemia, and PY-2 showed a similar trend (P = 0.06) (Table 1). Perfusion with the broad-spectrum MMPI CGS27023A and PD166793 yielded no significant changes in cTnI release (Table 1). cTnI release was not significantly different from controls when perfused with the negative control PICO-2 (Table 1). In hearts subjected to infarction (30-minute global ischemia), 5 μM 1,2-HOPO-2 significantly reduced troponin I release (data not shown). Creatine kinase activity, when measured at specific time points or as a total area under the curve (ie, total amount of damage), displayed no significant variations in the different MMPI-treated hearts versus controls (data not shown).
Effects of MMPI on Collagen Degradation
Results indicate that in hearts subjected to 30-minute global ischemia (infarction), damage occurred to the extracellular matrix, which was revealed by opalation assays. Collagen degradation was reduced by ~60% in hearts perfused with 5 μM 1,2-HOPO-2 (n = 3) versus controls (n = 3) (0.56 ± 0.29 vs. 1.53 ± 0.66 AFU, P = 0.056). Tissue from stunned hearts (20-minute GNFI) perfused with 5 μM 1,2-HOPO-2 (n = 10) did not demonstrate reductions in collagen damage versus controls (n = 5) (1.45 ± 0.89 vs. 1.49 ± 1.0 AFU, P = 0.93).
Identification of Coronary Effluent MMPs
Significant elevations of MMP-2 were detectable after 25-minute GNFI in coronary effluents collected during reperfusion from isolated stunned hearts (n = 12), whereas sham hearts (n = 3) demonstrated no elevations of MMP-2 at the equivalent time point (35-minute baseline perfusion) (1.143 ± 0.40 vs. 0.002 ± 0.005 ng/mL, P = 0.0003). In the same coronary effluents used for immunoassays, stunned hearts displayed ~95% elevation in global MMP activity within 1-minute reperfusion compared with baseline perfusion, whereas shams displayed no increases at the equivalent time point (data not shown). This finding suggests that ischemia promotes the activation and release of MMP-2 into the coronary effluent during reperfusion. Immunoassays failed to detect MMP-3 and -13 in coronary effluents.
DISCUSSION
The objective of this study was to assess the damage caused by MMPs during ischemia–reperfusion injury, identify the role of specific MMP subtypes, and contrast the protective effects exerted by novel semiselective versus broad-spectrum MMPI. Two broad-spectrum compounds were selected. One was CGS27023A, which is a hydroxamate-based, potent, orally active (ie, water soluble) MMPI that blocked the erosion of cartilage in a rabbit model of arthritis.18 The MMPI profile of this compound was verified in our laboratory. For MMPI characterization purposes, we selected a group of MMPs to test (total of 8), which have been reported to have a possible role in cardiac pathophysiology.34 Using fluorescent-based peptide substrates, we tested for MMP-1, -2, -3, -7, -8, -9, -12, and -13. Results confirmed that CGS27023A is a potent, broad-spectrum MMPI with IC50 values of 50 nM or less (except MMP-7 at 500 nM). These results are in agreement with previous reports.18,22 We also tested for evidence of cell toxicity using cocultured neonatal rat cardiac fibroblasts/myocytes. No evidence for reductions in cell viability was noted at concentrations of up to 100 μM.
A second broad-spectrum inhibitor, PD166793, was used. PD166793 is a carboxylate-based nanomolar inhibitor with a broad range of activity.12,35 IC50 values of 3 μM were noted for MMP-1, -7, and -9. IC50 values of <50 nM were determined for MMP-2, -3, -8, -12, and -13. Cell toxicity results with PD166793 indicate loss of cell viability only at doses greater than 10 μM (Fig. 2). Given that all compounds were tested at 5 μM, both CGS27023A and PD166793 qualify as broad spectrum. Our results are also in agreement with those of others.12
CGS27023A significantly increased RPP by ~8% over controls. However, the assessment of changes in left ventricular end diastolic pressure or peak systolic pressure did not yield differences. A lack of similar improvement in coronary flow rate, heart weight, and cTnI release was noted. The use of PD166793 compound yielded similar effects with a very modest improvement only in RPP. Thus, broad-spectrum inhibition failed to preserve cardiac structure–function. Interestingly, the notion that broad-spectrum inhibition may not be desirable under various pathological conditions has been proposed.11,13 This argument seems to be better suited for the long-term healing and remodeling of infarcted hearts.7,9,11,13,14 However, no previous study had examined this possibility under short-term conditions. The manner by which MMPs may be “beneficial” remains to be determined; however, scenarios include the possibility that MMPs may “activate” via their proteolytic action biological mediators of cardioprotection such as humoral factors, which would act in an autocrine and/or paracrine manner.36 One such possibility could be the cleavage of insulin-like growth factor or transforming growth factor-β1 by MMP-7. Such growth factors are known to exert cardioprotective effects at the time of myocardial reperfusion through the activation of the reperfusion injury salvage kinase pathway.37,38 Inhibition of MMP-7 could prevent cleavage of the cardioprotective growth factors blocking, in turn the activation of the reperfusion injury salvage kinase pathway.
