

Abstract
Pseudomonas aeruginosa produces copious amounts of the redoxactive tricyclic compound pyocyanin that kills competing microbes and mammalian cells, especially during cystic fibrosis lung infection. Cross-phylum susceptibility to pyocyanin suggests the existence of evolutionarily conserved physiological targets. We screened a Saccharomyces cerevisiae deletion library to identify presumptive pyocyanin targets with the expectation that similar targets would be conserved in humans. Fifty S. cerevisiae targets were provisionally identified, of which 60% have orthologous human counterparts. These targets encompassed major cellular pathways involved in the cell cycle, electron transport and respiration, epidermal cell growth, protein sorting, vesicle transport, and the vacuolar ATPase. Using cultured human lung epithelial cells, we showed that pyocyanin-mediated reactive oxygen intermediates inactivate human vacuolar ATPase, supporting the validity of the yeast screen. We discuss how the inactivation of VATPase may negatively impact the lung function of cystic fibrosis patients.
The most common pathogen during infection of the cystic fibrosis (CF) airways is Pseudomonas aeruginosa, which is also important in burn related sepsis, various nosocomial infections, and HIV-related illness (1, 2). The extensive host range and complex pathophysiology of infections associated with P. aeruginosa is due to its ability to produce a large repertoire of virulence determinants (1, 2). In addition to delivering proteins into eukaryotic cells by a type III secretion system to disrupt the host immune response and cause cytoskeletal reorganization (3), P. aeruginosa also produces a large number of toxic exoproducts, including proteases, rhamnolipids, and pyocyanin (PCN) (1, 2).
PCN, a blue redox-active secondary metabolite, is a member of a large family of tricyclic compounds known as phenazines (4). Mutational and biochemical analyses have identified two groups of gene products required for PCN biosynthesis. First, the MvfR transcription factor is required to activate the phnAB genes (5–7). The phnA-B gene products synthesize quinolone that regulate the phzRABCDEFG operons 1 and 2, the structural genes for phenazine synthesis (8).
Previous investigations have suggested that PCN contributes to the ability of P. aeruginosa to persist in the lungs of CF patients (9). In addition, in vitro studies have shown that PCN interferes with a myriad of mammalian cell functions. These include cell respiration, ciliary beating, epidermal cell growth (9, 10), calcium homeostasis (11), prostacyclin release from lung endothelial cells (12), apoptosis in neutrophils (13), release of interleukin-2 (limits growth of T lymphocytes), secretion of Ig by B-lymphocytes (14), and imbalance of protease-antiprotease activity in the airways of CF patients (15). However, the precise molecular mechanism underlying PCN-mediated pathology is unknown.
PCN also inhibits microbial growth by initiating a redox cascade that can occur nonenzymatically via NADH or NADPH (16). Several studies have suggested that PCN inhibits fungal growth in vivo in patients with (17) or without CF (18, 19). The antimicrobial properties of PCN enable P. aeruginosa to dominate other microbes in the CF airways. These properties, combined with the observation that levels of PCN capable of altering eukaryotic cell function are present in CF sputum (9), implicate PCN in epithelial cell dysfunction, altered pulmonary immunity, and proteolytic injury that allows P. aeruginosa to persist in CF lung.
Although the toxicity of PCN is wide spread (9–15), the molecular basis underlying its pathology is less clear. Based on our hypothesis that eukaryotic genes that confer susceptibility or resistance to PCN are evolutionarily conserved, even in distantly related organisms such as fungi and mammals, we exploited the ability of PCN to inhibit the growth of members of a mutant library of Saccharomyces cerevisiae as a strategy for discovering mammalian cellular pathways affected by this compound.
Materials and Methods
P. aeruginosa Strains, Growth Conditions, and PCN Purification. The P. aeruginosa strain PA14 (20) was grown in LB broth at 37°C. PCN was purified from late stationary phase culture with successive rounds of CHCl3/0.2 M HCl extraction as described (21). The final PCN preparations had no detectable levels of Pseudomonas lipopolysaccharide (LPS) as determined by the E-TOXATE assay (Sigma) or of autoinducer as measured by HPLC (8).
Yeast and A549 Cell Cultures. The S. cerevisiae Mat-A haploid deletion library (Invitrogen) was generated from strain BY4741 (MATα his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (22) by the Saccharomyces Genome Deletion Project (www-deletion.stanford.edu). Yeast strains were grown in yeast extract/peptone/dextrose (YPD) medium at 30°C. The human lung carcinoma alveolar type II cell line A549 (23) was purchased from American Type Culture Collection. A549 cells were cultured to ≈80% confluency in DMEM supplemented with 10% FCS, 2 mM glutamine, and 500 units/ml penicillin and streptomycin.
Screening for Yeast Deletion Mutants with Altered Sensitivity to PCN. To screen for yeast mutants with altered sensitivity to PCN, yeast cells cultured in microtiter plates (OD630 ≈ 0.3) were treated with PCN (75 μg/ml) and incubated for 18 h at 30°C while shaking at 150 rpm. The sensitivity of yeast strains to PCN was determined by the net growth between 0 and 18 h as measured by OD630. To ensure accuracy, the screening of each mutant library pool was performed in duplicate, with the OD630 measurement performed twice on each plate at indicated time points. The average readings were used to calculate growth rate.
Bioinformatics Analysis. A database containing the identity of the each deleted ORF in each yeast mutant is available from the Saccharomyces Genome Deletion Project. We used available nucleotide and protein databases (www.ncbi.nlm.nih.gov) to identify human orthologs of the yeast genes conferring altered sensitivity to PCN.
Catalase and Superoxide Dismutase (SOD) Assays. Actively growing log phase yeast cells (≈106 in 0.1 ml) were exposed to 50 or 100 μg of PCN for 30, 60, and 90 min. Cultured lung epithelial A549 cells were exposed to 20 or 30 μg of PCN for 30, 60, and 90 min. Yeast and A549 cells exposed to sterile water were used as controls. Protein was extracted from yeast cells disrupted with 0.3- to 0.4-mm glass beads, or from A549 cells by M-PER Mammalian Protein Extraction kit (Pierce). Total protein concentration was determined by using BCA Protein Assay kit (Pierce). Catalase and superoxide dismutase activity assays were performed as described (24, 25).
