

Abstract
The phosphatidylinositol 3-kinase (PI3K) signaling pathway is frequently upregulated in cancer. PIK3CA, the gene coding for the catalytic subunit p110α of PI3K, is mutated in about 12% of all human cancers. Most of these mutants are single amino acid substitutions that map to three positions (hot spots) in the helical or kinase domains of the enzyme. The mutant proteins show gain of enzymatic function, constitutively activate AKT signaling and induce oncogenic transformation in vitro and in animal model systems. We have shown previously that hot-spot mutations in the helical domain and kinase domain of the avian p110α have different requirements for interaction with the regulatory subunit p85 and with RAS-GTP. Here, we have carried out a genetic and biochemical analysis of these “hot-spot” mutations in human p110α. The present studies add support to the proposal that helical and kinase domain mutations in p110α trigger a gain of function by different molecular mechanisms. The gain of function induced by helical domain mutations requires interaction with RAS-GTP. In contrast, the kinase domain mutation is active in the absence of RAS-GTP binding, but depends on the interaction with p85.
Keywords: phosphatidylinositol 3-kinase (PI3K), p110α, p85, RAS, AKT, oncogenic transformation, hot-spot mutations
Introduction
The catalytic subunit p110α of class I PI3K is frequently mutated in human cancer. Particularly high incidences of mutation are found in cancers of the breast, the colon or the endometrium (Catalogue of Somatic Mutations in Cancer, http://www.sanger.ac.uk/genetics/CGP/cosmic). Genetic, biochemical and cell-based analyses suggest that such mutated p110α functions as an oncoprotein, playing an important role in tumorigenesis.1-13 About 80% of the mutations map to three hot-spots in the coding sequence of PIK3CA. Two of the hot spots, represented by the single amino acid substitutions E542K and E545K, are localized in the helical domain of the protein, the third, represented by the H1047R substitution, resides in the kinase domain. These mutations increase enzymatic activity, constitutively stimulate AKT signaling, induce growth factor- and anchorage-independent growth in culture, and cause tumors in vivo.4, 5, 9-13 We have demonstrated previously that helical domain and kinase domain mutations in the avian p110α have different requirements for interaction with the regulatory subunit p85 and with RAS-GTP.14, 15 We have now extended these studies to the human p110α. The human and avian proteins are closely related (96 % primary sequence identity), but minor sequence differences could affect mutant behavior. Here we report that the data on the human enzyme, though distinct in some respects, are qualitatively in accord with the previous results obtained with the avian protein. The gain of function seen with helical domain mutations depends on an interaction with RAS-GTP but is less affected by binding to p85. In contrast, the kinase domain mutation is active in the absence of RAS-GTP binding, but requires interaction with p85.
Results
Binding to p85 is dispensable for cellular transformation induced by the helical domain mutations E542K and E545K but is essential for the gain of function induced by the kinase domain mutation H1047R
Biochemical and structural studies support the proposal that the helical domain mutations, E542K and E545K, relieve an inhibitory interaction between the N-terminal SH2 domain of p85 and the helical domain of p110α.16-20 In order to examine the effect of p85-binding on mutant activity, we abolished the binding of human p110α to p85 by partial deletion (deletion of 72 N-terminal amino acids, Δ72) or total deletion (deletion of 108 N-terminal amino acids, Δ108) of the adaptor-binding domain (ABD) of human p110α. The expression level of the partial ABD deletion mutants (Δ72-p110α) was significantly decreased (Fig. 1A). This is in contrast to the results with the avian p110α, which was expressed at the same level as the wt protein.14, 21 The expression level of Δ108 was even lower which is in agreement with published information (Fig. 1A).9 However, these expression levels were sufficient to test for interaction with p85. We carried out co-immunoprecipitation experiments with HA-tagged p85 (Fig. 1A). The results show that both Δ72 and Δ108 of the human p110α failed to bind to p85. The bands seen with p110α antibody in the Δ72 and Δ108 lanes represent the endogenous p110α of CEF, their migration in the gel differs distinctly from that of the faster moving deletion mutants. The successful pull-down of the endogenous p110α also serves as a control, showing that the interaction between p85 and p110α is detectable at low levels of p110α expression.
Fig 1. Binding to p85 is essential for H1047R-induced cell transformation.
