

Abstract
Quiescent mouse embryonic C3H/10T½ cells are more resistant to different proapoptotic stimuli than are these cells in the exponential phase of growth. However, the exponentially growing 10T½ cells are resistant to inhibitors of RNA or protein synthesis, whereas quiescent cells die upon these treatments. Conditioned medium from quiescent 10T½ cells possesses anti-apoptotic activity, suggesting the presence of protein(s) that function as an inhibitor of the apoptotic program. Using differential display technique, we identified and cloned a cDNA designated sarp1 (secreted apoptosis-related protein) that is expressed in quiescent but not in exponentially growing 10T½ cells. Hybridization studies with sarp1 revealed two additional family members. Cloning and sequencing of sarp2 and sarp3 revealed 38% and 40% sequence identity to sarp1, respectively. Human breast adenocarcinoma MCF7 cells stably transfected with sarp1 or infected with SARP1-expressing adenovirus became more resistant, whereas cells transfected with sarp2 displayed increased sensitivity to different proapoptotic stimuli. Expression of sarp family members is tissue specific. sarp mRNAs encode secreted proteins that possess a cysteine-rich domain (CRD) homologous to the CRD of frizzled proteins but lack putative membrane-spanning segments. Expression of SARPs modifies the intracellular levels of β-catenin, suggesting that SARPs interfere with the Wnt–frizzled proteins signaling pathway.
Keywords: cloning, secreted proteins, frizzled homologue
Apoptosis, or programmed cell death, plays a significant role in normal development and functioning of multicellular organisms, and, when disregulated, is involved in pathogenesis of numerous diseases (1–4). Apoptosis is a result of an active cell response to physiological or damaging agents and numerous genes products are involved in signal transduction, triggering, and executive steps of the apoptotic pathway (2, 4, 5). Other proteins do not take part in the apoptotic cascade by themselves but they modify cell sensitivity to proapoptotic stimuli (6). Many genes and gene families that participate in different stages of apoptosis have been identified and cloned recently. However, because the apoptotic pathways have not been clearly delineated, it is probable that many novel apoptosis-regulating genes or gene families await discovery.
We observed that mouse embryonic 10T½ cells in the quiescent state become resistant to different proapoptotic treatments but readily die by apoptosis upon inhibition of RNA or protein synthesis. Conditioned medium (CM) from the quiescent 10T½ cells possesses anti-apoptotic activity. Thus, we suggested that in the quiescent state, 10T½ cells contain all the components of the apoptotic program but its onset is inhibited by a secreted protein(s) exhibiting anti-apoptotic activity. Using differential display, we cloned a gene that is expressed in quiescent but not in exponentially growing 10T½ cells. This gene and its two homologues screened out from human heart and pancreas cDNA libraries encode secreted proteins capable of modifying cell response to proapoptotic stimuli. All members of the secreted apoptosis-related protein (SARP) family have a cysteine-rich domain (CRD) homologous to the CRD of transmembrane frizzled proteins. Expression of SARPs modifies the intracellular content of β-catenin, which indicates their interference with the Wnt–frizzled proteins signaling pathway.
EXPERIMENTAL PROCEDURE
Cell Cultures.
C3H/10T½ (cl8) cells were grown in Eagle’s basal medium (BME) supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified 5% CO2 atmosphere without antibiotics. Cells were plated at 2 × 103 cells per ml and fed every 3–4 days. Approximately 2 weeks after the initial seeding, the cells were completely quiescent and few if any mitotic cells were present. To analyze the effect of serum deprivation or cycloheximide the exponentially proliferating (approximately 75% confluent) or quiescent cultures were maintained in serum-free medium or medium supplemented with 10 μg/ml cycloheximide, and then their viability was analyzed. MCF7 human breast adenocarcinoma cells were plated at 2 × 105 cells per ml and cultured in modified Eagle’s medium (MEM) supplemented with 10% FBS. The cytotoxic treatments of MCF7 cells were performed in culture media containing appropriate amounts of tumor necrosis factor (TNF) or drugs. Serum-free CM was obtained after 24-hr incubation of quiescent 10T½ or MCF7 cells in BME or MEM, respectively.