Measurement of Intracellular Reactive Oxygen Intermediates (ROI). The yeast BY4741 and the A549 cells grown on 96-well microtiter plates were incubated at 37°C for 1 h with DMEM/FCS containing 2 μM bafilomycin A1 (BA1) or 25, 50, and 100 μg/ml PCN. Intracellular oxidant stress was monitored by measuring changes in fluorescence resulting from oxidation of an intracellular probe at indicated time points. The probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA, 5 μM, Molecular Probes) enters cells and the acetate group on DCFH-DA is cleaved by cellular esterases, trapping the nonfluorescent 2′,7′-dichlorofluorescin (DCFH) inside. Subsequent oxidation by ROI, particularly hydrogen peroxide (H2O2) and hydroxyl radical (HO·), yields the fluorescent product DCF. Thus, increases in DCF fluorescence are suggestive of H2O2 or HO· generation (26). Dihydroethidine (5 μM, Molecular Probes) enters cells and can be oxidized by superoxide (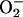 ) and/or HO· to yield fluorescent ethidium (Eth). Eth binds to DNA (Eth–DNA), further amplifying its fluorescence. Thus, increases in Eth–DNA fluorescence are suggestive of
) and/or HO· to yield fluorescent ethidium (Eth). Eth binds to DNA (Eth–DNA), further amplifying its fluorescence. Thus, increases in Eth–DNA fluorescence are suggestive of 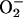 /HO- generation (26).
/HO- generation (26).
Acridine Orange (AO) Staining. The A549 cells grown on cover slips were incubated at 37°C for 1 h with DMEM/FCS containing 0.5 or 2 μM BA1 dissolved in 1/100 volume of DMSO or 25, 50, and 100 μg/ml PCN. Control cells were treated with DMSO alone. After incubation, the cells were treated with 5 μg/ml AO in Hanks' balanced salt solution (HBSS) at 37°C for 10 min (27). After four washings with HBSS, the coverslips were examined by using a Zeiss Axiophot 2 fluorescence microscope with excitation at 490 nm and emission at 590 nm. Fluorescence micrographs were taken with the same shutter speed for each experiment with Kodak Ektachrome 400 film. Trypan blue exclusion assays indicated that ≥99.7% of A549 cells remained viable after 6 h treatment with 100 μg/ml PCN (data not shown).
ATP Hydrolysis Assays. A549 cells were exposed for 1 h to either 15 μM H2O2, 0.1, 0.2, 1, and 2 μM BA1 or 12.5, 25, 50, and 100 μg/ml PCN or DMSO (control). After cell lysis, microsomes were isolated from cell fractions free of nuclei and mitochondria by centrifugation at 100,000 × g for 45 min. ATP hydrolysis was assayed for 10 min at 37°C by using 50-μg microsomes in 200 μl of reaction buffer (10 mM K-EPPS, pH 8.5/120 mM sucrose/15 mM KCl/60 mM NaCl/0.2 mM EDTA/5 mM MgSO4/5 mM ATP/≈2 × 105 cpm [γ32P]adenosine 5′-triphosphate). Reactions were stopped by adding 1 ml of 10% activated charcoal in 0.1 M KH2PO4, pH 2.0. After a 5-min centrifugation at 1,000 × g, 0.5 ml of supernatant was removed to determine the extent of ATP hydrolysis by scintillation counting.
Results and Discussion
PCN Kills Yeast and Injures Mammalian Cells by Causing Oxidative Stress. PCN has been implicated in the prevention of pulmonary mycoses in CF lungs (19). However, the mechanisms underlying the fungicidal activity of PCN are not well understood. To examine the mechanism(s) of fungal killing, we determined whether subinhibitory levels of PCN (18) could cause production of ROI by indirectly measuring the increase in intracellular fluorescence levels when DCFH-DA and dihydroethidine were oxidized to DCF and ethidium by H2O2 and 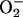 , respectively. As shown in Fig. 1 A and B, the levels of fluorescence increased rapidly and remained much higher than untreated cells throughout the experiments, suggesting that PCN caused the production of ROI. We next determined whether the increase in ROI was accompanied by increased activities of the protective antioxidant enzymes catalase and SOD. As shown in Fig. 1 C and D, yeast exposed to 50 μg/ml PCN exhibited higher catalase and SOD activities when compared with control cells at 30-, 60-, and 90-min time intervals. Exposure of yeast cells to 100 μg/ml PCN dramatically increased catalase and SOD activities at 30 and 60 min. The elevated activities of catalase and SOD decreased beyond 60 min.
, respectively. As shown in Fig. 1 A and B, the levels of fluorescence increased rapidly and remained much higher than untreated cells throughout the experiments, suggesting that PCN caused the production of ROI. We next determined whether the increase in ROI was accompanied by increased activities of the protective antioxidant enzymes catalase and SOD. As shown in Fig. 1 C and D, yeast exposed to 50 μg/ml PCN exhibited higher catalase and SOD activities when compared with control cells at 30-, 60-, and 90-min time intervals. Exposure of yeast cells to 100 μg/ml PCN dramatically increased catalase and SOD activities at 30 and 60 min. The elevated activities of catalase and SOD decreased beyond 60 min.
Fig. 1.

PCN causes oxidative stress in S. cerevisiae.(A and B) PCN increases the levels of H2O2 and 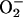 in yeasts. BY4741 cells were exposed to PCN at indicated concentrations. The production of H2O2 and
in yeasts. BY4741 cells were exposed to PCN at indicated concentrations. The production of H2O2 and 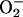 was observed by measuring fluorescence emitted by oxidation of DCFH-DA and dihydroethidine, respectively. (C and D) Increasing amounts of PCN-generated ROI are accompanied by higher level of catalase and SOD activities in yeasts. BY4741 cells were exposed to PCN, and total proteins were harvested at the indicated time intervals and concentrations to assay for catalase and SOD. The experiments were repeated five times with similar results. The results from one representative experiment are shown.
was observed by measuring fluorescence emitted by oxidation of DCFH-DA and dihydroethidine, respectively. (C and D) Increasing amounts of PCN-generated ROI are accompanied by higher level of catalase and SOD activities in yeasts. BY4741 cells were exposed to PCN, and total proteins were harvested at the indicated time intervals and concentrations to assay for catalase and SOD. The experiments were repeated five times with similar results. The results from one representative experiment are shown.