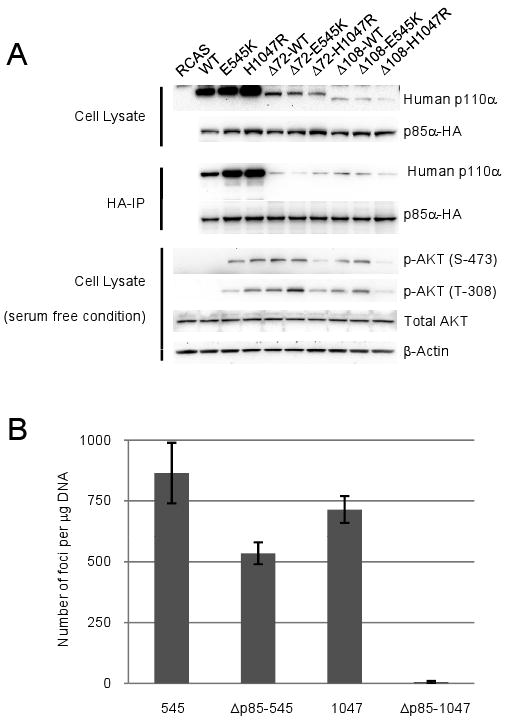
(A) Western blots comparing p110α expression levels, p110α and p85 binding, and the phosphorylation of AKT in serum-starved CEF. In the co-immunoprecipitation assay, the bands seen with anti-human p110α antibody in the Δ72 and Δ108 lanes represent the endogenous p110α of CEF. (B) Quantitative analysis of cell transformation induced by full-length or ABD deletion mutants of p110α. CEF were transfected with different p110α constructs. Cells were then cultured under nutrient agar. Foci of transformed cells were counted 14 days post transfection. 545 and 1047 signify the E545K and H1047R mutants respectively. Δp85-545 and Δp85-1047 mark the Δ108-E545K and Δ108-H1047R respectively.
Partial or total deletion of the ABD had different effects on the oncogenicity of helical and kinase domain mutations. Loss of p85 binding caused only a modest reduction in the efficiency of transformation of the helical domain mutation E545K (Fig. 1B). In contrast, the kinase domain mutation H1047R showed a more pronounced loss of oncogenicity if combined with Δ72 (4-fold, data not shown), combination with the Δ108 deletion caused a complete loss of transforming activity in H1047R (Fig. 1B). In human wt p110α, elimination of the ability to bind p85 activated the latent oncogenic activity of that protein (data not shown). This observation is in agreement with previous studies9, 14 and probably results from the removal of a p85-mediated inhibitory effect. The differential transforming potencies of the helical and kinase domain mutations that lack p85 binding are also reflected in their signaling to AKT. Δ108-wt and Δ108-E545K transformed cells showed constitutive activation of AKT, albeit at a lower level than cells expressing full-length E545K. In cells expressing the Δ108-H1047R double mutant, there was no constitutive activation of AKT (Fig. 1A).
RAS-binding is important for oncogenic transformation induced by helical domain mutants of human p110α; the kinase domain mutant is RAS-independent
The studies of Gupta and coworkers22 have identified point mutations in the RBD of human p110α, K227A or T208D, that block the interaction with RAS without affecting the basal activity of p110α. To test the effect of RAS binding on transforming activity, we combined K227A or T208D with the hot-spot mutations of p110α and tested these double mutants in focus assays. In contrast to the results obtained with the avian K227E, the single RBD mutations in the human protein did not affect oncogenic transformation induced by either the kinase domain mutant H1047R or the helical domain mutant E545K (results not shown). In cells expressing E545K/K227A, E545K/T208D, H1047R/K227A or H1047R/T208D, AKT was constitutively activated but at a significantly lower level as compared to cells expressing E545K or H1047R (Fig. 2A). The reduction in the activation of AKT did not affect the efficiencies of transformation seen with these mutants.
Fig 2. Oncogenic transformation by the helical domain mutation depends on binding to RAS.
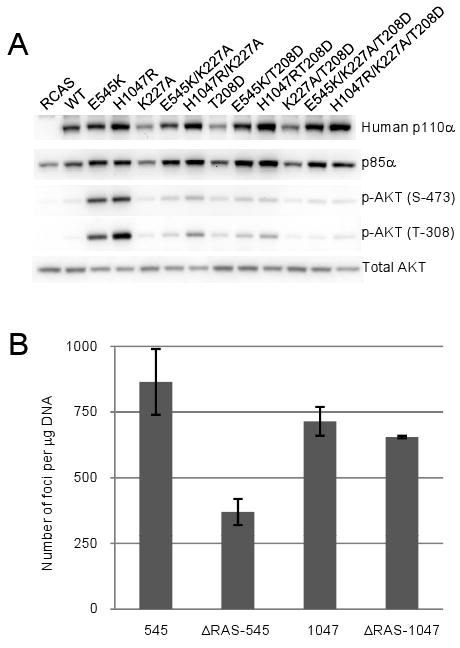
(A) Western blots comparing p110α expression levels and the phosphorylation of AKT in serum-starved CEF. (B) Quantitative analysis of cell transformation induced by full-length or RBD mutants of p110α. 545 and 1047 signify the E545K and H1047R mutants respectively. ΔRas-545 and ΔRas-1047 mark the E545K/K227A/T208D and H1047R/K227A/T208D mutants respectively.