Apoptosis Assay.
To estimate the relative amount of dead cells the apoptotic (i.e., nonadherent) and the nonapoptotic (i.e., adherent) cells were collected separately and their numbers were evaluated by using a Coulter Counter ZM. Cytotoxic effect of TNF or drugs on cell viability was assessed as percentage of adherent cells relative to the total cell number.
RNA Isolation and Northern Blot Analysis.
RNA was isolated by the guanidine isothiocyanate method of Chomczynski and Sacchi (7). Samples of total RNA (20 μg) were subjected to electrophoresis in a 1.2% agarose/formaldehyde gel (8). Hybridization was carried out according to standard protocols at high-stringency conditions (8), using cDNA probes labeled with [32P]dCTP in random priming reactions (9).
Differential Display Reaction.
The first-strand cDNA synthesis was primed from oligo(dT) by using SuperScript reverse transcriptase (GIBCO) according to the supplier’s protocol. The conditions and primers for the PCR were as originally described by Liang and Pardee (10).
cDNA Library Construction.
The mouse quiescent 10T½ fibroblast λZAP II (Stratagene) cDNA library was constructed essentially as described by Zapf et al. (11). The human heart and pancreas cDNA libraries were purchased from CLONTECH.
Transfection of MCF7 Cells.
MCF7 cells were transfected with the pcDNA3 mammalian expression vector (Invitrogen), containing no insert, msarp1 (mouse), or hsarp2 (human) cDNA, by using Lipofectamine reagent (GIBCO) according to manufacturer’s protocol. Stable transfectants and, 2–3 weeks later, single-cell-originated clones were selected with 1 mg/ml G418, and expression of the respective genes was confirmed by Northern hybridization.
Western Immunoblotting.
For Western analysis the samples of CM were concentrated by using Centriprep-10 concentrators (Amicon). Cells were harvested in extraction buffer consisting of 20 mM Tris⋅HCl (pH 7.8), 5 mM MgCl2, 250 mM sucrose, and 1% Nonidet P-40. After 1-hr incubation on ice, extracts were clarified by centrifugation. Protein concentrations of the cellular extracts were determined by using the DC protein assay kit (Bio-Rad). Equal amounts of proteins were subjected to SDS/PAGE (8), transferred onto nitrocellulose membranes, and probed with anti-glutathione S-transferase-mSARP1 polyclonal affinity-purified IgG (1 μg/ml) or anti-β-catenin monoclonal IgG (Transduction Laboratories, Lexington, KY).
Immunohistochemistry.
Paraformaldehyde-fixed cells grown on four-well Lab-Tek chamber slides were probed by anti-β-catenin monoclonal IgG (Transduction Laboratories). Staining was performed by avidin–biotin–peroxidase system (Vector Laboratories) using diaminobenzidine as a substrate. IgG isolated from preimmune serum was used as a negative control.
DNA and Protein Sequence Analysis.
Global alignment of DNA and protein sequences to the GenBank entries was performed by using the blast sequence analysis program (12). For multiple alignment of protein sequences we used clustal v software (13). Secretion signals were predicted by the signalase program (14).
Chromosome Mapping.
Chromosome mapping was performed by Southern hybridization of EcoRI-digested human/rodent somatic hybrid cell DNA (Mapping Panel no. 2, Corriel Cell Repositories) with sarp cDNA as a probe.
RESULTS
Differential Sensitivity of Exponentially Growing and Quiescent 10T½ Cells to Proapoptotic Stimuli.
Tomei et al. (15) have reported that exponentially proliferating 10T½ cells are especially sensitive to serum deprivation and die by apoptosis. Fig. 1A shows that after 24 hr in a serum-free medium about 50% of the cells detach and are apoptotic. When cultures reach density-dependent quiescence, cells become resistant to withdrawal of growth factors and other serum components. Similarly, quiescent cells are significantly more resistant to cytotoxic effects of staurosporine, menadione, and cisplatin, proapoptotic agents that have different mechanisms of action (data not shown).