PCN at concentrations as high as 27 μg/ml have been detected in sputum from CF patients (9), suggesting a role for PCN in CF pathogenesis and other airway infections. To correlate the results obtained with yeast to a mammalian counterpart, we examined whether PCN treatment evoked an increase in ROI in human lung epithelial A549 cells. As shown in Fig. 2 A and B, the intracellular levels of H2O2 and 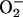 increased dramatically relative to untreated cells. We also assayed catalase and SOD activity in A549 cells after exposure to 20 and 30 μg/ml PCN. Consistent with the results in yeast, PCN-treated A549 cells showed elevated levels of both catalase and SOD activity (Fig. 2 C and D). However, as with the yeast system, prolonged exposure to PCN reduced the activities of both catalase and SOD activity at 90 min. Taken together, these data suggest that PCN injures mammalian and yeast cells by a similar mechanism that involves oxidative damage.
increased dramatically relative to untreated cells. We also assayed catalase and SOD activity in A549 cells after exposure to 20 and 30 μg/ml PCN. Consistent with the results in yeast, PCN-treated A549 cells showed elevated levels of both catalase and SOD activity (Fig. 2 C and D). However, as with the yeast system, prolonged exposure to PCN reduced the activities of both catalase and SOD activity at 90 min. Taken together, these data suggest that PCN injures mammalian and yeast cells by a similar mechanism that involves oxidative damage.
Fig. 2.

PCN causes oxidative stress in lung epithelial cells. (A and B) PCN increases the levels of H2O2 and 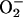 in A549 cells. A549 cells were exposed to PCN or BA1 at indicated concentrations. The production of H2O2 and
in A549 cells. A549 cells were exposed to PCN or BA1 at indicated concentrations. The production of H2O2 and 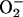 was observed by measuring fluorescence emitted by oxidation of DCFH-DA and dihydroethidine, respectively. (C and D) Increasing amounts of PCN-generated ROI are accompanied by higher level of catalase and SOD activities in the lung epithelial cells. A549 cells were exposed to PCN, and total proteins were harvested at the indicated time intervals and concentrations to assay for catalase and SOD. The experiments were repeated five times with similar results. The results from one representative experiment are shown.
was observed by measuring fluorescence emitted by oxidation of DCFH-DA and dihydroethidine, respectively. (C and D) Increasing amounts of PCN-generated ROI are accompanied by higher level of catalase and SOD activities in the lung epithelial cells. A549 cells were exposed to PCN, and total proteins were harvested at the indicated time intervals and concentrations to assay for catalase and SOD. The experiments were repeated five times with similar results. The results from one representative experiment are shown.
Yeast As a Surrogate For Screening Mammalian PCN Target Orthologs. Because species as distantly related as fungi and mammals are susceptible to PCN, we reasoned that the eukaryotic genes that confer sensitivity to PCN might be evolutionarily conserved. The haploid MAT-A yeast deletion library (≈5,000 strains) was screened for mutants that were either sensitive or resistant to PCN. Output from pool 11 represents a typical readout (Fig. 6, which is published as supporting information on the PNAS web site). The average density of input cells (time 0 h) was OD630 ≈ 0.3–0.4, whereas the average output cell density (time 16 h) was OD630 ≈ 0.6. Thus, the average positive net growth of OD630 ≈ 0.2 was achieved. A yeast deletion mutant with a negative net growth or a positive net growth that was well below the average value, OD630 ≤ 0.05, was considered to have increased sensitivity to PCN, whereas a yeast deletion mutant with a positive net growth of OD630 ≥ 0.35 was considered to have increased resistance to PCN. For example, strain 11E2 was more susceptible and strain 11A9 was more resistant to PCN. In addition, strain 11D2 has a growth defect, as it did not grow in either the presence or absence of PCN. Importantly, these data demonstrate that the PCN sensitivity assays give consistent cell turbidity readouts between duplicate microtiter plates.
Of the 5,000 yeast deletion strains screened, 50 mutants (≈1%) with altered sensitivity to PCN were recovered. Of these, 37 exhibited increased susceptibility to PCN, whereas the remaining 13 were more resistant to PCN. A detailed description of each mutant can be found in Tables 1 and 2, which are published as supporting information on the PNAS web site. In total, 60% of the genes have mammalian orthologs. We predict that the number of mammalian orthologs will increase as annotation of the human genome sequence improves. The majority of these targets have not been studied with respect to PCN toxicity. Most of these genes correspond to four cellular pathways: (i) vacuolar ATPase (V-ATPase), vesicle transport, and protein targeting; (ii) cell cycle; (iii) electron transport/oxidative stress/ apoptosis; and (iv) epidermal cell growth. The possible mechanisms by which PCN generated reactive oxygen intermediates may inhibit these pathways are briefly discussed below.
Lacking the Following Yeast Gene Products Causes Increased Susceptibility to PCN. V-ATPase synthesis and assembly, vesicle transport, and vacuolarprotein sorting. Four mutations in the yeast genes for V-ATPase synthesis and assembly, including VMA6, VMA7, VMA8, and VMA21 were identified (Table 1). The Vma6p is a nonintegral membrane component of the membrane pore domain and is required for V-ATPase complex assembly (28). Vma7p plays a critical role in stabilizing the V0 complex and bridging the V1 and V0 complexes to form a functional VATPase complex (29). Vma8p plays an important role in coupling of proton transport and ATP hydrolysis (30). Vma21p is a dilysine motif-containing integral membrane protein that is not a subunit of the purified V-ATPase complex. Instead, it resides in the endoplasmic reticulum (ER) and is required for assembly of the integral membrane segment of the V-ATPase (31).