We also assessed the effects that semiselective MMPI would have on myocardial structure–function during ischemia–reperfusion injury. In an effort to devise new strategies for the synthesis of semiselective inhibitors, different hydroxypyrone and hydroxypyridinone ZBGs were appended to a common biphenyl backbone and the inhibition efficiency of each inhibitor was determined.20 PY-2 and 1,2-HOPO-2 have similar selectivity profiles; however, 1,2-HOPO-2 has slightly stronger inhibitory capacities than PY-2 against MMP-2, -8, and -12. Both compounds demonstrated a lack of cytotoxicity at concentrations of <10 μM. PY-2 results at 5 μM indicated a lack of improvement in RPP, left ventricular end diastolic or peak systolic pressures, and ventricular weight. However, statistical trends (P = 0.06) were noted for coronary flow rate and cTnI release. Given these results, experiments were performed at 10 μM where PY-2 demonstrated improvements in RPP. Results with 5 μM 1,2-HOPO-2 showed a significant improvement in RPP, coronary flow rate, ventricular weight, and cTnI release. We also utilized a 30-minute GNFI protocol, which yielded evidence of a severe depression of RPP in untreated hearts. The use of 1,2-HOPO-2 improved RPP and reduced cTnI release.
1,2-HOPO-2 also yielded significant reductions (~10%) in myocardial edema. 1,2-HOPO-2 was thus noted as the most effective “cardioprotector.” Improvements were observed in muscle function (RPP) and cTnI. Thus, protection of the muscle compartment (cTnI) yielded a correlation with improved RPP. A preservation of the vascular compartment was noted by improvements in coronary flow rate, ventricular weight, and decreased left ventricular end diastolic pressure. Collagen degradation was also decreased by 1,2-HOPO-2 treatment, thus indicating protection of the extracellular matrix compartment. A recent publication reported the effectiveness of an MMPI that uses the same ZBG as 1,2-HOPO-2, which was able to reduce in vivo brain edema in a rodent model of stroke.39 Interestingly, in this study, they attributed the protective actions of the compound mostly to gelatinase inhibition.
Another important objective was to identify candidate MMP subtypes, which may be involved in mediating the loss of structure–function. MMP-1 is unlikely involved on the basis of its lack of existence in rats.34,40 MMP-7 is also an unlikely participant because the semiselective MMPI are not potent against this protease. MMP-8 is also a poor candidate, given that it is predominantly in neutrophils41–43 and our perfusion used a physiological buffer. A similar but weaker argument can be made for MMP-12 because it is mostly present in macrophages.41,42 MMP-9 can also be considered as unlikely, given the lack of potency for the semiselective MMPI used and also because it is most abundant in macrophages and neutrophils.3,6,41,42,44 Thus, the strongest candidates are MMP-2, -3, and -13.
Published studies have provided evidence for a critical role of MMP-2 in mediating ischemia–reperfusion injury in isolated perfused hearts.6,45–48 Cheung et al45 demonstrated that the function of isolated rat hearts subjected to ischemia–reperfusion was significantly improved upon the inclusion of MMP-2 neutralizing antibodies. Evidence for involvement of MMP-2 also comes from studies using overexpressing transgenic mice where findings indicate that MMP-2 impairs ventricular function in the absence of superimposed injury.32 To our knowledge, no studies have examined the participation of MMP-3 and/or MMP-13. To document the possible involvement of MMP-2, -3, and -13, we measured their release into coronary effluent. Results confirmed the release of MMP-2 within the first minute of reperfusion. No evidence for the release of MMP-3 and -13 was observed. The suitability of the enzyme-linked immunosorbent assay kits to detect the presence of MMP-3 and -13 in rat hearts was based on published reports.49,50
An important question that arises when using small molecules is their possible lack of specificity and other unrelated actions. A concern associated with the use of the novel semiselective MMPI was the possibility that they could act as antioxidants.51 The MMPI were tested in an in vitro assay, and results confirmed their lack of antioxidant actions versus known positive controls (Trolox and minocycline). A second issue was to establish that the action of the semiselective MMPI was dependent on their ability to chelate metals. For this purpose, a negative control compound (PICO-2) was devised. PICO-2 was tested in the same stunning conditions used and failed to demonstrate any cardio-protection. Furthermore, PICO-2 is not an effective MMPI in vitro.20 Thus, the ability to bind the zinc(II) ion in the MMPI active site is essential for the preservation of inhibitory activity.6,19,20,52,53 It is important to note that most MMPI (including hydroxamates, carboxylates, and novel ZBG based) can also chelate other metals and thus affect other metal-dependent proteases, which may play a role in mediating damage to myocardium.16
The implementation of the isolated rat heart system of ischemia–reperfusion injury was seen as necessary for the prelude for future in vivo studies. However, the ex vivo system of ischemia–reperfusion injury has several important limitations such as its dependence of a physiological (non–blood based) buffer for perfusion purposes. These limitations are well described in published literature and apply to any study that uses these conditions to evaluate cardioprotection.54–56 Other limitations include the fact that we did not measure the effects of inhibitors on membrane-type MMPs, nor did we account for interactions between MMPs and their physiological inhibitors, the tissue inhibitors of metalloproteinases.
