

1. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease that can affect any organ system. Pathogenic autoantibodies are a hallmark of this disease, and have been shown to play a necessary role in many of the manifestations of lupus. Although many molecular pathways can be abnormal in lupus and several cell types can become dysregulated, B cells have emerged as central players in this disease because autoantibodies are key to diagnosis and because autoantibodies arise years before there is any other evidence of immune dysregulation (Arbuckle, McClain et al. 2003). B cells are not only the cells that secrete autoantibodies; they also play crucial roles in antigen presentation and cytokine secretion.
In this review, we will examine the pathogenic role played by B cells in lupus, including a discussion of the importance of different B cell subsets in lupus development and flare. We will examine important pathways involved in the generation of autoreactive B cells, focusing on the role of B cell receptor (BCR) signaling in B cell escape from negative selection early in B cell development, and again in the censoring of autoreactive mature B cells that emerge from the germinal center (GC). We will examine how non-BCR mediated signaling in B cells can contribute to lupus. Because lupus is predominantly a disease of women, we will discuss the effect of estrogen on B cell tolerance. Lastly, we will briefly examine B-cell directed therapies in lupus.
2. Pathogenic role of B cells in lupus
B cells are important initiators and effectors of a normal immune response. In autoimmunity, B cells carry out those same roles, turning their arsenal towards self antigens. Autoantibodies are a defining characteristic of lupus, and many antibodies make a clearly delineated contribution to disease pathogenesis, such as anti-DNA antibodies, which we will discuss in detail and which contribute to kidney and brain disease, anti-β2 glycoprotein I and anti-cardiolipin antibodies that predispose to thrombosis, and anti-Ro antibodies that cause fetal heart block in the offspring of women with lupus (Tomer, Buskila et al. 1993). Other antibodies are of diagnostic use, such as the highly disease-specific anti-Smith (Sm) antibody.
Anti-DNA antibodies are the most extensively studied specificity in lupus. These antibodies have been shown to be present in 50-70% of SLE patients at some point in their disease and are a highly specific diagnostic marker (Pisetsky 2000). With few exceptions, these antibodies to double-stranded DNA (dsDNA) are detected only in lupus patients. A number of studies have shown that titers of anti-DNA antibodies tend to rise during flares of SLE disease activity, particularly lupus nephritis (ter Borg, Horst et al. 1990). In addition, in vivo murine studies have shown that passive transfer of some anti-DNA antibodies can deposit in glomeruli leading to inflammation and proteinuria (Ehrenstein, Katz et al. 1995; Gaynor, Putterman et al. 1997). It is important to note that not all anti-DNA antibodies are pathogenic; some anti-DNA antibodies have no pathogenic effect despite their binding DNA with affinities that are equal to those of pathogenic antibodies. Recent studies have suggested that certain isotypes and antigen binding properties are associated with pathogenicity. IgG anti-dsDNA antibodies, for example, are more closely associated with disease activity and tissue damage than IgM antibodies (Isenberg, Ravirajan et al. 1997). Indeed, there is increasing evidence that IgM anti-DNA antibodies may actually be protective (Witte 2008). Anti-dsDNA antibodies are more pathogenic than anti-single-stranded DNA antibodies (Okamura, Kanayama et al. 1993). Anti-DNA antibodies from SLE patients with renal lupus display a higher affinity for DNA (Williams, Malone et al. 1999). Anti-DNA antibodies extracted from kidney are more cationic than serum anti-DNA antibodies (Cabral and Alarcon-Segovia 1997). Furthermore, many display cross-reactivity to glomeruli even after DNase treatment of the glomeruli (Budhai, Oh et al. 1996). A recent understanding of DNA interactions with toll like receptor 9 (TLR9) (Krieg and Vollmer 2007), an innate receptor for DNA in monocytes, dendritic cells, B cells and other cell types, suggests that the particular DNA motif recognized by an anti-DNA antibody may also determine its pathogenicity. Distinguishing pathogenic anti-DNA antibodies from harmless ones will provide a useful diagnostic and prognostic tool.
Antibodies to naked dsDNA develop after anti-nucleosome antibodies in both murine and human disease (Hardin and Craft 1987). Recent studies have suggested that nucleosomes, which consist of DNA wrapped around a core of histone proteins, may in fact be more important antigenic targets in lupus than naked DNA. The presence of T helper cells specific for histone peptide has been demonstrated in both patients and murine models of SLE (Kaliyaperumal, Mohan et al. 1996; Lu, Kaliyaperumal et al. 1999). Furthermore, levels of circulating nucleosomes have been shown to be increased in the plasma of lupus patients (Williams, Malone et al. 2001). Nucleosomes are present in apoptotic blebs that form at the surface of dying cells (Radic, Marion et al. 2004). This is of great interest because a number of abnormalities that impair the clearance of apoptotic debris have been associated with lupus. These include deficiency of complement components C1q, C2, C4 (Truedsson, Bengtsson et al. 2007) and mannose-binding lectin (Monticielo, Mucenic et al. 2008), of DNAse I (Tsukumo and Yasutomo 2004; Martinez Valle, Balada et al. 2008), and of proteins expressed by macrophages that are necessary for their clearance of apoptotic debris, such as Macrophage Receptor with Collagenous Structure (MARCO) (Wermeling, Chen et al. 2007), Scavenger Receptor A (SR-A), and the Mer tyrosine kinase (Cohen, Caricchio et al. 2002). Recently, levels of anti-nucleosome antibodies have been shown to correlate highly with lupus disease activity (Min, Kim et al. 2002), particularly with renal flare (Simon, Cabiedes et al. 2004). Patients with higher titers of anti-nucleosome antibodies have a shorter time to first flare after a serologically active but clinically quiescent period (Ng, Manson et al. 2006). These studies suggest that titers of anti-nucleosome antibodies may be better than titers of anti-DNA antibodies in predicting flare.
How autoreactive antibodies develop has been intensely studied. Many pathogenic anti-DNA antibodies appear to be the products of a germinal center reaction: they exhibit heavy chain class-switching and have undergone somatic hypermutation (Diamond, Katz et al. 1992). Molecular analysis of anti-dsDNA antibodies from humans and mice suggests that there is an antigen-driven selection of these mutations in at least some antibodies. For example, a high frequency of replacement mutations to the amino acids Arginine, Asparagine and Lysine has been observed in the complementarity-determining regions of murine and human anti-dsDNA IgG antibodies (Radic and Weigert 1994). It has been postulated that these amino acid residues enhance the affinity for DNA, partially due to the positive charge of their side chains. Single-cell analysis of IgG memory B cells from SLE patients and healthy controls demonstrated that the majority of autoreactive IgG antibodies arise from nonautoreactive precursors, because most of these autoantibodies lost reactivity to tested self antigens when their sequences were back-mutated to their germ line configuration (Mietzner, Tsuiji et al. 2008). In another study, use of a tetrameric form of a peptide mimetope of dsDNA allowed identification of an IgM+ autoreactive B cell population in the peripheral blood of SLE patients (Zhang, Jacobi et al. 2008a). Interestingly, back-mutation analysis of three autoantibodies from this population revealed that two of them lost reactivity to self antigens, but one retained reactivity to DNA when reverted to the germ line sequence (Zhang, Jacobi et al. 2008b). This study demonstrates that autoantibodies can be generated from self-reactive or non self-reactive B cell precursors, and that chromatin is not the trigger for all anti-DNA antibodies.