Four yeast deletions in the vesicle transport machinery were found to be sensitive to PCN. These genes are SEC16, SEC22, SEC28, and YPT6. Sec16p binds to major–minor mix liposomes and facilitates the recruitment of coatomer (COP) complex II (COPII) proteins and vesicle budding in a reaction that is stimulated by Sar1p and guanylimide diphosphate (32). Sec22p, a v-SNARE (soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor) is required for transport between the ER and early Golgi apparatus (33). Sec28 epsilon-COP plays a critical role in maintaining the structural integrity of α-COP and the COPI-coatomer complex (34). YPT6 is a GTPase that participates in early Golgi transport and ribosome biosynthesis (35).
Mutations in five different components of protein sorting machinery were found to enhance susceptibility to PCN. These components are VPS4, VPS20, VPS28, VPS36, and DOA4. Vps4p is an AAA-type ATPase (36), which catalyzes the release of “class E Vps proteins,” among which, Vps28p, Vps36p and Vps20p, form the ESCRT-I (endosomal sorting complex required for transport), ESCRT-II, and ESCRT-III complexes, respectively. These ESCRT complexes are required to deliver transmembrane proteins, such as activated cell surface receptors to the lumen of the vacuole/lysosome, either for degradation/ down regulation, or for proper localization (37). Finally, Doa4 is a deubiquitinating enzyme that localizes to endosomal membranes by associating with ESCRT-III, where it plays an important role in the efficient deubiquitination of cargo and the recycling of ubiquitin molecules (38).
In mammalian cells, the V-ATPase has been shown to be extremely sensitive to micromolar concentrations of H2O2 (39). Mechanistically, this is based on a H2O2-mediated oxidation of a reactive cysteine sulfhydryl group in the ATP-binding site of the V1A1 subunit (39). Precisely how ROI inactivate vesicle transport and vacuolar protein sorting machineries are not well understood. However, it was shown that H2O2 causes tyrosine phosphorylation of clathrin heavy chain and affects intracellular vesicular trafficking via the alteration of clathrin-dependent vesicular trafficking (40). Thus, it is conceivable that COPI- and COPII-mediated vesicle transport pathways, as well as vacuolar protein sorting pathways, are affected by PCN mediated oxidative stress.
Electron transport/oxidative stress/apoptosis. The catalase and SOD assays showed that activity of these antioxidative enzymes was predictably increased in response to PCN exposure (Fig. 1 C and D). Thus, we predicted that some of the mutants would exhibit defects in electron transport and oxidative stress. In addition, ROI generated by PCN could induce both apoptosis and necrosis through the activation of caspase 3-like proteases. Indeed, nine yeast mutants had deletions in genes that may directly affect the mitochondrial respiration/electron transport chain or participate in the oxidative stress response. The YSA1, BNA2, and COQ1 gene products are required for nicotinic acid and ubiquinone biosynthesis. Nicotinic acid and ubiquinone are both electron carriers involved in mitochondrial respiration, which are targeted by PCN (21). SOD1 is a cytoplasmic Cu,Zn–SOD involved in detoxifying 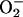 . YDL119C encodes a mitochondrial uncoupling protein with three mitochondrial carrier motifs that diverts energy from ATP synthesis by catalyzing a regulated leak of protons across the inner membrane (41). Bsd2p is a metal homeostasis regulator that prevents hyperaccumulation of metals by negatively regulating both the SMF1 and SMF2 metal transport systems and minimizing oxidative stress (42). BSD2 is similar in function to Fur-like proteins in bacteria that regulate iron homeostasis and, when mutated, organisms are more susceptible to oxidative stress. Mutation of yeast EGR4 lowers the content of ergosterol and causes enhanced sensitivity to H2O2 and paraquat (43). ELO2 mediates the elongation of very long chain fatty acids and subsequent ceramide and inositol sphingolipid synthesis, as well as the trafficking of secretory vesicles in yeast. Disruption of ELO2 reduces cellular sphingolipid levels and results in the accumulation of phytosphingosine and ceramides that induce caspase-independent apoptosis observed in mammalian systems (44). OSH2 encodes an oxysterol-binding protein, which is believed to control cholesterol homeostasis, vesicle transport, and apoptosis (45).
. YDL119C encodes a mitochondrial uncoupling protein with three mitochondrial carrier motifs that diverts energy from ATP synthesis by catalyzing a regulated leak of protons across the inner membrane (41). Bsd2p is a metal homeostasis regulator that prevents hyperaccumulation of metals by negatively regulating both the SMF1 and SMF2 metal transport systems and minimizing oxidative stress (42). BSD2 is similar in function to Fur-like proteins in bacteria that regulate iron homeostasis and, when mutated, organisms are more susceptible to oxidative stress. Mutation of yeast EGR4 lowers the content of ergosterol and causes enhanced sensitivity to H2O2 and paraquat (43). ELO2 mediates the elongation of very long chain fatty acids and subsequent ceramide and inositol sphingolipid synthesis, as well as the trafficking of secretory vesicles in yeast. Disruption of ELO2 reduces cellular sphingolipid levels and results in the accumulation of phytosphingosine and ceramides that induce caspase-independent apoptosis observed in mammalian systems (44). OSH2 encodes an oxysterol-binding protein, which is believed to control cholesterol homeostasis, vesicle transport, and apoptosis (45).