In conclusion, our studies were able to characterize a model of ischemia–reperfusion-induced stunning where the integrity of muscular, vascular, and extracellular matrix compartment was compromised. We also determined the activation of MMPs and their very early release into the coronary effluent. The strongest evidence was provided by the ability of 1,2-HOPO-2 to reduce collagen degradation, a known substrate for MMPs. However, the lack of correlation between the apparent potency of the various compounds used and improvement of end points measured suggests that either specific compound properties affect their ability to exert “effective” MMPI actions or that other nonspecific effects of the drugs are responsible for their actions; these issues await further investigation. On the basis of the results obtained from the use of novel ZBG-based compounds such as 1,2-HOPO-2, these small molecule inhibitors may be considered suitable for testing in in vivo models of ischemia–reperfusion injury. Evidence supports an important role for the activation of MMPs, in particular MMP-2.
Acknowledgments
We would like to acknowledge the support provided by the NHLBI to Dr. Francisco Villarreal (HL-43617, HL-67922) and Dr. Seth Cohen (HL-80049). Dr. Diego Romero-Perez was supported by a Doctoral Fellowship from UC-MEXUS CONACYT.
Footnotes
The authors report no conflicts of interest.
References
- 1.Day SM, Westfall MV, Fomicheva EV, et al. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med. 2006;12:181–189. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 2.Sawicki G, Leon H, Sawicka J, et al. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury. A new intracellular target for matrix metalloproteinase-2. Circulation. 2005;112:544–552. doi: 10.1161/CIRCULATIONAHA.104.531616. [DOI] [PubMed] [Google Scholar]
- 3.Chow AK, Cena J, Schulz R. Acute actions and novel targets of matrix metalloproteinases in the heart and vasculature. Br J Pharmacol. 2007;152:189–205. doi: 10.1038/sj.bjp.0707344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rork TH, Hadzimichalis NM, Kappil MA, et al. Acetaminophen attenuates peroxynitrite-activated matrix metalloproteinase-2-mediated troponin I cleavage in the isolated guinea pig myocardium. J Mol Cell Cardiol. 2006;40:553–561. doi: 10.1016/j.yjmcc.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Sung MM, Schulz CG, Wang W, et al. Matrix metalloproteinase-2 degrades the cytoskeletal protein α-actinin in peroxynitrite mediated myocardial injury. J Mol Cell Cardiol. 2007;43:429–436. doi: 10.1016/j.yjmcc.2007.07.055. [DOI] [PubMed] [Google Scholar]
- 6.Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol. 2007;47:211–242. doi: 10.1146/annurev.pharmtox.47.120505.105230. [DOI] [PubMed] [Google Scholar]
- 7.Spinale FG. Matrix metalloproteinases. Regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–530. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 8.Lindsey ML. MMP induction and inhibition in myocardial infarction. Heart Fail Rev. 2004;9:7–19. doi: 10.1023/B:HREV.0000011390.44039.b7. [DOI] [PubMed] [Google Scholar]
- 9.Creemers EE, Cleutjens JP, Smits JF, et al. Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001;89:201–210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 10.D’Armiento J. Matrix metalloproteinase disruption of the extracellular matrix and cardiac dysfunction. Trends Cardiovasc Med. 2002;12:97–101. doi: 10.1016/s1050-1738(01)00160-8. [DOI] [PubMed] [Google Scholar]
- 11.Hu J, Van den Steen PE, Sang QX, et al. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular disease. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 12.Kaludercic N, Lindsey ML, Tavazzi B, et al. Inhibiting metalloproteases with PD 166793 in heart failure: impact on cardiac remodeling and beyond. Cardiovasc Ther. 2008;26:24–37. doi: 10.1111/j.1527-3466.2007.00034.x. [DOI] [PubMed] [Google Scholar]
- 13.Fingleton B. Matrix metalloproteinases as valid clinical targets. Curr Pharm Des. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22:1–9. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- 15.Hein S, Schaper J. The extracellular matrix in normal and diseased myocardium. J Nucl Cardiol. 2001;8:188–196. doi: 10.1067/mnc.2001.113331. [DOI] [PubMed] [Google Scholar]