We then tested the effect of the double mutation (K227A/T208D) in the RBD on the transforming activity of E545K and of H1047R. In E545K/K227A/T208D, the efficiency of transformation was reduced substantially compared to E545K, whereas the transforming activity of H1047R/K227A/T208D did not differ from that of H1047R (Fig. 2B). Similarly, in the wt p110α protein carrying the Δ108-K227A/T208D mutations, the efficiency of transformation was reduced but a residual activity remained (data not shown). This latter result is surprising and may reflect subliminal RAS binding to the K227A/T208D mutants in vivo, although such binding is not evident in in vitro assays.22 All mutant constructs were expressed at approximately equal levels (Fig. 2A). The quantitative differences in transforming potency between the H1047R/K227A/T208D and the E545K/K227A/T208D mutants are also reflected in the phosphorylation level of AKT at T-308. Quantitative analysis of the pAKT (T-308) bands shows that the pAKT(T-308) level in cells expressing the H1047R/K227A/T208D mutant is at least twice as high as that in cells expressing E545K/K227A/T208D.
Discussion
Gain of function and oncogenic activity make the cancer-specific mutants of p110α promising therapeutic targets. In order to take advantage of this unique situation and design mutant-specific interventions, it is necessary to understand the molecular mechanisms that induce the gain of function. As a first step toward this goal, we have previously studied the hot-spot mutations in avian p110α and have shown that helical and kinase domain mutations cause the gain of function in p110α by different molecular mechanisms.14 Helical domain mutations function independently of p85 but require RAS. The kinase domain mutation depends on p85 but is unaffected by a loss of RAS binding. In the present study, we have extended this analysis to human p110α.
The p110α protein occurs in the cell as a heterodimer bound to p85.23, 24 We generated p110α constructs that are defective in p85 binding by partial or total deletion of the ABD of p110α. These constructs do not interact with p85 as determined by co-immunoprecipitation. The deletion of the full ABD has contrasting effects on helical and kinase domain mutations. The oncogenic and signaling activities of the helical domain mutations show an only minor reduction, whereas these same activities in the kinase domain mutation are completely extinguished. As reported previously, the deletion of the ABD in the wt p110α leads to activation of cell transformation and of signaling as a result of removing p85-mediated inhibition (see below).9, 14, 21
Biochemical and structural studies have provided evidence for an interaction between the N-terminal SH2 domain of p85 and the helical domain of p110α.16-18, 25 This interaction is responsible for the p85-induced inhibition of p110α. The helical domain mutations probably weaken this p85-p110α interaction and thus remove the inhibition. Such a weakened interaction between the N-terminal SH2 domain and p110α is also suggested by the failure of the phosphorylated insulin receptor substrate to further activate lipid kinase activity of the helical domain mutants.19 In contrast, the kinase domain mutation remains susceptible to this growth factor-mediated enhancement.
p110α is an important RAS effector and has a role in mediating the proliferative, survival, and tumorigenic functions of RAS.22, 26, 27 Conversely, direct interaction between GTP-bound RAS and p110α augments the activity of p110α, possibly by inducing a conformational change at the substrate binding site or by mediating a closer interaction with the plasma membrane.28, 29 Indeed, the activity of p110γ that is defective in RAS binding can be restored with a myristylation signal. We have previously shown that the gain of function induced by the H1047R mutant of avian p110α is independent of RAS binding, whereas that of the helical domain mutants requires RAS interaction.14 In this study, we disabled RAS binding by introducing the double mutation, K227A/T208D, and found a modest inhibition of the oncogenic activity of the helical domain mutant but no effect on the kinase domain mutant. This result is in accord with our previous studies, although the effect of inactivating RAS binding on the helical domain mutant is not as extreme as seen with the avian protein. This result raises the question of whether the K227A/T208D mutations fully disable RAS binding in the human protein. The interaction between p110α and RAS is difficult to demonstrate by pull-down experiments, but we have observed that constitutively active RAS, which induces a strong stimulation of PI3K signaling, is ineffective in constructs carrying the K227A/T208D mutations (results not shown). This observation suggests that the mutations in the RBD do reduce the interaction, in agreement with previous reports22, but they may not completely eliminate it.