Cell cycle. PCN has been shown to inhibit lymphocyte proliferation (10). Thus, it is relevant and consistent that we recovered six yeast mutations responsible for cell cycle control: CDC50, CLN2, SSD1, NBP2, HRS1, SIR4, and PSP1. Mutation of CDC50 arrests the cell cycle at START in G1 phase (46). Cln2p positively regulates the initiation of DNA replication during the cell cycle (47). Ssd1p is required for stable maintenance of yeast chromosomes (48). Hrs1p functions in initiation of direct-repeat recombination (49). Sir4p has been shown to participate in DNA repair (50) and regulates the expression of cell cycle genes CTS1 and EGT2. Finally, Psp1p is required for both proper DNA replication and mitosis (51). There are multiple means by which PCN may disrupt the cell cycle and inhibit lymphocyte proliferation. For example, we have shown that PCN generates H2O2 and 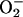 (Figs. 1 and 2) that are capable of causing oxidative damage to components of the cell cycle, as well as direct damage to DNA. Damage to cell cycle components, such as DNA repair and recombination machinery, would prevent cell proliferation. However, we acknowledge that there are additional factors that could inhibit lymphocyte proliferations, such as inhibition of cellular respiration (by interfering with electron transport in mitochondria), as well as inhibiting V-ATPase. At present, we do not know which pathway is more critical.
(Figs. 1 and 2) that are capable of causing oxidative damage to components of the cell cycle, as well as direct damage to DNA. Damage to cell cycle components, such as DNA repair and recombination machinery, would prevent cell proliferation. However, we acknowledge that there are additional factors that could inhibit lymphocyte proliferations, such as inhibition of cellular respiration (by interfering with electron transport in mitochondria), as well as inhibiting V-ATPase. At present, we do not know which pathway is more critical.
Lacking the Following Yeast Gene Products Causes Increased Resistance to PCN. Most of the PCN-resistant yeast mutants displayed slow-growth phenotypes. They are deleted in genes encoding mitochondrial ribosomal proteins, genes involved in electron transport, and a gene encoding a rhomboid-like serine protease (Table 2).
Mitochondrial electron transport chain/oxidative stress. We have identified yeast mutants that correspond to genes that are involved in the synthesis of mitochondrial-specific ribosomal proteins (RSM18, RSM27, YNL177C, MRPL17) and a nuclear-encoded mitochondrial protein (PET130). Mutations in these genes cause respiratory defects (52). Interestingly, it was reported that exposure of Caenorhabditis elegans to oxidative stress strongly induced transcription of 18S, 5.8S, and 26S rRNAs (53), suggesting an attempt by the cells to replace damaged ribosomal proteins or to compensate for a reduction in or the loss of the electron transport chain. Two additional mutations are in genes required for mitochondrial coenzyme-Q (CAT5) and cytochrome b (CBP6) biosynthesis. Coenzyme-Q is an essential electron transport component and a lipid soluble antioxidant (54). Cytochrome b is a component of the cytochrome bc (1) electron transport complex (55). Mutation of either gene causes a modest to pronounced defect in respiration. Finally, trehalose-6-phosphate synthase (TPS1) participates in resistance to variety of stresses, including oxidative stress (56).
Epidermal cell growth. YGR101W encodes a rhomboid-like serine protease that cleaves TGF-α to activate the EGF receptor. This is relevant because PCN was previously shown to inhibit epithermal cell growth (10), possibly by inactivating a rhomboid-like serine protease.
Unknown genes. We have also identified 13 additional mutants with altered sensitivity to PCN (Tables 1 and 2). The function(s) of these gene products is unknown. Unraveling the identity of these genes may add to our understanding of the precise molecular basis behind PCN-mediated injury.
Confirmation of Selected Yeast Targets in Lung Epithelial Cells: PCN Inactivates Human V-ATPase. Multiple V-ATPase mutants that showed increased susceptibility to PCN were recovered from our yeast screen (Table 1). To examine whether the enhanced sensitivity to PCN was caused by impaired yeast proliferation or enhanced yeast death, yeast V-ATPase mutants were exposed to 50 μg/ml PCN for 21 h, and growth was monitored. Untreated cells were used as controls. By 21 h, all V-ATPase yeast mutants grew to the same cell density as wild-type BY4741 in the absence of PCN (Fig. 3A). In contrast, V-ATPase mutants showed varying degrees of increased sensitivity to PCN. Strains vma6 and vma21 lost viability throughout the incubation with PCN. Strain vma8 was unable to proliferate in the presence of PCN. Interestingly, strain vma7 was able to proliferate as well as the parental isolate BY4741. However, viability decreased upon prolonged incubation in the presence of PCN, particularly in late stationary phase. By 21 h, the viable cell counts of the parental strain BY4741 increased by 1 log. In contrast, the vma7 mutant only grew 0.4 log, whereas the viability of vma6, vma8 and vma21 mutants decreased by 1.4, 0.5 and 2.5 logs, respectively. These results suggest that yeast V-ATPase plays a crucial role in combating PCN-mediated oxidative stress, perhaps by regulating proper localization of antioxidative enzymes.
Fig. 3.

PCN alters the growth phenotype of yeast V-ATPase mutants. Yeast strains with mutations in V-ATPase were followed for 21 h while shaking at 150 rpm in yeast extract/peptone/dextrose (YPD) or YPD supplemented with 100 μg/ml PCN. (A) Cell density of parental strain BY4741 according to OD630 over time. (B) Viability of PCN-treated BY4741 and V-ATPase mutants as determined by colony-forming unit counts on YPD agar plates. Similar results were obtained from six independent experiments. Results from one representative experiment are shown.
To determine whether PCN generated ROI inactivate VATPase in lung epithelial cells, we determined the activity of V-ATPase by performing AO staining. When protonated in acidic vacuolar compartments, AO is retained and imparts an orange color to vacuoles. As shown in Fig. 4, a 1-h exposure to 2 μM BA1, a V-ATPase-specific inhibitor, inhibited AO staining, indicating that V-ATPase was inactivated and that the pH of acidic compartments within A549 cells was destabilized. In contrast, control cells treated with DMSO or with 0.5 μM BA1 stained orange, indicating a functional V-ATPase. When A549 cells were treated with 25 μg/ml PCN, the level of vacuole staining was reduced dramatically, staining some of the cells yellow rather than the characteristic bright orange color of normal cells (Fig. 4). These results suggest that physiological levels of PCN isolated from sputum of CF patients (27 μg/ml) are capable of interfering with V-ATPase activities. The AO staining of vacuoles was completely abolished in A549 cells treated with 50 or 100 μg/ml PCN.
Fig. 4.