The antigens other than chromatin that trigger production of anti-DNA antibodies in lupus remain a mystery. Cross-reactivity of anti-DNA antibodies with other antigens has clearly been established. Immunization of mice with phosphorylcholine, a molecule found in various membranes, was shown to induce generation, through somatic mutation, of DNA-reactive B cells, although these cells never move to the memory B cell compartment (Kuo, Bynoe et al. 1999). This is important because phosphorylcholine is expressed by a number of bacteria, including Streptococcus, Haemophilus and Mycoplasma, and anti-phosphorylcholine antibodies are protective in mice against a lethal pneumococcal infection (Trolle, Chachaty et al. 2000), demonstrating that antibodies that cross-react with DNA can arise in the course of a protective response to an infectious agent. Epstein Barr virus has been a favored candidate trigger as some anti-DNA antibodies cross-react with EBNA1 protein (Sundar, Jacques et al. 2004). Environmental triggers of lupus are known to be important, but no study has clearly established that a particular pathogen is needed to trigger the disease.
Anti-DNA antibodies can also cross-react with other self-antigens. It has been demonstrated that certain anti-DNA antibodies can bind to N-methyl D Aspartate (NMDA) receptors on neurons (DeGiorgio, Konstantinov et al. 2001). Binding of these antibodies to NMDA receptors was shown to induce excitation-mediated neuronal death and cerebrospinal fluid levels of the cross-reactive antibodies correlate with central nervous system manifestations of lupus. Furthermore, in mice, these antibodies were shown to pass through the placenta during pregnancy and deposit in the developing fetal brain, resulting in behavioral abnormalities that persist through adulthood (to be published in Nature Medicine).
In additon to secreting pathogenic antibodies, B cells also play other important roles in lupus. B cells are known to be professional antigen presenting cells, and secrete both pro-and anti-inflammatory cytokines. The role of B cells as antigen-presenting cells and cytokine secretors in autoimmunity was demonstrated in the MRL/lpr murine model, where elimination of B cells completely abrogates disease (Shlomchik, Madaio et al. 1994). The presence of B cells that are unable to secrete antibody but can still function as antigen presenting cells and still secrete cytokine, results in some kidney and vascular disease, albeit less than when fully functional B cells are present (Chan, Hannum et al. 1999). Thus, B cells contribute more than autoantibody to autoimmune pathogenesis. B cells activate T cells by surface expression of peptide-MHC complexes that interact with the T cell receptor (TCR), as well as a number of other molecules on the B cell surface, such as C80 and CD86, CD40, Inducible Costimulator (ICOS) ligand, and OX40 ligand, which bind to CD28, CD154 (CD40 ligand), ICOS and OX40, respectively, on the T cell surface. In addition, activated B cells themselves express CD154, and B cell to B cell CD40-CD154 interactions have been shown to be necessary for normal memory B cell differentiation and development of plasma cells from memory B cells (Grammer and Lipsky 2003). This is of interest because CD154 has been found to be over-expressed by B cells of lupus-prone mice and some lupus patients.
Another central function of B cells is cytokine secretion. B cells have been shown to produce Interleukin (IL)-4, IL-6, IL-10, Interferon (IFN)-γ, Transforming Growth Factor-β and Lymphotoxin-α (Anolik 2007). Lymphotoxin-α is important for the formation of tertiary lymphoid tissue. This tissue consists of organized collections of lymphocytes in non-lymphoid peripheral organs, where such immune aggregates are not normally found. In lupus, tertiary lymphoid tissue has been demonstrated in the kidney. Tertiary lymphoid tissue can also be seen in rheumatoid arthritis (RA), Sjogren’s syndrome, inflammatory bowel disease, Type I diabetes, and autoimmune thyroid disease. Lymphotoxin-α secreted by B cells has been shown to be necessary for the formation of these tertiary lymphoid tissues.
While IL-6 and IFN-γ are pro-inflammatory cytokines secreted by B cells, B cells are also able to secrete IL-10, which, in many cases, has been shown to dampen inflammation. Hence, the B cells that primarily secrete IL-10 have been termed regulatory B cells. Early evidence for regulatory B cells in autoimmune diseases came from the observation that B10.PL mice lacking B cells developed a more severe and chronic form of experimental autoimmune encephalomyelitis (EAE) (Wolf, Dittel et al. 1996). Delineation of the underlying mechanism revealed that B cells regulate disease severity through production of IL-10. The transitional B cell subset that comprises marginal zone precursors is a major B cell subset producing IL10 (Yanaba, Bouaziz et al. 2008). This may be a mechanism by which a reconstituting B cell compartment might be anti-inflammatory. IL-10-producing regulatory B cells were also found to play a role in suppressing other murine models of autoimmune disease, such as inflammatory bowel disease (Mizoguchi, Mizoguchi et al. 2002) and collagen-induced arthritis (Mauri, Gray et al. 2003). Studies have shown that B cell-derived IL-10 plays a beneficial role in murine lupus models by inhibiting Th1 cytokine production and ensuing tissue damage. IL-10 deficient mice with SLE develop more severe lupus associated with higher levels of Th1 cytokines (Yin, Bahtiyar et al. 2002). It is worth noting, however, that IL-10 is also a suppressor of Th2-mediated immune pathology such ulcerative colitis and schistosomiasis (Hoffmann, Cheever et al. 2000). Therefore, immune deviation toward a Th2 response cannot fully explain the regulatory role of IL-10 producing B cells. Unfortunately, the role of IL-10 in human lupus remains controversial and paradoxically, most data suggest that IL-10 enhances, rather than prevents, disease. Blocking IL-10 with antibody was shown to reduce disease activity in refractory cases of SLE (Llorente, Richaud-Patin et al. 2000). Also, an IL-10 promoter polymorphism leading to increased cytokine expression is associated with higher susceptibility to SLE (Chong, Ip et al. 2004). These conflicting results highlight the need to better understand the role of IL-10 and IL-10 producing B cells in lupus.
3. Contribution of different B cell subsets to lupus
Mature B cells are designated as either B1 or B2 cells, and the latter are further divided into follicular and marginal zone B cells. While all three subsets are able to secrete anti-DNA antibodies (Schiffer, Hussain et al. 2002), a major focus has been on the role of follicular B cells, as these are the B cells that were known to participate in T-dependent immune responses that involve germinal center reactions, and early studies of murine lupus emphasized the role of the germinal center reaction. Recent evidence, however, also points to an important contribution of marginal zone B cells. Marginal zone B cells are expanded in NZB/W mice, and have been found to produce 25 times higher levels of anti-DNA IgM than non-marginal zone B cells (Zeng, Lee et al. 2000). In the context of high BAFF levels (Bossen and Schneider 2006) or TLR9-activating DNA (Jegerlehner, Maurer et al. 2007), these B cells may switch to production of IgG antibodies independently of T cell stimulation. Mice that over-express B cell activator of the TNF family (BAFF), an important B cell survival factor, show a lupus-like phenotype and expansion of marginal zone B cells (Mackay, Silveira et al. 2007). Estrogen has been implicated in the pathogenesis of lupus (discussed below). In an estrogen-induced model of lupus, marginal zone B cells are expanded and these cells display a ten-fold increased frequency of DNA reactivity than follicular B cells (Grimaldi, Michael et al. 2001). In addition to secreting anti-DNA antibodies, marginal zone B cells appear to play an important role in antigen presentation, as demonstrated by the recent finding that these B cells are not confined to the marginal zone and frequently shuttle back and forth between the marginal zone and the follicular area, where helper T cells and follicular dendritic cells reside (Cinamon, Zachariah et al. 2008).