Our data add further support to the conclusion that helical and kinase domain hot-spot mutations of p110α induce a gain of function by different molecular mechanisms. Kinase and helical domain mutants show opposite requirements for p85 and RAS interactions. The kinase domain mutant is RAS-independent but needs p85 binding, the helical domain mutants are p85-independent but require RAS for full activity. Release from p85-induced inhibition provides an explanation for the behavior of the helical domain mutations.16-20 They mimic a growth-factor-induced activated state that results from a weakened interaction with p85. The additional, mutational deletion of p85 binding recapitulates this same mechanism and therefore has no effect on these mutants. However, the helical domain mutants remain dependent on an interaction with RAS. The kinase domain mutant mimics RAS-induced activation of p110α.14, 20 The dependence of this mutant on p85 binding could be explained by referring to the crystal structure of the p110α-p85 complex18 which reveals an unexpected interaction between the ABD domain and the kinase domain. This interaction might be important for the active conformation induced by H1047R and could account for the sensitivity of H1047R to a loss of p85 binding.
Genetic analysis can suggest molecular mechanisms, but a more definitive understanding of the functional effects of the hot-spot mutations in p110α requires high-resolution structures. Such structural data have led to a novel interpretation of the H1047R kinase domain mutation. Although H1047R maps close to the activation loop, it appears unlikely that H1047R affects the position of the loop.18, 30-32 Rather, this mutation shows an unexpected effect on the interaction of the protein with membrane lipids. This altered membrane affinity could explain the mutation-induced gain of function. Such a mechanism would be distinct from the disinhibition that is the probable cause for the enhanced function of the helical domain mutations.
We speculate that the conformational re-arrangements induced by the kinase and helical domain hot spot mutations in p110α are unique and of sufficient magnitude to be exploitable for the identification of small molecule inhibitors that are mutant-specific and do not affect the wt protein. Because of their exclusive effects on cancer tissue, such inhibitors could have therapeutic properties that are superior to those of pan-specific PI3K inhibitors.
Materials and Methods
Plasmid Construction
Construction of the pBSFI vector KOZ-cp3k encoding wt, E545K and H1047R human p110α has been described.11, 33 To generate the ABD deletion mutant, the deletion constructs were PCR amplified and cloned into pBSFI cloning vector using NotI and BamHI restriction sites. The forward primers used for amplification of Δ72-hp110α and Δ108-hp110α were NotI(F) GCC GCG GCC GCA CCA TGA GTG TTA CTC AAG AAG CAG AAA GGG AAG AAT TTT TTG and NotI(F) GCC GCG GCC GCA CCA TGC GTG AAG AAA AGA TCC TCA ATC GAG AAA TTG GTT TTG, respectively. The reverse primer used for amplification of Δ72-hp110α and Δ108-hp110α was BamHI(R) TAT CGG ATC CTC AGT TCA ATG CAT GCT GTT TAA TTG TGT GGA AGA TC. To generate the RBD mutant constructs, pBSFI vector KOZ-cp3k encoding wt, E545K, H1047R were used as templates and the primers used were: T208D 5′-CAA ATA ATG ACA AGC AGA AGT ATG ATC TGA AAA TCA ACC ATG AC-3′ and K227A 5′-GCT GAA GCA ATC AGG GCA AAA ACT AGA AGT ATG TTG-3′ (only the forward primer is listed). The mutated genes were subsequently cloned into the avian retrovirus vector RCAS.Sfi.34 All mutations were confirmed by sequencing. The p85α-HA expression construct was generated by PCR amplification of pCAGGS-p85α, a generous gift from Dr. Tomoichiro Asano (Tokyo University, Tokyo, Japan), and subcloned into the avian retroviral vector RCAS.Sfi.
Cell Culture and Transfection
Fertilized chicken eggs (white Leghorn) were obtained from Charles River Breeding Laboratories (Wilmington, MA). Preparation and cultivation of primary CEF have been described previously.35 For focus assays, DNA was transfected into CEF by using the dimethyl sulfoxide/Polybrene method as described previously.14 For serum starvation, CEF were first maintained in Ham's F-10 medium with 0.25% fetal bovine serum and 0.05% chicken serum for 40–44 hours, followed by additional 2 hours in Ham's F-10 medium, and harvested for protein analyses.