PCN inactivates V-ATPase and prevents vacuole acidification in lung epithelial cells. The A549 cells were treated for 1 h with 0.5 and 2 μM BA1 or with 25, 50, and 100 μg/ml PCN or DMSO as control. Treated cells were stained with 5 μg/ml AO and examined with a fluorescence light microscope. (Original magnification, ×40.)
To test whether loss of AO staining was directly caused by the loss of V-ATPase activities, we assayed for V-ATPase activity in microsomes isolated from A549 cells treated with PCN. Because PCN triggers the production of ROI, we examined whether an externally supplied oxidizing agent would be able to inhibit V-ATPase. A 1-h treatment with 15 μM H2O2 inhibited 74% of the V-ATPase activity (Fig. 5). As expected, BA1, the VATPase-specific inhibitor, inhibited V-ATPase activity in a dose-dependent manner (Fig. 5). Accordingly, exposing A549 cells to increasing levels of PCN caused a corresponding decrease in V-ATPase activity, indicating the production of higher levels of intracellular H2O2 generated (Fig. 2). Specifically, V-ATPase activity in the microsomes isolated from A549 cells exposed to 12.5, 25, 50, and 100 μg/ml PCN decreased by 32%, 52%, 71%, and 83%, respectively. Thus, PCN inhibits V-ATPase by generating ROI, particularly H2O2, in a dose-dependent manner, and it is as effective in inhibiting V-ATPase as BA1. The saturation level for V-ATPase inactivation appears to be 50–100 μg/ml PCN, which inhibits V-ATPase to the levels evoked by 2 μM BA1 and 15 μM H2O2. H2O2, BA1, and PCN did not completely abolish ATP hydrolysis, presumably because microsome preparations contained another ATPase not inactivated by these compounds. Taken together, these results showed that PCN inactivates V-ATPase within lung epithelial cells in a dose-dependent manner by generating ROI, particularly H2O2.
Fig. 5.

PCN inhibits ATP hydrolysis of V-ATPase from lung epithelial cells. Shown are ATP hydrolysis assays with microsomes isolated from A549 cells after1hof exposure to DMSO (control) or to indicated concentrations of H2O2, PCN, or BA1. The percent of ATP hydrolysis was calculated relative to the DMSO control. The mean of three independent experiments and standard deviation are shown. All data were statistically significant at P < 0.05.
Conclusions
Yeasts have a rapid growth rate, a short life, cycle and are amenable to genetic studies. These factors represent a distinct advantage over mammalian systems, especially when it comes to dissecting the internetworking of various PCN targets. Here, by exploiting the genetic simplicity of S. cerevisiae as a surrogate host, we have provided insights into the mammalian cellular pathways that are disrupted by PCN that may ultimately lead to novel therapeutic interventions for preventing and treating infection by P. aeruginosa. Based on our screening data, recovering multiple yeast V-ATPase mutants with increased susceptibility to PCN, we showed that PCN inactivates human VATPase in lung epithelial cells. Thus, we have provided support for our hypothesis that a yeast screen can be used to identify mammalian orthologs conferring sensitivity to PCN.
To date, there has not been any report of V-ATPase inactivation by PCN-mediated oxidative stress. V-ATPase is the major source of ATP generation and consumption, but also regulates receptor-mediated endocytosis, intracellular targeting of lysosomal enzymes, protein processing and degradation, vesicular transport, viral entry, and coupled transport of small molecules such as neurotransmitters (57). Within the plasma membrane, V-ATPases function in such processes as bone resorption, renal acidification, pH homeostasis, and K+ secretion (57). The vacuole is also the major storage site of intracellular Ca2+ in yeast and functions to maintain cytosolic Ca2+ levels within a narrow physiological range via a Ca2+ pump and a H+/Ca2+ antiporter driven by the V-ATPase (58). Furthermore, VATPase is also critical for the bactericidal activity of alveolar macrophages (59). Because V-ATPase regulates multiple cellular processes, its inactivation by PCN may be of huge medical significance. However, no study to date has examined whether PCN inactivates V-ATPase of mammalian cells.
Inactivation of V-ATPase by PCN may have wider implication in CF pathogenesis. Particularly, its role in regulating Ca2+ homeostasis may be critical to regulate ciliary motility (60), a process that is disrupted by PCN (9, 10). V-ATPase is also critical for the proliferation of various cultured cell lines, because nanomolar concentrations of the V-ATPase inhibitor BA1 inhibits cell proliferation (61). Thus, inactivation of V-ATPase by PCN-generated H2O2 (or “downstream” HO·), may account for many of the previously reported PCN-mediated mammalian injuries such as cilia dyskinesis (9, 10), disruption of Ca2+ homeostasis (9, 10), and inhibition of lymphocyte proliferations (14). V-ATPase also regulates vesicle transport and protein sorting via pH gradients among various cellular compartments such as the ER and Golgi (62). When coupled with the fact that PCN also inactivates vesicle transport and protein sorting machineries (Table 1), it is tantalizing to postulate that PCN may inhibit or exacerbate the reduced cystic fibrosis transmembrane conductance regulator (CTFR) localization in CF lungs chronically infected with PCN-producing P. aeruginosa.
Our findings are not limited in scope to only PCN mediated inhibition of the V-ATPase. Because oxidation and phosphorylation are coupled in mitochondria, inhibition of its ATPase by PCN would result in an increased reduced pyridine pool within this organelle (e.g., NADH, NADPH). These molecules can readily reduce PCN nonenzymatically to either mono- or dication radicals, which readily react with molecular oxygen to generate 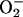 and/or H2O2. Recall that two yeast mutants that were found to be resistant to PCN had mutations in loci affecting ubiquinone (CAT5) and cytochrome b biosynthesis (CBP6) (Table 2). The standard reduction potential for these molecules are 0.04 and 0.07 V, respectively, and that of PCN is -0.034 V (63). Because of the high capacity for PCN autoxidation, it is reasonable to assume that both ubiquinone and cytochrome b can reduce PCN, thereby increasing the level of oxidative stress in cells. An absence or reduced amount of these molecules would almost certainly, in turn, reduce the amount of concomitant oxidative stress.
and/or H2O2. Recall that two yeast mutants that were found to be resistant to PCN had mutations in loci affecting ubiquinone (CAT5) and cytochrome b biosynthesis (CBP6) (Table 2). The standard reduction potential for these molecules are 0.04 and 0.07 V, respectively, and that of PCN is -0.034 V (63). Because of the high capacity for PCN autoxidation, it is reasonable to assume that both ubiquinone and cytochrome b can reduce PCN, thereby increasing the level of oxidative stress in cells. An absence or reduced amount of these molecules would almost certainly, in turn, reduce the amount of concomitant oxidative stress.