Different B cell subsets may contribute differentially to disease flare. After antigen activation, B cells can become short-lived plasma cells, long-lived plasma cells, or memory cells. The former usually develop after T-independent activation, while the latter two are typically T-cell-dependent. Short lived plasma cells live weeks to months, reside in the tissue where they are generated, and, in vitro, are unable to secrete antibody in the presence of anti-proliferative drugs (Grammer and Lipsky 2003). Long-lived plasma cells, on the other hand, home to the bone marrow where they live and secrete antibody for many years. These plasma cells have been shown to secrete antibody even in the presence of anti-proliferative drugs. This is of interest because different autoantibodies in lupus show different patterns of expression. Anti-RNP antibodies, for example, show a stable pattern of expression over a patient’s lifetime, and their levels are not typically affected by immune suppression. This pattern is suggestive of antibody secretion by long-lived plasma cells. In contrast, anti-DNA antibodies fluctuate significantly in titer, and often rise in association with clinical flares and diminish with immunosuppressive treatment. This suggests that these antibodies, which are the one most closely associated with pathology in lupus, are produced by short-lived plasma cells, and suggests that flares may arise either with a new wave of activation of naïve autoreactive B cells or with activation of memory B cells.
4. The role of BCR signaling in predisposition to lupus
B cells pass through various developmental stages before they become mature cells that can be activated by encounter with antigen. Of central importance to this maturation is the process of negative selection, which eliminates immature B cells that react to self antigen with high affinity. B cells form immunoglobulin by random rearrangements of a set of genes, which for the heavy chain include the V (variability), D (diversity) and J (joining) gene segments, and for the light chains include V and J segments. This process generates a wide diversity of receptors, and hence provides protection against a tremendous number of foreign pathogens. However, this process also generates receptors that recognize self antigens. In fact, experimental evidence suggests that the majority of B cells at the immature stage have receptors that recognize self antigen (Wardemann, Yurasov et al. 2003). During the immature stage in the bone marrow and the transitional stage within the spleen and tonsils, B cells are susceptible to negative selection, wherein an encounter with antigen that triggers BCR signaling beyond a certain threshold results in receptor editing (generation of a new light chain rearrangement), anergy, or deletion. It follows that abnormalities that affect BCR signaling can predispose to autoimmune disease.
The majority of research until recently has focused on increased BCR signaling as a pathway to autoimmunity, with the demonstration that over-expression of molecules that enhance the strength of BCR signaling, such as CD19 (Tsubata and Honjo 2000), or deletion/dysfunction of molecules that reduce BCR signaling, such as SHP-1 (Shultz, Rajan et al. 1997) and FcγRIIb (Kono, Kyogoku et al. 2005), lead to autoimmunity. This model depends on the same exposure to antigen delivering a stronger signal in mature B cells than in immature B cells, such that exposure to antigen does not mediate tolerance in immature cells but does mediate activation of mature B cells. This model requires that the strength of BCR signaling differs at different stages of B cell development and indeed, experimental data confirm some differences in signaling mechanisms between immature and mature B cells (Koncz, Bodor et al. 2002).
A growing body of literature is now demonstrating that diminished BCR signaling can also lead to autoimmunity. Given that negative selection depends on BCR signaling (Monroe, Bannish et al. 2003), it is clear that diminished signaling allows B cells that express self-reactive immunoglobulins to escape negative selection, and hence become mature immunocompetent cells that can contribute to autoimmune disease. Evidence for this model was provided by the study of B cells in patients with X-linked agammaglobulinemia (XLA), a disease caused by defects in Bruton’s tyrosine kinase, a mediator of BCR signaling (Ng, Wardemann et al. 2004). This study found that peripheral B cells from XLA patients were enriched with autoreactive clones, demonstrating a link between diminished BCR signaling and increased autoreactivity in the naïve B cell population. A similar study of B cells from lupus patients shows a higher frequency of autoreactive cells in the immature, transitional and naïve B cell compartments compared to non-autoimmune individuals (Yurasov, Wardemann et al. 2005), demonstrating impaired negative selective and consistent with diminished BCR signaling strength. Compelling evidence for diminished signaling in autoimmunity comes from the study of the PTPN22 gene. PTPN22 is a phosphatase that inhibits BCR and TCR signaling. A polymorphism in this gene, R620W, has been associated with autoimmunity (Vang, Miletic et al. 2007). It has been shown that the risk allele causes a gain of function (Vang, Congia et al. 2005), and inhibits TCR and BCR signaling to a greater extent than the protective allele (Rieck, Arechiga et al. 2007). An important prediction of the model wherein diminished BCR signaling leads to B cell escape from negative selection is that this defect would predispose to multiple forms of autoimmunity; this is borne out by the association of the PTPN22 risk allele with lupus, rheumatoid arthritis, type I diabetes, and autoimmune thyroid disease, all of which have critical contributions from B cells, either as antibody-producing cells or antigen-presenting cells. A recent genome-wide association study has found that alleles of the gene BLK are associated with lupus (Hom, Graham et al. 2008). BLK is a mediator of BCR signaling, and the alleles associated with lupus risk correlate with decreased BLK expression, furthering the link between diminished BCR signaling and lupus.
Diminished BCR signaling has also been implicated in mouse models of lupus. The NZM2410 is a strain that has highly penetrant autoimmune disease that includes glomerulonephritis. Various genetic susceptibility loci have been identified in these mice, and the Ly108 gene was recently identified as a likely culprit gene within the SLE1b locus (Kumar, Li et al. 2006). The autoimmune-associated variant of this gene was shown to decrease BCR signaling, and cause diminished BCR-mediated apoptosis of an immature B cell line, mirroring a failure of negative selection.
Our laboratory has worked on a murine model of lupus in which diminished BCR signaling is associated with a naïve B cell repertoire enriched for DNA-reactivity. While Balb/c mice do not develop any spontaneous autoimmune disease, they can be induced to produce anti-DNA antibodies by immunization with a peptide mimetope for DNA, named DWEYS-MAP (Putterman and Diamond 1998). The antibodies generated in this way have been shown to deposit in glomeruli and contribute to lupus central nervous system disease (DeGiorgio, Konstantinov et al. 2001). Comparison of Balb/c to DBA/2 mice, which are resistant to the induction of autoimmunity by MAP-DWEYS, reveals that Balb/c B cells have a lower BCR signaling amplitude, less apoptosis in transitional B cells in response to BCR cross-linking, and a higher prevalence of naïve, mature splenic DNA-reactive B cells (Wang, Khalil et al. 2003). These studies further support the model where a defect that attenuates BCR signaling strength allows autoreactive immature or transitional B cells to escape negative selection and hence become immunocompetent cells that can contribute to the pathogenesis of lupus (Figure 1).
Figure 1.