Figure 1.
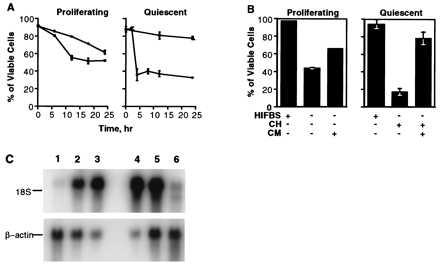
(A) Serum deprivation (□) induces death of the exponentially growing (Proliferating) 10T½ cells and cycloheximide (•) inhibits cell death. The cells become resistant to the serum deprivation (□) and extremely sensitive to cycloheximide (•) once they reach density-dependent quiescence (Quiescent). (B) The CM from quiescent 10T½ cells protects the exponentially growing and quiescent cells from apoptosis induced by the serum removal and cycloheximide (CH) treatment, respectively. (C) Northern hybridization of the differentially displayed DNA fragment to the RNA samples isolated from the 10T½ cells at different phases of growth: lanes 1–3, exponentially growing, 90–95% confluent, and quiescent (G0) cells, respectively; lanes 4–6, the quiescent cells were replated at lower density and harvested after 0, 2, and 6 hr, respectively.
In many cases of apoptosis cell death can be partially prevented or delayed by the inhibition of RNA or protein synthesis. Fig. 1A shows that during exponential proliferation, apoptosis of 10T½ cells is delayed by the addition of cycloheximide. In contrast, inhibition of protein synthesis rapidly induces death in quiescent cells arrested in G0 (Fig. 1A). Apoptosis of G0 cells is also induced by RNA synthesis inhibitors actinomycin D or α-amanitin (data not shown). These results imply that quiescent 10T½ cell cultures possess all the components of the apoptotic pathway but their activation is suppressed by quiescent state-specific protein(s). Furthermore, CM from quiescent 10T½ cells can inhibit apoptotic death of both serum-deprived exponentially growing and cycloheximide-treated quiescent 10T½ cells (Fig. 1B). Given the facts presented, we suggest that the increased viability of quiescent 10T½ cells may be attributed to secreted anti-apoptotic protein(s) exhibiting paracrine action.
Cloning of a Gene That Is Differentially Expressed in Quiescent 10T½ Cells.
In an attempt to identify the gene(s) whose differential expression changes the susceptibility of 10T½ cells to apoptosis we used the mRNA differential display strategy (10). We identified a differentially displayed DNA fragment that hybridized to RNA from quiescent but not to RNA from proliferating 10T½ cells (Fig. 1C). The 2.2-kb mRNA is induced when cells reach about 90% confluence, increases as the culture becomes quiescent, and disappears by the sixth hour after the quiescent cells have been replated at a lower density. The cDNA corresponding to this mRNA species was isolated from a cDNA library of quiescent 10T½ cells and designated msarp1. DNA sequence analysis revealed a single extended ORF encoding a predicted protein product of 295 amino acids, 252 bp of 5′ untranslated sequence, and 891 bp of 3′ untranslated sequence with two putative polyadenylation signals positioned at 637 bp and 234 bp from the 3′ end. The 3′ untranslated region contains 11 conserved 3′-UTR/HMG motifs proposed to be involved in posttranscriptional degradation of mRNA (16).
Analysis with the signalase software (14) indicates the mSARP1 contains an N-terminal hydrophobic region with an acceptable signal peptidase cleavage site following Gly-20. The global alignment of the msarp1 sequence to the GenBank database was performed by using blast software (12) and revealed a significant homology to the receptor-like proteins referred to as the homologues to Drosophila melanogaster frizzled (fz) gene product. However, unlike fz proteins, the mSARP1 does not contain a transmembrane region and the C-terminal region is rich with basic amino acids.
Cloning of Human sarps.