- 16.Singh RB, Dandekar SP, Elimban V, et al. Role of proteases in the pathophysiology of cardiac disease. Mol Cell Biochem. 2004;263:241–256. doi: 10.1023/B:MCBI.0000041865.63445.40. [DOI] [PubMed] [Google Scholar]
- 17.Peterson JT. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail Rev. 2004;9:63–79. doi: 10.1023/B:HREV.0000011395.11179.af. [DOI] [PubMed] [Google Scholar]
- 18.MacPherson LJ, Bayburt EK, Capparelli MP, et al. Discovery of CGS 27023A, a non-peptidic, potent, and orally active stromelysin inhibitor that blocks cartilage degradation in rabbits. J Med Chem. 1997;40:2525–2532. doi: 10.1021/jm960871c. [DOI] [PubMed] [Google Scholar]
- 19.Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem. 2007;2:152–171. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 20.Agrawal A, Romero-Perez D, Jacobsen JA, et al. Zinc-binding groups modulate selective inhibition of MMPs. Chem Med Chem. 2008;3:812–820. doi: 10.1002/cmdc.200700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson JT. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc Res. 2005;69:677–687. doi: 10.1016/j.cardiores.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 22.Schafers M, Riemann B, Kopka K, et al. Scintigraphic imaging of matrix metalloproteinase activity in the arterial wall in vivo. Circulation. 2004;109:2554–2559. doi: 10.1161/01.CIR.0000129088.49276.83. [DOI] [PubMed] [Google Scholar]
- 23.Puerta DT, Griffin MO, Lewis JA, et al. Heterocyclic, zinc-binding groups for use in next-generation matrix metalloproteinase inhibitors: potency, toxicity, and reactivity. J Biol Inorg Chem. 2006;11:131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 24.Tiwari M, Hemalatha T, Ganesan K, et al. Myocardial ischemia and reperfusion injury in rats: lysosomal hydrolases and matrix metalloproteinases mediated cellular damage. Mol Cell Biochem. 2008;312:81–91. doi: 10.1007/s11010-008-9723-7. [DOI] [PubMed] [Google Scholar]
- 25.Palmer BS, Hadziahmetovic M, Veci T, et al. Global ischemic duration and reperfusion function in the isolated perfused rat heart. Resuscitation. 2004;62:97–106. doi: 10.1016/j.resuscitation.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 26.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 27.Kloner RA, Bolli R, Marban E, et al. Medical and cellular implications of stunning, hibernation, and preconditioning. Circulation. 1998;97:1848–1867. doi: 10.1161/01.cir.97.18.1848. [DOI] [PubMed] [Google Scholar]
- 28.Hansen PR. Myocardial reperfusion injury: experimental evidence and clinical relevance. Eur Heart J. 1995;16:734–740. doi: 10.1093/oxfordjournals.eurheartj.a060991. [DOI] [PubMed] [Google Scholar]
- 29.Grund F, Treiman C, Ilebekk A. How to discriminate between hibernating and stunned myocardium. J Cardiovasc Surg. 2006;47:323–328. [PubMed] [Google Scholar]
- 30.Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68:1905–1912. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- 31.Villarreal FJ, Kim NN, Ungab GD, et al. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88:2849–2861. doi: 10.1161/01.cir.88.6.2849. [DOI] [PubMed] [Google Scholar]
- 32.Wang GY, Bergman MR, Nguyen AP, et al. Cardiac transgenic matrix metalloproteinase-2 expression directly induces impaired contractility. Cardiovasc Res. 2006;69:688–696. doi: 10.1016/j.cardiores.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 33.Go K, Horikawa Y, Garcia R, et al. Fluorescent method for detection of cleaved collagens using O-phthaldialdehyde (OPA) J Biochem Biophys Methods. 2008;70:878–882. doi: 10.1016/j.jbbm.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Simon H, Bocan TM, et al. MMP/TIMP expression in spontaneously hypertensive heart failure rats: the effect of ACE- and MMP-inhibition. Cardiovasc Res. 2000;46:298–306. doi: 10.1016/s0008-6363(00)00028-6. [DOI] [PubMed] [Google Scholar]