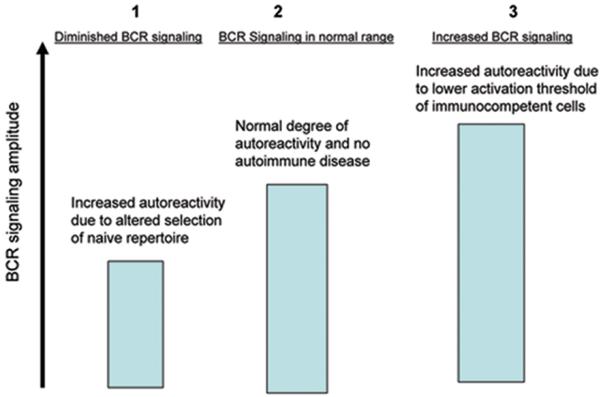
BCR signaling strength can also affect B cell fate after antigen activation. In mice transgenic for the heavy chain of an antibody to the hapten nitrophenyl (NP), B cells with low affinity BCRs participate in germinal center reactions and produce both memory and long-lived plasma B cells after antigen challenge, whereas B cells with high affinity BCRs differentiate into short-lived plasma cells and do not enter germinal center reactions (O’Connor, Vogel et al. 2006). This is of note because, as previously discussed, many pathogenic anti-DNA antibodies in lupus demonstrate class switch to IgG and affinity maturation, both of which are hallmarks of the germinal center reaction.
5. Tolerance induction in antigen-activated B cells
In recent years, a series of peripheral tolerance checkpoints have been identified in antigen-experienced B cells, both in humans and in mice. Single-cell studies showed that in humans, up to 20% of IgM+ naïve mature B cells express low-affinity self-reactive antibodies (Tsuiji, Yurasov et al. 2006). However, these self-reactive B cell clones are removed from the IgM+ memory compartment before the onset of somatic mutation. The exclusion from the memory compartment seems to be specific for self-reactive B cells, because cells expressing antibodies specific for bacterial polysaccharides are allowed to enter the memory pool. This suggests that a checkpoint exists at the junction between naïve and memory B cells. Studies of human tonsillar B cells have identified a tolerance checkpoint before entry into the germinal center response. 9G4 is an anti-idiotype antibody that delineates B cells that are reactive to self glycoproteins and apoptotic cells (Cappione, Anolik et al. 2005). 9G4 positive B cells represent 5-10% of the mature, naïve B cell repertoire in normal individuals. In healthy individuals, 9G4 B cells are prevented from participating in the germinal center response. In contrast, germinal center exclusion of 9G4 B cells is impaired in SLE patients and the autoreactive B cells are able to expand through the germinal center response and differentiate into IgG-expressing memory and plasma cells. Interestingly, the faulty regulation of 9G4 B cells seems to be specific for human SLE, because it is not observed in RA patients. Thus, germinal center exclusion represents an important mechanism in peripheral B cell tolerance and loss of this checkpoint may be implicated in the pathogenesis of SLE.
Interestingly, a recent study reported that approximately 50% IgG+ memory B cells from healthy humans express self-reactive antibodies, including anti-nuclear antibodies, and many of them are polyreactive (Tiller, Tsuiji et al. 2007). The self-reactivity in the IgG+ memory B cell compartment appears to represent a by-product of somatic mutation in B cells responding to foreign antigens (Diamond and Scharff 1984; Ray, Putterman et al. 1996), as the germ line encoded precursor antibodies are often not autoreactive. Moreover, it is not known how many of these self-reactive IgG+ memory B cells eventually develop into plasma cells. Given the fact that non-autoimmune humans do not routinely express pathogenic autoantibodies, there might be a checkpoint that prevents activation and expansion of these autoreactive IgG+ memory cells.
B cells may acquire autoreactivity as a result of somatic hypermutation in the germinal center. These antigen-activated autoreactive B cells are tightly regulated by a number of checkpoints along the differentiation pathway toward antibody-secreting plasma cells. Newly generated antigen-specific memory B cells have been shown to pass through a window of tolerance susceptibility (Linton, Rudie et al. 1991). This tolerance can be induced by exposure to soluble antigen. Soluble antigen is able to reduce the primary antigen-specific antibody response when it is administered to mice several days after immunization (Nossal, Karvelas et al. 1993). Other studies have identified the germinal center as an important checkpoint for antigen-activated autoreactive B cells. At the peak of the germinal center response, administration of a large amount of soluble antigen induces extensive apoptosis of light zone B cells in mice immunized with the same antigen or a cross-reactive antigen (Han, Zheng et al. 1995; Pulendran, Kannourakis et al. 1995; Shokat and Goodnow 1995). B cell death in the germinal center can be inhibited completely or partially by constitutive overexpression of a Bcl-2 transgene. The deletion induced by soluble antigen is antigen specific and is thought to be a mechanism for peripheral tolerance of B cell clones that acquire self reactivity through the process of somatic hypermutation.
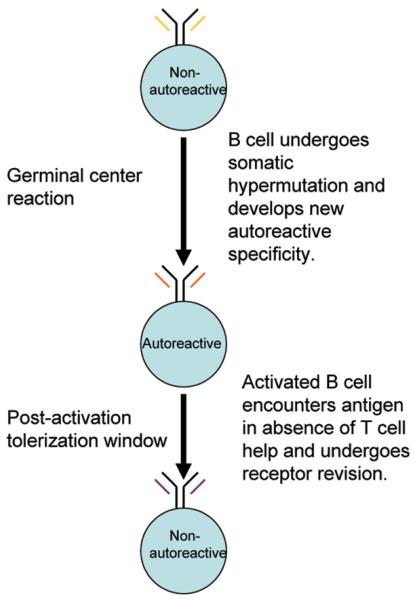
Using the DWEYS-MAP model, we have identified a tolerance checkpoint in early memory/pre-plasma cells at a post-germinal center stage (Wang and Diamond 2008). We observed de novo expression of recombinase-activating genes (RAG) and secondary light chain rearrangement, termed receptor revision, in post-germinal center autoreactive B cells. Expression of RAG is antigen dependent and required IL-7R signaling. The production of autoantibody is markedly elevated when receptor editing is suppressed by inhibiting IL-7 signaling. Therefore, receptor revision in the early memory population plays a potent role in restricting autoantibody production during an ongoing immune response. Interestingly, we observed that the potential for induction of receptor revision is impaired in aged NZBW F1 mice but intact in young mice (unpublished data). This suggests that defects in receptor revision in antigen-activated B cells may be implicated in the breach of self tolerance and contribute to pathogenesis in lupus (Figure 2).
Figure 2.
Studies of immunoglobulin transgenic mice have revealed several additional tolerance checkpoints in antigen-activated B cells in the immune system. In rheumatoid-factor (RF) transgenic mice, for instance, RF-expressing autoreactive B cells are subject to regulation at two checkpoints after their initial activation (William, Euler et al. 2006). The RF B cells participate in germinal center formation and undergo somatic mutation in both autoimmune and non-autoimmune backgrounds. In non-autoimmune mice, however, RF B cells neither differentiate into plasma cells nor clonally expand, thus preventing the generation of pathologic autoantibodies. In autoimmune-prone mice the regulation of RF B cells in germinal centers is abrogated, leading to production of high titers of autoantibody.