Low-stringency Northern blot analysis of human tissue mRNA by using the msarp1 cDNA as a probe indicated strong hybridization with heart and pancreas mRNA (data not shown). Screening of cDNA libraries made from these tissues (CLONTECH), using the msarp1 cDNA as a probe, yielded two clones from human pancreas, designated hsarp1 and hsarp3, and a single cDNA from the heart library, designated hsarp2.
hsarp1 is most likely the human homologue of msarp1. The incomplete, 890-bp cDNA encodes a polypeptide starting at the ATG at position 203 with about 95% sequence identity to mSARP1. The hsarp2 clone, a 2,094-bp cDNA fragment, has one major ORF. The ATG start site is found at position 303, and the termination site is at position 1248, and thus, hsarp2 encodes a polypeptide of 314 amino acids. hsarp3 is a 1,923-bp long cDNA fragment encoding a full-length protein of 316 amino acids. hSARP2 and hSARP3 share 38% and 40% sequence identity with mSARP1, respectively. N-terminal hydrophobic regions with acceptable signal peptidase cleavage sites are found in all hSARPs (Fig. 2).
Figure 2.
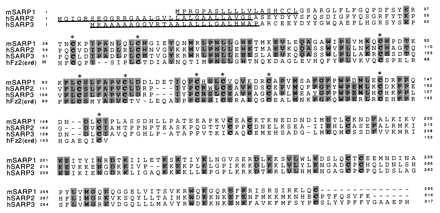
Multiple alignment of the SARP family members and CRD of human frizzled2. The gaps created by the software in the sequences corresponding to secretion signals were removed manually. The cysteines that are invariant in SARPs are shown in boldface. Ten cysteines in the CRD are marked by asterisks. The underlined polypeptide fragments represent the putative secretion sequences.
Multiple alignment of the proteins predicted by the sequences of cDNA clones and CRD extracellular portion of one of the frizzled-like proteins is presented in Fig. 2. SARPs share several common structural properties. Starting from the N terminus, the hydrophobic putative signal peptides are followed by the mature protein sequences, 270–300 amino acids in length with 16 invariant cysteines. Of these, 10 cysteines are located in the N-terminal 110- to 120-amino acid segments, which are 25–35% identical to the extracellular CRD of frizzled-like proteins (Fig. 2). None of the SARP group contains transmembrane regions which are characteristic of frizzled-like proteins (17).
Hybridization of sarp cDNA probes with DNA of individual human chromosomes revealed that sarp1, sarp2, and sarp3 genes are localized on chromosomes 4, 8, and 15, respectively (data not shown).
Expression of sarps Is Tissue Specific.
The pattern of the expression of human sarp genes shown by Northern hybridization appears to be tissue specific (Fig. 3). The highest level of hsarp1 expression is found in colon and small intestine, where two mRNA species of 2.2 and 1.3 kb have been observed. It is likely that this is a result of alternative polyadenylation of the message at two poly(A) signals found in the 3′ untranslated region. The human sarp2 probe reveals an ≈5 kb mRNA with maximal expression in heart, and a 2.2-kb message for hsarp3 has been found only in pancreas. None of the three sarp genes are expressed in lung, liver, or peripheral blood leukocytes.
Figure 3.
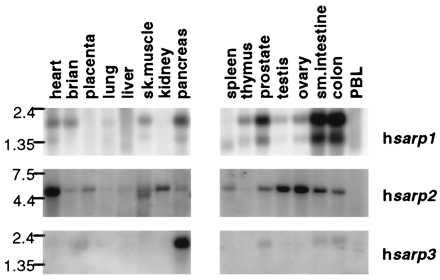
Tissue-specific expression of sarp genes.
SARPs Modify Cell Response to Proapoptotic Signals.
The MCF7 breast adenocarcinoma cell line was used as a model to study the possible involvement of SARP proteins in the regulation of apoptosis. The programmed death of these cells induced by different agents has been well characterized (18), and we have also found that this cell type does not express either sarp1 or sarp2 (data not shown). MCF7 cells were stably transfected with a pcDNA3 mammalian expression construct bearing full-length msarp1 or hsarp2 cDNAs or vector (no insert) and selected on G418. Four single-cell-originated clones expressing SARP2 and two clones expressing SARP1 were isolated and analyzed. Expression of mSARP1 and hSARP2 by transfectants was confirmed by Northern blot analysis.