- 35.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 36.Roztocil E, Nicholl SM, Davies MG. Insulin-induced epidermal growth factor activation in vascular smooth muscle cells is ADAM-dependent. Surgery. 2008;144:245–251. doi: 10.1016/j.surg.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Housenloy DJ, Yellon DM. Reperfusion injury salvage kinase signaling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–234. doi: 10.1007/s10741-007-9026-1. [DOI] [PubMed] [Google Scholar]
- 38.Housenloy DJ, Yellon DM. Clinical translation of cardioprotective strategies: report and recommendations of the Hatter Institute 5th International Workshop on Cardioprotection. Basic Res Cardiol. 2008;103:493–500. doi: 10.1007/s00395-008-0736-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang YM, Fan X, Chakaravarty D, et al. 1-hydroxy-2-pyridinone-based MMP inhibitors: synthesis and biological evaluation for the treatment of ischemic stroke. Bioorg Med Chem Lett. 2008;18:409–413. doi: 10.1016/j.bmcl.2007.10.045. [DOI] [PubMed] [Google Scholar]
- 40.Peterson JT, Li H, Dillon L, et al. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted heart. Cardiovasc Res. 2000;46:307–315. doi: 10.1016/s0008-6363(00)00029-8. [DOI] [PubMed] [Google Scholar]
- 41.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 42.Kadoglou NP, Liapis CD. Matrix metalloproteinases: contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr Med Res Opin. 2004;20:419–432. doi: 10.1185/030079904125003143. [DOI] [PubMed] [Google Scholar]
- 43.Herman MP, Sukhova GK, Libby P, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 44.Lindsey ML, Escobar GP, Dobrucki LW, et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290:H232–H239. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- 45.Cheung P, Sawicki G, Wozniak M, et al. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101:1833–1839. doi: 10.1161/01.cir.101.15.1833. [DOI] [PubMed] [Google Scholar]
- 46.Lalu MM, Pasini E, Schulze CJ, et al. Ischaemia-reperfusion injury activates matrix metalloproteinases in the human heart. Eur Heart J. 2005;26:27–35. doi: 10.1093/eurheartj/ehi007. [DOI] [PubMed] [Google Scholar]
- 47.Wang W, Schulze CJ, Suarez-Pinzon WL, et al. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106:1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 48.Fert-Bober J, Leon H, Sawicka J, et al. Inhibiting matrix metal-loproteinase-2 reduces protein release into coronary effluent from isolated rat hearts during ischemia-reperfusion. Basic Res Cardiol. 2008;103:431–443. doi: 10.1007/s00395-008-0727-y. [DOI] [PubMed] [Google Scholar]
- 49.Swann K, Berger J, Sprague SM, et al. Peripheral thermal injury causes blood-brain barrier dysfunction and matrix metalloproteinase (MMP) expression in rat. Brain Res. 2007;1129:26–33. doi: 10.1016/j.brainres.2006.10.061. [DOI] [PubMed] [Google Scholar]
- 50.Vishnubhotla R, Sun S, Hug J, et al. ROCK-II mediates colon cancer invasion via regulation of MMP-2 and MMP-13 at the site of invadopodia as revealed by multiphoton imaging. Lab Invest. 2007;87:1149–1158. doi: 10.1038/labinvest.3700674. [DOI] [PubMed] [Google Scholar]
- 51.Ferrari R, Guardigli G, Mele D, et al. Oxidative stress during myocardial ischemia and heart failure. Curr Pharm Des. 2004;10:1699–1711. doi: 10.2174/1381612043384718. [DOI] [PubMed] [Google Scholar]
- 52.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26:587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu X, Parks WC, Heinecke JW. Activation and silencing of matrix metalloproteinases. Semin Cell Dev Biol. 2008;19:2–13. doi: 10.1016/j.semcdb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Sutherland FJ, Hearse DJ. The isolated blood and perfusion fluid perfused heart. Pharmacol Res. 2000;41:613–627. doi: 10.1006/phrs.1999.0653. [DOI] [PubMed] [Google Scholar]
- 55.Skrzypiec-Spring M, Grotthus B, Szelag A, et al. Isolated heart perfusion according to Langendorff-still viable in the new millennium. J Pharmacol Toxicol Methods. 2007;55:113–126. doi: 10.1016/j.vascn.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 56.Ytrehus K. The ischemic heart-experimental models. Pharmacol Res. 2000;42:193–203. doi: 10.1006/phrs.2000.0669. [DOI] [PubMed] [Google Scholar]