Tolerance induction has also been reported at the early pre-plasma cell stage in anti-Sm heavy chain transgenic B cells (Culton, O’Conner et al. 2006). Anti-Sm B cells are present at a high frequency in the spleen and bone marrow of the transgenic mice and express the plasma cell marker CD138; however, these cells do not differentiate into antibody secreting cells in normal mice. Regulation of anti-Sm B cells occurs before the expression of Blimp1, the transcriptional repressor required for plasma cell differentiation. In addition, these anti-Sm B cells display a higher turnover rate than B cells not binding Sm, suggesting that they have a shorter lifespan. It has been shown that IL-6 contributes to sustained non-responsiveness in these B cells. Thus, IL-6 appears to be a mechanism for sustaining B cells in an anergic state after antigen activation.
These studies, together with earlier studies inducing tolerance with soluble antigen, suggest that antigen activated B cells are susceptible to tolerance induction. However, the mechanisms by which they are tolerized remains to be elucidated. Clonal deletion cannot account all the tolerance induction because many autoreactive B cells are not eliminated. Other mechanisms that appear to be operative in preventing the cells from further evolving into plasma cells or memory cells, include the induction of anergy, and alteration of the BCR specificity through secondary V (D)J rearrangement, but details of these processes are lacking.
6. Non-BCR-mediated signaling in autoreactive B cells
Non-BCR pathways of note in lupus include the Toll-like receptor (TLR), FcγRIIb, and BAFF signaling pathways. Contributing to their ability to induce inflammation in lupus, B cells express of a number of Toll-like receptors, in particular TLR7 and TLR9, which recognize single-stranded RNA and DNA rich in unmethylated CpG, respectively. Both DNA and RNA are found in the apoptotic blebs that are thought to be important to lupus pathogenesis. B cells that express DNA-reactive BCRs can be activated by DNA simultaneously through the BCR and TLR9 signaling pathways (Viglianti, Lau et al. 2003), which leads to augmented activation compared to signaling by either pathway alone. Similarly, B cells with receptors specific for RNA can be activated by ribonucleoproteins simultaneously through the BCR and TLR7 signaling pathways (Krieg and Vollmer 2007). TLR7 is of further interest because the gene is duplicated in the Yaa chromosomal abnormality found in the BXSB murine lupus model, and this increased gene dosage has been shown to contribute to autoimmunity in this mouse (Fairhurst, Hwang et al. 2008). Another pathway of note is that of FcγRIIb, which is the only Fcγ receptor is activated by the Fc portion of cross-linked IgG molecules, and dampens B cell activation by the recruitment of the phosphatase SHIP, which dephosphorylates and thus inactivates mediators of BCR signaling. Recently, lupus-prone strains of mice were shown to have low levels of FcγRIIb expression and over-expression of FcγRIIb in B cells was shown to diminish anti-DNA antibody levels and proteinuria in the NZM2410 and BXSB lupus-prone mouse models (McGaha, Sorrentino et al. 2005). FcγRIIb has been shown to be critical for increasing the signaling threshold for memory B cell activation. FcγRIIb is upregulated on memory B cells in normal humans, but this upregulation is significantly decreased in SLE patients (Mackay, Stanevsky et al. 2006). Accordingly, there is a decreased FcγRIIb-mediated suppression of BCR activation in B cells from lupus patients. The abnormally low expression of FcγRIIb may impair tolerance induction in memory B cells or lead to activation by a diminished concentration of antigen and contribute to disease development.
BAFF is a molecule of great relevance to normal B cell physiology and autoreactivity (Mackay, Silveira et al. 2007). BAFF can either be expressed on the cell surface or can be secreted as a homotrimer. The cells that typically express BAFF include monocytes, macrophages, dendritic cells and activated T cells, but recently, other cell types have been shown to produce BAFF, including astrocytes, bone marrow stromal cells, osteoclasts and epithelial cells. Inflammatory cytokines such as IFN-γ, as well as TLR agonists such as LPS, upregulate expression of BAFF. BAFF has three receptors: BAFF-R, TACI (transmembrane activator and CAML [calcium modulator and cyclophilin ligand] interactor), and BCMA (B cell maturation antigen). All of these receptors are expressed on B cells, but at different levels depending on developmental stage. BAFF signaling has been shown in vitro to promote survival of B cells after the T1 transitional stage of development. Consistent with this finding, BAFF deficient mice lack B cell development past the T1 transitional stage. One pathway by which BAFF has been shown to promote B cell survival is the induction of NF-κB activation through the alternate NF-κB pa thway. BAFF-transgenic mice develop an expansion of the peripheral B cell pool, in particular marginal zone B cells, and spontaneously produce autoantibodies. Elevated BAFF levels have been found in the serum of various autoimmune mouse models, and in 20–50% of patients with various autoimmune diseases. BAFF-R-Ig and TACI-Ig, soluble receptors for BAFF, diminish serum BAFF levels and have shown promise in treating mouse models of lupus. Studies in humans are ongoing.
Women are nine times more likely to be afflicted with lupus than men (Grimaldi, Hill et al. 2005), and the common age of onset is between menarche and menopause. Yet prepubertal girls are only three times more likely to develop lupus than boys (Buoncompagni, Barbano et al. 1991). Because of these data, a role for sex hormones in lupus has been postulated. In a large, randomized-controlled study, post-menopausal women with lupus were found to have a higher rate of flare if they received hormone replacement compared to those who received placebo (Buyon, Petri et al. 2005). In animal models, estrogen has been found to accelerate disease in both NZB/W (Roubinian, Talal et al. 1978) and MRL/lpr mice (Carlsten, Tarkowski et al. 1990). The mechanism by which estrogen induces or aggravates autoimmunity has been studied using Balb/c mice transgenic for the heavy chain of an anti-DNA antibody. The transgenic heavy chain is able to assoicate with endogenous light chains to form BCRs of varying affinity for DNA. These mice do not spontaneously develop autoimmunity as B cells with high affinity DNA-reactive receptors are able to undergo normal tolerization. However, when treated with estradiol, these mice develop high titers of anti-DNA antibodies and glomerular IgG deposition (Bynoe, Grimaldi et al. 2000). Estradiol was found to allow B cells to escape negative selection at both the immature and transitional checkpoints (Grimaldi, Jeganathan et al. 2006). This failure of negative selection is associated with a decrease in BCR-mediated signaling and an increase in the expression of CD22 and SHP-1, which negatively regulate the BCR (Grimaldi, Cleary et al. 2002). These studies further strengthen the association between diminished BCR signaling and lupus. Estrogen also causes an expansion of the marginal zone population, which is corroborated by the body of data that suggests that B cell fate is in part determined by BCR signaling amplitude, with lower signaling promoting the differentiation of immature and transitional B cells into marginal zone B cells (Pillai, Cariappa et al. 2005).