The growth rate and cell cycle of transfected MCF7 cells were not significantly different from those of the parental cells (data not shown). Thus, overexpression of msarp1 or hsarp2 in MCF7 cells does not affect cell survival and proliferation. However, the cellular response to proapoptotic agents is significantly different in MCF7 cells overexpressing SARPs. The results presented in Fig. 4A demonstrate that the expression of mSARP1 and hSARP2 had opposite effects on cell sensitivity to cytotoxic stimuli. Whereas the expression of mSARP1 resulted in a higher resistance, expression of hSARP2 sensitized the cells to apoptosis induced by TNF and by ceramide, a secondary messenger in the apoptotic pathways induced by various agents (19, 20). We also infected MCF7 cells with SARP1- or SARP2-expressing adenovirus and obtained similar results (data not shown). Sensitivity of the clones to proapoptotic agents correlated with the levels of SARP expression (Fig. 4B). SARP2 clones 1–3 that displayed a high level of transfected gene expression are more sensitive to serum deprivation and adriamycin than are MCF7 cells transfected with vector only or clone 4, which expresses SARP2 much less effectively.
Figure 4.
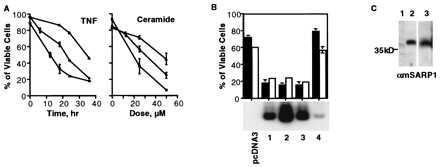
(A) MCF7 cells transfected with msarp1 (▴) are more resistant to the cytotoxic effects of TNF and ceramide, whereas hsarp2 (□) makes them more sensitive to the same agents compared with pcDNA3-transfected control (•). (B) Viability of MCF7 transfectants with different levels of hsarp2 expression upon serum deprivation (filled bars) and adriamycin (open bars) treatment. Lanes 1–4, four different single-cell-originated hsarp2 transfectants. (C) Western blotting of CM from MCF7 cells, containing the pcDNA3 control (lane 1) and msarp1 vectors (lane 2), and from the 10T½ quiescent cells (lane 3). The anti-mSARP1 polyclonal affinity-purified antibodies were used as a probe.
mSARP1 Is a Secreted Protein.
Because SARPs have the signal sequences but no transmembrane domains (see Fig. 2), it is probable that they are secreted proteins. Using affinity-purified anti-mSARP1 antibodies obtained against recombinant protein, we detected the secreted protein in the CM from both the transformed MCF7 cells and untransformed quiescent 10T½ cells (Fig. 4C). The mSARP antibodies fail to interact with hSARP2.
SARPs Interfere with the Wnt–Frizzled Proteins Signaling Pathway.
Recently, it was shown that frizzled proteins function as receptors for members of the Wnt protein family (21–24). Interaction of Wnt family members with their respective frizzled receptor causes inactivation of glycogen synthase kinase 3β (GSK-3) or its Drosophila homologue Zw-3 (25–27). In the absence of Wnt, GSK-3β phosphorylates β-catenin (Armadillo is its Drosophila homologue). Phosphorylated β-catenin or Armadillo is degraded more rapidly than nonphosphorylated forms of the proteins (24, 27–29). As a result, Wnt signaling causes changes in intracellular concentration of β-catenin or Armadillo, and this parameter has been used to register Wnt–frizzled protein interaction and signal transduction (22). Because SARPs are soluble proteins possessing a domain homologous to CRD of frizzled proteins we suggested that they can function by interference with Wnt–frizzled protein interaction. To check this hypothesis we compared expression of β-catenin in MCF7-transfectants. Fig. 5 shows that expression of SARP2 decreases the intracellular concentration of β-catenin. The effect of SARP1 on the levels of β-catenin is more complicated. By Western blotting we could not detect significant changes, but immunohistochemical data revealed a higher concentration of β-catenin in the SARP1 transfectants. Because β-catenin is a multifunctional protein, a more detailed study of the intracellular concentration of the free and bound protein should be done in the future.