Supplementary Material
Acknowledgments
We thank Dr. Gary Dean (University of Cincinnati) for technical help with the ATP hydrolysis assays. This work was partially supported by a research grant from the American Lung Association.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PCN, pyocyanin; SOD, superoxide dismutase; ROI, reactive oxygen intermediates; DCFH, 2′,7′-dichlorofluorescin; DCFH-DA, DCFH diacetate; AO, acridine orange; V-ATPase, vacuolar ATPase.
References
- 1.Tatterson, L. E., Poschet, J. F., Firoved, A., Skidmore, J. & Deretic, V. (2001) Front. Biosci. 6, D890-D897. [DOI] [PubMed] [Google Scholar]
- 2.Govan, J. R. & Deretic, V. (1996) Microbiol. Rev. 60, 539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank, D. W. (1997) Mol. Microbiol. 26, 621-629. [DOI] [PubMed] [Google Scholar]
- 4.Frank, L. H. & Demos, R. D. (1959) J. Bacteriol. 77, 776-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahajan-Miklos, S., Tan, M.-W., Rahme, L. G. & Ausubel, F. M. (1999) Cell 96, 47-56. [DOI] [PubMed] [Google Scholar]
- 6.Cao, H., Krishnan, G., Goumnerov, B., Tsongalis, J., Tompkins, R. & Rahme, L. (2001) Proc. Natl. Acad. Sci. USA 98, 14613-14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallagher, L. A., McKnight, S. L., Kuznetsova, M. S., Pesci, E. C. & Manoil, C. (2002) J. Bacteriol. 184, 6472-6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mavrodi, D. V., Bonsall, R. F., Delaney, S. M., Soule, M. J., Phillips, G. & Thomashow, L. S. (2001) J. Bacteriol. 183, 6454-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson, R., Sykes, D. A., Watson, D., Rutman, A., Taylor, G. W. & Cole, P. J. (1988) Infect. Immun. 56, 2515-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorensen, R. U. & Klinger, J. D. (1987) Antibiot. Chemother. 39, 113-124. [DOI] [PubMed] [Google Scholar]
- 11.Denning, G. M., Railsback, M. A., Rasmussen, G. T., Cox, C. D. & Britigan, B. E. (1998) Am. J. Physiol. 274, L893-L900. [DOI] [PubMed] [Google Scholar]
- 12.Kamath, J. M., Britigan, B. E., Cox, C. D. & Shasby, D. M. (1995) Infect. Immun. 63, 4921-4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Usher, L. R., Lawson, R. A., Geary, I., Taylor, C. J., Bingle, C. D., Taylor, G. W. & Whyte, M. K. (2002) J. Immunol. 168, 1861-1868. [DOI] [PubMed] [Google Scholar]
- 14.Muhlradt, P. F., Tsai, H. & Conradt, P. (1986) Eur. J. Immunol. 16, 434-440. [DOI] [PubMed] [Google Scholar]
- 15.Britigan, B. E., Railsback, M. A. & Cox, C. D. (1999) Infect. Immun. 67, 1207-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassan, H. M. & Fridovich, I. (1980) J. Bacteriol. 141, 156-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes, W. T. & Kim, H. K. (1973) Mycopathol. Mycol. Appl. 50, 261-269. [DOI] [PubMed] [Google Scholar]
- 18.Kerr, J. (1994) J. Infect. 28, 305-310. [DOI] [PubMed] [Google Scholar]
- 19.Kerr, J. R., Taylor, G. W., Rutman, A., Hoiby, N., Cole, P. J. & Wilson, R. (1999) J. Clin. Pathol. 52, 385-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahme, L. G., Stevens, E. J., Wolfort, S. F., Shao, J., Tompkins, R. G. & Ausubel, F. M. (1995) Science 268, 1899-1902. [DOI] [PubMed] [Google Scholar]
- 21.Hassett, D. J., Charniga, L., Bean, K., Ohman, D. E. & Cohen, M. S. (1992) Infect. Immun. 60, 328-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. & Boeke, J. D. (1998) Yeast 14, 115-132. [DOI] [PubMed] [Google Scholar]
- 23.Giard, D. J., Aaronson, S. A., Todaro, G. J., Arnstein, P., Kersey, J. H., Dosik, H. & Parks, W. P. (1973) J. Natl. Cancer Inst. 51, 1417-1423. [DOI] [PubMed] [Google Scholar]
- 24.Beer, R. F. & Sizer, I. W. (1952) J. Biol. Chem. 195, 133-140. [PubMed] [Google Scholar]
- 25.McCord, J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6056-6063. [PubMed] [Google Scholar]
- 26.Vanden Hoek, T. L., Li, C., Shao, Z., Schumacker, P. T. & Becker, L. B. (1997) J. Mol. Cell. Cardiol. 29, 2571-2583. [DOI] [PubMed] [Google Scholar]
- 27.Geisow, M. J., Beaven, G. H., Hart, P. D. & Young, M. R. (1980) Exp. Cell. Res. 126, 159-165. [DOI] [PubMed] [Google Scholar]
- 28.Bauerle, C., Ho, M. N., Lindorfer, M. A. & Stevens, T. H. (1993) J. Biol. Chem. 268, 12749-12757. [PubMed] [Google Scholar]
- 29.Graham, L. A., Hill, K. J. & Stevens, T. H. (1998) J. Biol. Chem. 269, 25974-25977. [PubMed] [Google Scholar]