7. B-cell-directed therapies
As this review emphasizes, B cells have been proven to play a critical role in both human lupus and in mouse models. Thus, the rationale clearly exists for therapies that target B cells. Rituximab, a monoclonal antibody against CD20, was initially developed to treat B cell lymphomas(Marwick 1997), but its application has grown to the treatment of autoimmune disease. CD20 is expressed on immature and mature B cells, but is not expressed on plasma cells (Glennie, French et al. 2007). Rituximab has been shown to be effective in a randomized-controlled trial in rheumatoid arthritis (Edwards, Szczepanski et al. 2004). Recently, the EXPLORER (Jayne 2008) study was undertaken to test the efficacy of Rituximab in lupus. EXPLORER was a phase II/III randomized trial that treated patients with moderate to severe lupus, excluding those with renal disease. This study exhibited no therapeutic effect. Another study, the LUNAR trial, to examine the effect of Rituximab on lupus nephritis, is currently ongoing. The lack of plasma cell targeting may have contributed to the failure of Rituximab in EXPLORER, especially given the direct role of some autoantibodies in tissue injury. Also, depletion of B cells by Rituximab has been shown to cause elevations in serum BAFF levels (Lavie, Miceli-Richard et al. 2007). Hence, when the B cell repertoire reconstitutes, autoreactive B cells, which normally are deleted or tolerized due to limited levels of BAFF, may survive in the presence of elevated levels of BAFF. Thus, while B cells clearly remain essential mediators of lupus, an understanding of how to tackle them remains to be fine-tuned.
8. Conclusion
The failure of B cell depletion to demonstrate an effect of a B-cell targeted therapy in SLE crystallizes our need to better understand the role B cells are playing in this disease. B cells are clearly important in lupus, and a tremendous number of B cell abnormalities may precipitate this disease. For example, in some murine models of lupus, increased BCR-mediated signaling leads to autoimmunity, while in other cases, diminished BCR signaling does the same. In some cases, marginal zone B cells play an important role, whereas in others, follicular B cells appear more important. Currently, treatments for lupus are administered, developed and tested in trials without a focus on the heterogeneity that clearly exists in this disease. This heterogeneity at the molecular level may in fact be “built in” to lupus because the diagnosis includes such a wide variety of symptoms. We need to understand the abnormalities that lead to lupus in humans at the molecular and cellular level. By determining which of those abnormalities an individual patient has, we can consider customized therapy. We would surely have greater success in treating disease in this fashion, rather than attempting to treat all lupus patients with the same medications. Hence, B cells still have many mysteries yet to reveal with respect to how they mediate SLE and how we can successfully negate those effects.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anolik JH. B cell biology and dysfunction in SLE. Bull NYU Hosp Jt Dis. 2007;65(3):182–6. [PubMed] [Google Scholar]
- Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18(5):263–75. doi: 10.1016/j.smim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Budhai L, Oh K, Davidson A. An in vitro assay for detection of glomerular binding IgG autoantibodies in patients with systemic lupus erythematosus. J Clin Invest. 1996;98(7):1585–93. doi: 10.1172/JCI118952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buoncompagni A, Barbano GC, Pistoia V, Fasce L, Micalizzi C, Gusmano R, Cordone G, Cottafava F, Mori PG, Franchini E, et al. Childhood systemic lupus erythematosus: a review of 30 cases. Clin Exp Rheumatol. 1991;9(4):425–30. [PubMed] [Google Scholar]
- Buyon JP, Petri MA, Kim MY, Kalunian KC, Grossman J, Hahn BH, Merrill JT, Sammaritano L, Lockshin M, Alarcon GS, Manzi S, Belmont HM, Askanase AD, Sigler L, Dooley MA, Von Feldt J, McCune WJ, Friedman A, Wachs J, Cronin M, Hearth-Holmes M, Tan M, Licciardi F. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142(12 Pt 1):953–62. doi: 10.7326/0003-4819-142-12_part_1-200506210-00004. [DOI] [PubMed] [Google Scholar]
- Bynoe MS, Grimaldi CM, Diamond B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A. 2000;97(6):2703–8. doi: 10.1073/pnas.040577497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral AR, Alarcon-Segovia D. Autoantibodies in systemic lupus erythematosus. Curr Opin Rheumatol. 1997;9(5):387–92. doi: 10.1097/00002281-199709000-00003. [DOI] [PubMed] [Google Scholar]
- Cappione A, 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005;115(11):3205–16. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten H, Tarkowski A, Holmdahl R, Nilsson LA. Oestrogen is a potent disease accelerator in SLE-prone MRL lpr/lpr mice. Clin Exp Immunol. 1990;80(3):467–73. doi: 10.1111/j.1365-2249.1990.tb03311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189(10):1639–48. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong WP, Ip WK, Wong WH, Lau CS, Chan TM, Lau YL. Association of interleukin-10 promoter polymorphisms with systemic lupus erythematosus. Genes Immun. 2004;5(6):484–92. doi: 10.1038/sj.gene.6364119. [DOI] [PubMed] [Google Scholar]
- Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr., Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9(1):54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–40. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culton DA, O’Conner BP, Conway KL, Diz R, Rutan J, Vilen BJ, Clarke SH. Early preplasma cells define a tolerance checkpoint for autoreactive B cells. J Immunol. 2006;176(2):790–802. doi: 10.4049/jimmunol.176.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7(11):1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- Diamond B, Katz JB, Paul E, Aranow C, Lustgarten D, Scharff MD. The role of somatic mutation in the pathogenic anti-DNA response. Annu Rev Immunol. 1992;10:731–57. doi: 10.1146/annurev.iy.10.040192.003503. [DOI] [PubMed] [Google Scholar]
- Diamond B, Scharff MD. Somatic mutation of the T15 heavy chain gives rise to an antibody with autoantibody specificity. Proc Natl Acad Sci U S A. 1984;81(18):5841–4. doi: 10.1073/pnas.81.18.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, Isenberg DA. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995;48(3):705–11. doi: 10.1038/ki.1995.341. [DOI] [PubMed] [Google Scholar]
- Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, Casco J, Li QZ, Connolly JE, Wakeland EK. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. 2008;38(7):1971–8. doi: 10.1002/eji.200838138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor B, Putterman C, Valadon P, Spatz L, Scharff MD, Diamond B. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proc Natl Acad Sci U S A. 1997;94(5):1955–60. doi: 10.1073/pnas.94.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–37. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- Grammer AC, Lipsky PE. B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther. 2003;5(Suppl 4):S22–7. doi: 10.1186/ar1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109(12):1625–33. doi: 10.1172/JCI14873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi CM, Hill L, Xu X, Peeva E, Diamond B. Hormonal modulation of B cell development and repertoire selection. Mol Immunol. 2005;42(7):811–20. doi: 10.1016/j.molimm.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Grimaldi CM, Jeganathan V, Diamond B. Hormonal regulation of B cell development: 17 beta-estradiol impairs negative selection of high-affinity DNA-reactive B cells at more than one developmental checkpoint. J Immunol. 2006;176(5):2703–10. doi: 10.4049/jimmunol.176.5.2703. [DOI] [PubMed] [Google Scholar]