Figure 5.
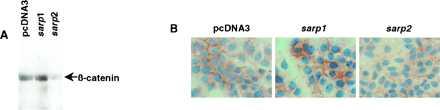
Detection of β-catenin in MCF7 cells transfected with pcDNA3, msarp1, or hsarp2. (A) Western analysis. (B) Immunohistochemical staining. (×400.)
DISCUSSION
We have identified a family of genes capable of modulating cellular responses to cytotoxic signals. The following data indicate that SARP1 possesses anti-apoptotic activity. First, SARP1 is expressed in quiescent 10T½ cells, which are resistant to various cytotoxic treatments, but is not expressed in exponentially growing cells, which are much more sensitive to proapoptotic agents. Second, CM from quiescent 10T½ cells that contain mSARP1 has anti-apoptotic activity. Third, overexpression of SARP1 in MCF7 cells decreases their sensitivity to TNF and ceramide. Experiments with SARP2 show that this member of the family has the opposite effect: it increases sensitivity of MCF7 cells to cytotoxic treatments. From this viewpoint, it is interesting that another apoptosis-modulating gene family, ced-9/bcl2 gene, also includes members with opposite effects on cell survival (1–4).
Several features of SARPs seem to be interesting: (i) SARPs are secreted proteins and from this viewpoint resemble growth factors and cytokines that now are more and more widely used for treatment of various diseases; (ii) SARP expression is tissue specific, and future experiments should show if the effect of these proteins is tissue specific (also see below); (iii) SARP expression is dependent on the physiological state of cells; and (iv) aside from viral soluble receptors (30), it is unusual that a family of transmembrane receptors (frizzled proteins) is homologous to a family of secreted proteins.
The mechanism of SARPs activity on cell sensitivity to cytotoxic agents is unknown. It is possible that SARPs also have other functions unrelated to the regulation of apoptosis. However, it is important to note the high degree of sequence homology of SARP CRDs to the similar regions of the frizzled proteins, a class of cellular membrane receptors with seven transmembrane domains. In D. melanogaster, frizzled proteins are involved in the regulation of bristle and hair polarity (31). Recently, the ability of Dfz2, a frizzled protein family member, to function as a receptor for Wingless protein was reported (22). Wingless is a member of the Wnt gene family, whose products are involved in cell–cell and cell–extracellular matrix interaction (32). Interesting similarities between SARP, frizzled, and Wnt families include tissue specificity (17, 32) and the existence of members with opposite physiological effects (33).
Interaction of Wingless with its corresponding frizzled receptor stabilizes Armadillo, a key effector of signal transduction in this system, and accumulation of Armadillo is used as an indicator of Wingless–frizzled signaling (22). In our experiments expression of SARPs modulated intracellular levels of β-catenin, a homologue of Armadillo in vertebrates, suggesting that SARPs interfere with Wnt–frizzled interaction. It is interesting that SARP1 and SARP2 induced opposite changes in β-catenin concentration. This can mean that SARP1 and SARP2 interact with different members of the Wnt family or their binding to the same Wnt causes opposite effects. It is unclear if modulation of intracellular levels of β-catenin by SARPs is coupled to their effects on cell response to proapoptotic stimuli. β-Catenin is a multifunctional protein and each of its functions, involvement in cell–cell junction through interaction with E-cadherin (34, 35) and transcription regulation in a complex with members of the TCF/Lef-1 family (36–38), can be involved in the apoptotic pathway. Recently it was shown that β-catenin accumulated in colon cancer (39, 40) and melanomas (41) that had mutations in the tumor suppressor APC. Moreover, regulation of β-catenin is critical to APC’s tumor-suppressive effect (40). In our experiments a correlation between the levels of β-catenin and expression of the SARP family members, which possess pro- or anti-apoptotic activity, has been found. Thus, a higher level of β-catenin in tumors can cause the reduction in apoptotic cell death, a feature characteristic of carcinogenesis (3).