- 30.Xu, T. & Forgac, M. (2000) J. Biol. Chem. 275, 22075-22081. [DOI] [PubMed] [Google Scholar]
- 31.Hill, K. J. & Stevens, T. H. (1994) Mol. Biol. Cell 5, 1039-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Supek, F., Madden, D. T., Hamamoto, S., Orci, L. & Schekman, R. (2002) J. Cell. Biol. 158, 1029-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sacher, M., Stone, S. & Ferro-Novick, S. (1997) J. Biol. Chem. 272, 17134-17138. [DOI] [PubMed] [Google Scholar]
- 34.Duden, R., Kajikawa, L., Wuestehube, L. & Schekman, R. (1998) EMBO J. 17, 985-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, B. & Warner, J. R. (1999) J. Biol. Chem. 271, 16813-16819. [DOI] [PubMed] [Google Scholar]
- 36.Babst, M., Sato, T. K., Banta, L. M. & Emr, S. D. (1997) EMBO J. 16, 1820-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Babst, M., Katzmann, D. J., Estepa-Sabal, E. J., Meerloo, T. & Emr, S. D. (2002) Dev. Cell. 3, 271-282. [DOI] [PubMed] [Google Scholar]
- 38.Swaminathan, S., Amerik, A. Y. & Hochstrasser, M. (1999) Mol. Biol. Cell 10, 2583-2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, Y. & Floor, E. (1998) J. Neurochem. 70, 646-652. [DOI] [PubMed] [Google Scholar]
- 40.Ihara, Y., Yasuoka, C., Kageyama, K., Wada, Y. & Kondo, T. (2002) Biochem. Biophys. Res. Commun. 297, 353-360. [DOI] [PubMed] [Google Scholar]
- 41.Echtay, K. S., Roussel, D., St-Pierre, J., Jekabsons, M. B., Cadenas, S., Stuart, J. A., Harper, J. A., Roebuc, S. J., Morrison, A., Pickering, S., et al. (2002) Nature 415, 96-99. [DOI] [PubMed] [Google Scholar]
- 42.Liu, X. F., Supek, F., Nelson, N. & Culotta, V. C. (1997) J. Biol. Chem. 272, 11763-11769. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt, C. L., Grey, M., Schmidt, M., Brendel, M. & Henriques, J. A. (1999) Yeast 15, 1503-1510. [DOI] [PubMed] [Google Scholar]
- 44.Cheng, J., Park, T. S., Chio, L. C., Fischl, A. S. & Ye, X. S. (2003) Mol. Cell. Biol. 23, 163-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroepfer, G. J., Jr. (2000) Physiol. Rev. 80, 361-554. [DOI] [PubMed] [Google Scholar]
- 46.Radji, M., Kim, J. M., Togan, T, Yoshikawa, H. & Shirahige, K. (2001) Yeast 18, 195-205. [DOI] [PubMed] [Google Scholar]
- 47.Bartlett, R. & Nurse, P. (1990) BioEssays 12, 457-463. [DOI] [PubMed] [Google Scholar]
- 48.Uesono, Y., Fujita, A., Tohe, A. & Kikuchi, Y. (1994) Gene 143, 135-138. [DOI] [PubMed] [Google Scholar]
- 49.Santos-Rosa, H., Clever, B., Heyer, W. D. & Aguilera, A. (1996) Genetics 142, 705-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsukamoto, Y., Kato, J. & Ikeda, H. (1997) Nature 388, 900-903. [DOI] [PubMed] [Google Scholar]
- 51.Jang, Y. J., Won, M., Chung, K. S., Kim, D. U., Hoe, K. L., Park, C. & Yoo, H. S. (1997) J. Biol. Chem. 272, 19993-20002. [DOI] [PubMed] [Google Scholar]
- 52.Fearon, K. & Mason, T. L. (1992) J. Biol. Chem. 267, 5162-5170. [PubMed] [Google Scholar]
- 53.Yanase, S. & Ishi, N. (1999) J. Radiat. Res. 40, 39-47. [DOI] [PubMed] [Google Scholar]
- 54.Jonassen, T., Proft, M., Randez-Gil, F., Schultz, J. R., Marbois, B. N., Entian, K. D. & Clarke, C. F. (1998) J. Biol. Chem. 273, 3351-3357. [DOI] [PubMed] [Google Scholar]
- 55.Dieckmann, C. L. & Tzagoloff, A. (1985) J. Biol. Chem. 260, 1513-1520. [PubMed] [Google Scholar]
- 56.Fillinger, S., Chaveroche, M. K., van Dijck, P., de Vries, R., Ruijter, G., Thevelein, J. & d'Enfert, C. (2001) Microbiology 147, 1851-1862. [DOI] [PubMed] [Google Scholar]
- 57.Nishi, T. & Forgac, M. (2002) Nat. Rev. Mol. Cell Biol. 3, 94-103. [DOI] [PubMed] [Google Scholar]
- 58.Forster, C. & Kane, P. M. (2000) J. Biol. Chem. 275, 38245-38253. [DOI] [PubMed] [Google Scholar]
- 59.Bidani, A., Reisner, B. S., Haque, A. K., Wen, J., Helmer, R. E., Tuazon, D. M. & Heming, T. A. (2000) Lung 178, 91-104. [DOI] [PubMed] [Google Scholar]
- 60.Lansley, A. B. & Sanderson, M. J. (1999) Biophys. J. 77, 629-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manabe, T., Yoshimori, T., Henomatsu, N. & Tashiro, Y. (1993) J. Cell Physiol. 157, 445-452. [DOI] [PubMed] [Google Scholar]
- 62.Wu, M. M., Grabe, M., Adams, S., Tsien, R. Y., Moore, H.-P. H. & Machen, T. E. (2001) J. Biol. Chem. 276, 33027-33035. [DOI] [PubMed] [Google Scholar]
- 63.Friedheim, E. & Michaelis, L. (1931) J. Biol. Chem. 91, 355-368. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}