Immunoprecipitation and Western blotting
Western blotting was performed as described14 with minor modifications. Cells were lysed in 1 × Passive Lysis buffer (Promega, Madison, WI) containing 1× protease Inhibitor cocktail (Roche, Indianapolis, IN), with 1 mM DTT/1 mM PMSF/50 mM β-glycerophosphate/50 mM NaF/1 mM Na3VO4. Whole cell lysates were incubated with mouse monoclonal anti-HA agarose (Sigma, St. Louis, MO) at 4°C overnight. The immune complexes were pelleted and washed three times with cold lysis buffer in the presence of protease and phosphatase inhibitor. For Western-blot analysis, the bound proteins were eluted by boiling in SDS loading buffer, resolved on SDS-PAGE gel, then transferred to Immobilon-P membranes (Millipore, Billerica, MA). After the membranes were blocked with 5% BSA in TBS-T (Tris-buffered saline with 0.05% Tween-20) for 1 hour at room temperature, they were incubated overnight at 4°C with primary antibodies. Anti-HA, anti-p85α, anti-AKT, anti-phospho-AKT (S-473), anti-phospho-AKT (T-308), and anti-β-actin were purchased from Cell Signaling Technology (Beverly, MA). Membranes were washed three times in TBS-T and incubated with secondary antibody (Pierce, Rockford, IL) in 5% non-fat dry milk in TBS-T for 1 hour at room temperature. The reactive bands were visualized by chemiluminescence (Pierce, Rockford, IL).
Acknowledgments
We thank Lynn Ueno for expert and skilled technical assistance. This work was supported by grants from the National Cancer Institute and by The Stein Foundation. Dr. Li Zhao is the recipient of a postdoctoral fellowship from the National Cancer Institute (F32CA130304). This is manuscript no. 20291-MEM from The Scripps Research Institute.
Abbreviations
- ABD
adaptor binding domain
- AKT
cellular homolog of murine thymoma virus akt8 oncogene, also referred to as protein kinase B
- CEF
chicken embryo fibroblasts
- N-SH2
N-terminal SH2 domain of p85
- PI3K
phosphatidylinositol 3-kinase
- RBD
RAS-binding domain
- SH2
Src homology 2
- wt
wild-type
References
- 1.Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–50. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 2.Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–81. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 3.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 4.Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–7. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 5.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–7. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- 7.Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–8. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 8.Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–9. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 9.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–8. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475–9. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104:5569–74. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–7. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008;27:5486–96. doi: 10.1038/onc.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shekar SC, Wu H, Fu Z, Yip SC, Nagajyothi, Cahill SM, et al. Mechanism of constitutive phosphoinositide 3-kinase activation by oncogenic mutants of the p85 regulatory subunit. J Biol Chem. 2005;280:27850–5. doi: 10.1074/jbc.M506005200. [DOI] [PubMed] [Google Scholar]
- 17.Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 18.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 19.Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409:519–24. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 20.Chaussade C, Cho K, Mawson C, Rewcastle GW, Shepherd PR. Functional differences between two classes of oncogenic mutation in the PIK3CA gene. Biochem Biophys Res Commun. 2009;381:577–81. doi: 10.1016/j.bbrc.2009.02.081. [DOI] [PubMed] [Google Scholar]
- 21.Aoki M, Schetter C, Himly M, Batista O, Chang HW, Vogt PK. The catalytic subunit of phosphoinositide 3-kinase: requirements for oncogenicity. J Biol Chem. 2000;275:6267–75. doi: 10.1074/jbc.275.9.6267. [DOI] [PubMed] [Google Scholar]
- 22.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 23.Hiles ID, Otsu M, Volinia S, Fry MJ, Gout I, Dhand R, et al. Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell. 1992;70:419–29. doi: 10.1016/0092-8674(92)90166-a. [DOI] [PubMed] [Google Scholar]
- 24.Hu P, Mondino A, Skolnik EY, Schlessinger J. Cloning of a novel, ubiquitously expressed human phosphatidylinositol 3-kinase and identification of its binding site on p85. Mol Cell Biol. 1993;13:7677–88. doi: 10.1128/mcb.13.12.7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–87. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. Embo J. 1996;15:2442–51. [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 28.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–43. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 29.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2007 doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- 30.Gabelli S, Huang CH, Mandelker D, Schmidt-Kittler O, Vogelstein B, Amzel LM. Current Topics in Microbiology and Immunology. Springer; Structural Effects of Oncogenic PI3Kα Mutations. in press. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang CH, Mandelker D, Gabelli SB, Amzel LM. Insights into the oncogenic effects of /PIK3CA/ mutations from the structure of p110alpha/p85alpha. Cell Cycle. 2008;7 doi: 10.4161/cc.7.9.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–7001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289–94. doi: 10.1073/pnas.0510772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci U S A. 1998;95:14950–5. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogt PK. Focus Assay of Rous Sarocma virus. In: Habel K, Salzman NP, editors. Fundamental techniques in virology. New York, N.Y.: Academic Press; 1969. pp. 198–211. [Google Scholar]