The SDF5 gene, which is identical to msarp1, was recently cloned among other genes by technique designed for cloning secreted proteins (42). Another group cloned frzb, a gene possibly involved in skeletal morphogenesis (43). This gene is homologous to sarps and seems to be another member of the family.
Future investigations should answer many intriguing questions. Wnt proteins can be divided into several functional groups (33, 44, 45). Is the opposite effect of different SARP family members on the apoptotic pathway associated with their ability to bind different Wnt proteins? Are biological activities of SARPs in embryogenesis and morphogenesis related to their anti- or proapoptotic effects? Are SARPs, as well as other proteins regulating the intracellular level of β-catenin, involved in carcinogenesis? Some data indicate the existence of other receptors for Wnt besides frizzled proteins (24). Can SARPs function as modulators of Wnt interaction with these receptors?
Acknowledgments
We thank the members of Cell Biology, Molecular Biology, and Protein Chemistry groups for discussions of results; Dr. Vaclav Prochazka for DNA sequencing; Bryce Baker and Anna Logvinova for help in analysis of β-catenin; Virginia C. Powers for comments and help in manuscript preparation; and Dr. Eugene M. Shekhtman for help with software.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: SARP, secreted apoptosis-related protein; CM, conditioned medium; CRD, cysteine-rich domain; TNF, tumor necrosis factor.
References
- 1.Wyllie A H, Kerr J F R, Currie A R. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaux D L, Strasser A. Proc Natl Acad Sci USA. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson C B. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 4.Umansky S R. Mol Biol (Moscow) 1996;30:285–295. [Google Scholar]
- 5.Korsmeyer S. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 6.Raff M C. Nature (London) 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 7.Chomczynski P, Sacchi N. Anal Biochem. 1987;161:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 8.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 9.Feinberg A P, Vogelstein B. Anal Biochem. 1982;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 10.Liang P, Pardee A B. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 11.Zapf J, Kiefer M, Merryweather J, Musiarz F, Bauer D, Born W, Fischer J A, Froesch E R. J Biol Chem. 1990;265:14892–14898. [PubMed] [Google Scholar]
- 12.Altschul S F, Gish W, Miller W, Myers E, Lipman J L. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 13.Higgins D G, Bleasby A J, Fuchs R. Comput Appl Biosci. 1991;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- 14.von Heijne G. Nucleic Acids Res. 1986;14:4683–4690. doi: 10.1093/nar/14.11.4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomei L D, Shapiro J P, Cope F O. Proc Natl Acad Sci USA. 1993;90:853–857. doi: 10.1073/pnas.90.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reeves R, Elton T S, Nissen M S, Lehn D, Johnson K R. Proc Natl Acad Sci USA. 1987;84:6531–6535. doi: 10.1073/pnas.84.18.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Macke J P, Abella B S, Andreasson K, Worley P, Gilbert D J, Copeland N G, Jenkins N A, Nathans J. J Biol Chem. 1996;271:4468–4476. doi: 10.1074/jbc.271.8.4468. [DOI] [PubMed] [Google Scholar]
- 18.Zyad A, Benard J, Tursz T, Clarke R, Chouaib S. Cancer Res. 1994;54:825–831. [PubMed] [Google Scholar]
- 19.Hannun Y A, Obeid L M. Trends Biochem Sci. 1995;20:73–77. doi: 10.1016/s0968-0004(00)88961-6. [DOI] [PubMed] [Google Scholar]
- 20.Kolesnick R, Fuks Z. J Exp Med. 1995;181:1949–1952. doi: 10.1084/jem.181.6.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang-Snyder J, Miller J R, Brown J D, Lai C J, Moon R T. Curr Biol. 1996;6:1302–1306. doi: 10.1016/s0960-9822(02)70716-1. [DOI] [PubMed] [Google Scholar]