- Grimaldi CM, Michael DJ, Diamond B. Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001;167(4):1886–90. doi: 10.4049/jimmunol.167.4.1886. [DOI] [PubMed] [Google Scholar]
- Han S, Zheng B, Dal Porto J, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. IV. Affinity-dependent, antigen-driven B cell apoptosis in germinal centers as a mechanism for maintaining self-tolerance. J Exp Med. 1995;182(6):1635–44. doi: 10.1084/jem.182.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin JA, Craft JE. Patterns of autoimmunity to nucleoproteins in patients with systemic lupus erythematosus. Rheum Dis Clin North Am. 1987;13(1):37–46. [PubMed] [Google Scholar]
- Hoffmann KF, Cheever AW, Wynn TA. IL-10 and the dangers of immune polarization: excessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J Immunol. 2000;164(12):6406–16. doi: 10.4049/jimmunol.164.12.6406. [DOI] [PubMed] [Google Scholar]
- Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, Ballinger DG, Kosoy R, Demirci FY, Kamboh MI, Kao AH, Tian C, Gunnarsson I, Bengtsson AA, Rantapaa-Dahlqvist S, Petri M, Manzi S, Seldin MF, Ronnblom L, Syvanen AC, Criswell LA, Gregersen PK, Behrens TW. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358(9):900–9. doi: 10.1056/NEJMoa0707865. [DOI] [PubMed] [Google Scholar]
- Isenberg DA, Ravirajan CT, Rahman A, Kalsi J. The role of antibodies to DNA in systemic lupus erythematosus--a review and introduction to an international workshop on DNA antibodies held in London, May 1996. Lupus. 1997;6(3):290–304. doi: 10.1177/096120339700600316. [DOI] [PubMed] [Google Scholar]
- Jayne D. Role of Rituximab Therapy in Glomerulonephritis. J Am Soc Nephrol. 2008 doi: 10.1681/ASN.2008070786. [DOI] [PubMed] [Google Scholar]
- Jegerlehner A, Maurer P, Bessa J, Hinton HJ, Kopf M, Bachmann MF. TLR9 signaling in B cells determines class switch recombination to IgG2a. J Immunol. 2007;178(4):2415–20. doi: 10.4049/jimmunol.178.4.2415. [DOI] [PubMed] [Google Scholar]
- Kaliyaperumal A, Mohan C, Wu W, Datta SK. Nucleosomal peptide epitopes for nephritis-inducing T helper cells of murine lupus. J Exp Med. 1996;183(6):2459–69. doi: 10.1084/jem.183.6.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koncz G, Bodor C, Kovesdi D, Gati R, Sarmay G. BCR mediated signal transduction in immature and mature B cells. Immunol Lett. 2002;82(1–2):41–9. doi: 10.1016/s0165-2478(02)00017-2. [DOI] [PubMed] [Google Scholar]
- Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, Tokunaga K, Honda Z. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. 2005;14(19):2881–92. doi: 10.1093/hmg/ddi320. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–69. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312(5780):1665–9. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- Kuo P, Bynoe M, Diamond B. Crossreactive B cells are present during a primary but not secondary response in BALB/c mice expressing a bcl-2 transgene. Mol Immunol. 1999;36(7):471–9. doi: 10.1016/s0161-5890(99)00052-8. [DOI] [PubMed] [Google Scholar]
- Lavie F, Miceli-Richard C, Ittah M, Sellam J, Gottenberg JE, Mariette X. Increase of B cell-activating factor of the TNF family (BAFF) after rituximab treatment: insights into a new regulating system of BAFF production. Ann Rheum Dis. 2007;66(5):700–3. doi: 10.1136/ard.2006.060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton PJ, Rudie A, Klinman NR. Tolerance susceptibility of newly generating memory B cells. J Immunol. 1991;146(12):4099–104. [PubMed] [Google Scholar]
- Llorente L, Richaud-Patin Y, Garcia-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, Alcocer-Varela J, Grangeot-Keros L, Alarcon-Segovia D, Wijdenes J, Galanaud P, Emilie D. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000;43(8):1790–800. doi: 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Lu L, Kaliyaperumal A, Boumpas DT, Datta SK. Major peptide autoepitopes for nucleosome-specific T cells of human lupus. J Clin Invest. 1999;104(3):345–55. doi: 10.1172/JCI6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Silveira PA, Brink R. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr Opin Immunol. 2007;19(3):327–36. doi: 10.1016/j.coi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med. 2006;203(9):2157–64. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Valle F, Balada E, Ordi-Ros J, Vilardell-Tarres M. DNase 1 and systemic lupus erythematosus. Autoimmun Rev. 2008;7(5):359–63. doi: 10.1016/j.autrev.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Marwick C. Monoclonal antibody to treat lymphoma. Jama. 1997;278(8):616–618. [PubMed] [Google Scholar]
- Mauri C, Gray D, Mushtaq N, Londei M. Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003;197(4):489–501. doi: 10.1084/jem.20021293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307(5709):590–3. doi: 10.1126/science.1105160. [DOI] [PubMed] [Google Scholar]
- Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, Gonzalez JB, Pascual V, Stichweh D, Wardemann H, Nussenzweig MC. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A. 2008;105(28):9727–32. doi: 10.1073/pnas.0803644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min DJ, Kim SJ, Park SH, Seo YI, Kang HJ, Kim WU, Cho CS, Kim HY. Anti-nucleosome antibody: significance in lupus patients lacking anti-double-stranded DNA antibody. Clin Exp Rheumatol. 2002;20(1):13–8. [PubMed] [Google Scholar]
- Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16(2):219–30. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- Monroe JG, Bannish G, Fuentes-Panana EM, King LB, Sandel PC, Chung J, Sater R. Positive and negative selection during B lymphocyte development. Immunol Res. 2003;27(2–3):427–42. doi: 10.1385/IR:27:2-3:427. [DOI] [PubMed] [Google Scholar]
- Monticielo OA, Mucenic T, Xavier RM, Brenol JC, Chies JA. The role of mannose-binding lectin in systemic lupus erythematosus. Clin Rheumatol. 2008;27(4):413–9. doi: 10.1007/s10067-008-0838-8. [DOI] [PubMed] [Google Scholar]
- Ng KP, Manson JJ, Rahman A, Isenberg DA. Association of antinucleosome antibodies with disease flare in serologically active clinically quiescent patients with systemic lupus erythematosus. Arthritis Rheum. 2006;55(6):900–4. doi: 10.1002/art.22356. [DOI] [PubMed] [Google Scholar]
- Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J Exp Med. 2004;200(7):927–34. doi: 10.1084/jem.20040920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossal GJ, Karvelas M, Pulendran B. Soluble antigen profoundly reduces memory B-cell numbers even when given after challenge immunization. Proc Natl Acad Sci U S A. 1993;90(7):3088–92. doi: 10.1073/pnas.90.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor BP, Vogel LA, Zhang W, Loo W, Shnider D, Lind EF, Ratliff M, Noelle RJ, Erickson LD. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J Immunol. 2006;177(11):7723–32. doi: 10.4049/jimmunol.177.11.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura M, Kanayama Y, Amastu K, Negoro N, Kohda S, Takeda T, Inoue T. Significance of enzyme linked immunosorbent assay (ELISA) for antibodies to double stranded and single stranded DNA in patients with lupus nephritis: correlation with severity of renal histology. Ann Rheum Dis. 1993;52(1):14–20. doi: 10.1136/ard.52.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–96. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- Pisetsky DS. Anti-DNA and autoantibodies. Curr Opin Rheumatol. 2000;12(5):364–8. doi: 10.1097/00002281-200009000-00002. [DOI] [PubMed] [Google Scholar]