- 22.Bhanot P, Brink M, Samos C H, Hsieh J C, Wang Y, Macke J P, Andrew D, Nathans J, Nusse R. Nature (London) 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 23.Orsulic S, Peifer M. Curr Biol. 1996;6:1363–1367. doi: 10.1016/s0960-9822(96)00731-2. [DOI] [PubMed] [Google Scholar]
- 24.Perrimon N. Cell. 1996;86:513–516. doi: 10.1016/s0092-8674(00)80124-5. [DOI] [PubMed] [Google Scholar]
- 25.Pai L M, Orsulic S, Bejsovec A, Peifer M. Development. 1997;124:2255–2266. doi: 10.1242/dev.124.11.2255. [DOI] [PubMed] [Google Scholar]
- 26.Cook D, Fry M J, Hughes K, Sumathipala R, Woodgett J R, Dale T C. EMBO J. 1996;15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- 27.Siegfried E, Wilder E L, Perrimon N. Nature (London) 1994;367:76–80. doi: 10.1038/367076a0. [DOI] [PubMed] [Google Scholar]
- 28.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 29.Yost C, Torres M, Miller J R, Huang E, Kimelman D, Moon R T. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- 30.Alcami A, Smith G L. Immunol Today. 1995;16:474–478. doi: 10.1016/0167-5699(95)80030-1. [DOI] [PubMed] [Google Scholar]
- 31.Adler P N. BioEssays. 1992;14:735–741. doi: 10.1002/bies.950141103. [DOI] [PubMed] [Google Scholar]
- 32.Nusse R, Varmus H E. Cell. 1992;69:1073–1087. doi: 10.1016/0092-8674(92)90630-u. [DOI] [PubMed] [Google Scholar]
- 33.Moon R T, Brown J D, Torres M. Trends Genet. 1997;13:157–162. doi: 10.1016/s0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- 34.Gambiner B M. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 35.Hulsken J, Birchmeier W, Behrens J. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhl M, Wedlich D. BioEssays. 1997;19:101–104. doi: 10.1002/bies.950190204. [DOI] [PubMed] [Google Scholar]
- 37.Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann B G, Kemler R. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- 38.Behrens J, von Kries J P, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Nature (London) 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 39.Korinek V, Barker N, Morin P J, van Wichen D, de Weger R, Kinzler K W, Vogelstein B, Clevers H. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 40.Morin P J, Sparks A B, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler K W. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 41.Rubinfeld B, Robbins P, El G M, Albert I, Porfiri E, Polakis P. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- 42.Shirozu M, Tada H, Tashiro K, Nakamura T, Lopez N D, Nazarea M, Hamada T, Sato T, Nakano T, Hanjo T. Genomics. 1996;37:273–280. doi: 10.1006/geno.1996.0560. [DOI] [PubMed] [Google Scholar]
- 43.Hoang B, Moos M, Jr, Vukicevic S, Luyten F P. J Biol Chem. 1996;271:26131–26137. doi: 10.1074/jbc.271.42.26131. [DOI] [PubMed] [Google Scholar]
- 44.Wong G T, Gavin B J, McMahon A. Mol Cell Biol. 1994;14:6278–6286. doi: 10.1128/mcb.14.9.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du S J, Purcell S M, Christian J L, McGrew L L, Moon R T. Mol Cell Biol. 1995;15:2625–2634. doi: 10.1128/mcb.15.5.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leyns L, Bouwmeester T, Kim S H, Piccolo S, De R E. Cell. 1997;88:747–756. doi: 10.1016/s0092-8674(00)81921-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Krinks M, Lin K, Luyten F P, Moos M J. Cell. 1997;88:757–766. doi: 10.1016/s0092-8674(00)81922-4. [DOI] [PubMed] [Google Scholar]
- 48.Finch P W, He X, Kelly M J, Üren A, Schaudies R P, Popescu N C, Rudikoff S, Aaronson S A, Varmus H A, Rubin J S. Proc Natl Acad Sci USA. 1997;94:6770–6775. doi: 10.1073/pnas.94.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rattner A, Hsieh J C, Smallwood P M, Gilbert D J, Copeland N G, Jenkins N A, Nathans J. Proc Natl Acad Sci USA. 1997;94:2859–2863. doi: 10.1073/pnas.94.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]