- Pulendran B, Kannourakis G, Nouri S, Smith KG, Nossal GJ. Soluble antigen can cause enhanced apoptosis of germinal-centre B cells. Nature. 1995;375(6529):331–4. doi: 10.1038/375331a0. [DOI] [PubMed] [Google Scholar]
- Putterman C, Diamond B. Immunization with a peptide surrogate for double-stranded DNA (dsDNA) induces autoantibody production and renal immunoglobulin deposition. J Exp Med. 1998;188(1):29–38. doi: 10.1084/jem.188.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172(11):6692–700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. 1994;12:487–520. doi: 10.1146/annurev.iy.12.040194.002415. [DOI] [PubMed] [Google Scholar]
- Ray SK, Putterman C, Diamond B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: a paradigm for autoimmune disease. Proc Natl Acad Sci U S A. 1996;93(5):2019–24. doi: 10.1073/pnas.93.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179(7):4704–10. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- Roubinian JR, Talal N, Greenspan JS, Goodman JR, Siiteri PK. Effect of castration and sex hormone treatment on survival, anti-nucleic acid antibodies, and glomerulonephritis in NZB/NZW F1 mice. J Exp Med. 1978;147(6):1568–83. doi: 10.1084/jem.147.6.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer LE, Hussain N, Wang X, Huang W, Sinha J, Ramanujam M, Davidson A. Lowering anti-dsDNA antibodies--what’s new? Lupus. 2002;11(12):885–94. doi: 10.1191/0961203302lu311rr. [DOI] [PubMed] [Google Scholar]
- Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180(4):1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokat KM, Goodnow CC. Antigen-induced B-cell death and elimination during germinal-centre immune responses. Nature. 1995;375(6529):334–8. doi: 10.1038/375334a0. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15(8):302–7. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- Simon JA, Cabiedes J, Ortiz E, Alcocer-Varela J, Sanchez-Guerrero J. Anti-nucleosome antibodies in patients with systemic lupus erythematosus of recent onset. Potential utility as a diagnostic tool and disease activity marker. Rheumatology (Oxford) 2004;43(2):220–4. doi: 10.1093/rheumatology/keh024. [DOI] [PubMed] [Google Scholar]
- Sundar K, Jacques S, Gottlieb P, Villars R, Benito ME, Taylor DK, Spatz LA. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J Autoimmun. 2004;23(2):127–40. doi: 10.1016/j.jaut.2004.06.001. [DOI] [PubMed] [Google Scholar]
- ter Borg EJ, Horst G, Hummel EJ, Limburg PC, Kallenberg CG. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum. 1990;33(5):634–43. doi: 10.1002/art.1780330505. [DOI] [PubMed] [Google Scholar]
- Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity. 2007;26(2):205–13. doi: 10.1016/j.immuni.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomer Y, Buskila D, Shoenfeld Y. Pathogenic significance and diagnostic value of lupus autoantibodies. Int Arch Allergy Immunol. 1993;100(4):293–306. doi: 10.1159/000236429. [DOI] [PubMed] [Google Scholar]
- Trolle S, Chachaty E, Kassis-Chikhani N, Wang C, Fattal E, Couvreur P, Diamond B, Alonso J, Andremont A. Intranasal immunization with protein-linked phosphorylcholine protects mice against a lethal intranasal challenge with streptococcus pneumoniae. Vaccine. 2000;18(26):2991–8. doi: 10.1016/s0264-410x(00)00089-x. [DOI] [PubMed] [Google Scholar]
- Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40(8):560–6. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- Tsubata T, Honjo T. B cell tolerance and autoimmunity. Rev Immunogenet. 2000;2(1):18–25. [PubMed] [Google Scholar]
- Tsuiji M, Yurasov S, Velinzon K, Thomas S, Nussenzweig MC, Wardemann H. A checkpoint for autoreactivity in human IgM+ memory B cell development. J Exp Med. 2006;203(2):393–400. doi: 10.1084/jem.20052033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukumo S, Yasutomo K. DNaseI in pathogenesis of systemic lupus erythematosus. Clin Immunol. 2004;113(1):14–8. doi: 10.1016/j.clim.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37(12):1317–9. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- Vang T, Miletic AV, Bottini N, Mustelin T. Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity. 2007;40(6):453–61. doi: 10.1080/08916930701464897. [DOI] [PubMed] [Google Scholar]
- Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19(6):837–47. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- Wang C, Khalil M, Ravetch J, Diamond B. The naive B cell repertoire predisposes to antigen-induced systemic lupus erythematosus. J Immunol. 2003;170(9):4826–32. doi: 10.4049/jimmunol.170.9.4826. [DOI] [PubMed] [Google Scholar]
- Wang YH, Diamond B. B cell receptor revision diminishes the autoreactive B cell response after antigen activation in mice. J Clin Invest. 2008;118(8):2896–907. doi: 10.1172/JCI35618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, Ravetch JV, Tryggvason K, Karlsson MC. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007;204(10):2259–65. doi: 10.1084/jem.20070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William J, Euler C, Primarolo N, Shlomchik MJ. B cell tolerance checkpoints that restrict pathways of antigen-driven differentiation. J Immunol. 2006;176(4):2142–51. doi: 10.4049/jimmunol.176.4.2142. [DOI] [PubMed] [Google Scholar]
- Williams RC, Jr., Malone C, Blood B, Silvestris F. Anti-DNA and anti-nucleosome antibody affinity--a mirror image of lupus nephritis? J Rheumatol. 1999;26(2):331–46. [PubMed] [Google Scholar]
- Williams RC, Jr., Malone CC, Meyers C, Decker P, Muller S. Detection of nucleosome particles in serum and plasma from patients with systemic lupus erythematosus using monoclonal antibody 4H7. J Rheumatol. 2001;28(1):81–94. [PubMed] [Google Scholar]
- Witte T. IgM antibodies against dsDNA in SLE. Clin Rev Allergy Immunol. 2008;34(3):345–7. doi: 10.1007/s12016-007-8046-x. [DOI] [PubMed] [Google Scholar]
- Wolf SD, Dittel BN, Hardardottir F, Janeway CA., Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996;184(6):2271–8. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF. B-lymphocyte contributions to human autoimmune disease. Immunol Rev. 2008;223:284–99. doi: 10.1111/j.1600-065X.2008.00646.x. [DOI] [PubMed] [Google Scholar]
- Yin Z, Bahtiyar G, Zhang N, Liu L, Zhu P, Robert ME, McNiff J, Madaio MP, Craft J. IL-10 regulates murine lupus. J Immunol. 2002;169(4):2148–55. doi: 10.4049/jimmunol.169.4.2148. [DOI] [PubMed] [Google Scholar]
- Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005;201(5):703–11. doi: 10.1084/jem.20042251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng D, Lee MK, Tung J, Brendolan A, Strober S. Cutting edge: a role for CD1 in the pathogenesis of lupus in NZB/NZW mice. J Immunol. 2000;164(10):5000–4. doi: 10.4049/jimmunol.164.10.5000. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jacobi AM, Mackay M, Aranow C, Wang T, Chinnasamy P, Diamond B. Identification of DNA-reactive B cells in patients with systemic lupus erythematosus. J Immunol Methods. 2008a;338(1–2):79–84. doi: 10.1016/j.jim.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Jacobi AM, Wang T, Diamond B. Pathogenic autoantibodies in systemic lupus erythematosus are derived from both self-reactive and non-self-reactive B cells. Mol Med. 2008b;14(11–12):675–81. doi: 10.2119/2008-00066.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]