

Abstract
The asymmetric distribution of the amino-containing phospholipids, phosphatidyl-serine (PS) and phosphatidyl-ethanolamine (PE), across the two leaflets of red blood cell (RBC) membrane is essential to the function and survival of the cell. PS and PE are sequestered in the inner leaflet by an ATP-dependent transport activity of a membrane protein known as the RBC flippase that specifically moves amino-phospholipids from the outer to the inner leaflet. The enucleated RBC lacks the means to replace damaged enzymes and inactivation of the flippase can lead to the unwarranted exposure of PS on the cell surface. Loss in the ability to maintain phospholipid asymmetry is exacerbated in RBC disorders and PS-exposing RBCs present in the circulation play a significant role in the pathology of hemoglobinopathies. We identified the Atp8a1 protein, a member of the family of the P4-type ATPases, as a RBC flippase candidate. Atp8a1 is expressed in RBC precursors and is present in the membrane of mature red cells. The flippase activity of the protein was established in purified secretory vesicles of Saccharomyces cerevisiae. ATPase activity was stimulated by PS and PE. In addition, Atp8a1 can move PS molecules across the leaflets of the vesicle membrane in presence of ATP.
Keywords: flippase, ATP8A1, PS transport, red blood cell, sickle cell disease
Introduction
The sequestration of amino-glycerophospholipids, phosphatidyl-serine (PS) and phosphatidyl-ethanolamine (PE), on the cytofacial leaflet of the plasma membrane is essential to the function and the survival of the red blood cell (RBC). Due to the passive diffusion of lipids and the activity of unidentified protein(s) that can distribute lipids across the two leaflets, the asymmetry of the membrane could not be maintained without a mechanism to transport the exposed PS and PE back to the inner layer (Pomorski et al 2004; Holthuis and Levine 2005; Paulusma and Oude Elferink 2005; Ikeda et al 2006; Pomorski and Menon 2006). This inward movement of PS is driven by an ATP-dependent transporter, with selectivity for amino-phospholipids, known as the flippase. Hydrolysis of one ATP is needed for each molecule of PS transported (Beleznay et al 1993). Exposure of PS on the surface of cells is a normal event of many different physiological processes, such as apoptosis, spermatozoa capitation, platelet activation, membrane fusion, and cell adhesion (Pomorski et al 2004; Wang et al 2004; Holthuis and Levine 2005; Paulusma and Oude Elferink 2005; Ikeda et al 2006; Pomorski and Menon 2006). RBCs with PS present on their surface are rarely found in circulation as they are rapidly recognized and removed by macrophages. However, in sickle cell disease, increased numbers of PS-exposing RBCs are commonly detected. They are also found in splenectomized thalassemia patients. In addition of having a shortened survival, these cells can induce pathophysiologic responses such as imbalanced hemostasis, interactions with other blood cells, and with endothelial cells of the vascular wall (Kuypers and De Jong 2004). In addition, PS-exposing RBCs can be the target of secretory phospholipase A2 and a source of plasma LPA, which can lead to vascular dysfunction (Neidlinger et al 2006). The occurrence of PS-exposing RBCs in hemoglobinopathies is in part due to the activation of the scramblase activity, which randomizes lipids across the leaflets, and the inability of the cells to transport PS back to the inner leaflet.
We have previously established that the P-type ATPase Atp8a1, a membrane-embedded protein of ≈ 120 kDa, is expressed in erythrocyte precursors and present in the plasma membrane of mature RBC (Soupene and Kuypers 2006a). Also known as ATPaseII, we have proposed that Atp8a1 represents the protein carrying the flippase activity identified in the RBC membrane at the biochemical level (for reviews see [Pomorski et al 2004; Holthuis and Levine 2005; Paulusma and Oude Elferink 2005; Ikeda et al 2006; Pomorski and Menon 2006]). The establishment of its function as a flippase in the membrane of this enucleated cell is complicated by the presence of other activities and factors that may affect its activity. In addition, purification and solubilization of this membrane protein in detergents is not preferred due to their potential effects on the structure and/or enzymatic characteristics of the protein. We decided to determine the activity of Atp8a1 in an in vivo membrane system that does not require purification or solubilization in detergent. We successfully established both the ATPase and transport activities of Atp8a1 in purified post-Golgi secretory vesicles of a sec6 Saccharomyces cerevisiae mutant strain. These vesicles can accumulate to high yield in the cytosol of the cells and can be used to study the activity of transport proteins as an alternative method to in vitro made proteo-liposomes (Walworth and Novick 1987; Nakamoto et al 1991; Alder-Baerens et al 2006). Contrary to the formation of proteo-liposomes that requires solubilization in detergents of both the lipids and the enzyme, insertion of the protein in the lipid bilayer of the vesicles occurs in the cells. In addition, in contrast to liposome preparations, which are often characterized by an undefined population of multi-lamellar structures of various sizes, yeast transit vesicles are homogenous in size (100 to 200 nm in diameter) and are composed of a single lipid bilayer. They are enriched in the membrane protein over-produced by the cells, the recombinant foreign protein, and they can be isolated. The lipid composition of their single membrane is defined and can be modified (Zinser et al 1991; Schneiter et al 1999). We report that Atp8a1 protein was successfully expressed in the yeast mutant strain and accumulated in the post-Golgi vesicles. The protein was produced in an active form and its ATPase activity was stimulated by PS and to a lesser degree by PE. In addition, Atp8a1 uni-directionally transported a fluorescently labeled PS molecule, NBD-PS, in presence of ATP. Thus, the P4-type ATPase Atp8a1, which is present in the RBC membrane, displays the enzymatic properties that define the red cell flippase activity.
Materials and methods
Materials
Concanavalin A, sodium ortho vanadate, Ouabain, N-Ethylmaleimide, C12E9, and the lipids L-α phosphatidyl serine (bovine brain), phosphatidylethanolamine (bovine brain) and phosphatidylcholine (egg yolk) were obtained from Sigma-Aldrich, Inc. (St. Louis, MO, USA). NBD-PS (1-Palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-Glycero-3-Phospho-L-Serine) was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA).
DNA manipulations
A cDNA clone encoding the full-length product of mouse Atp8a1 (GenBank accession no. AK045367; RefSeq no. NP_001034088) was identified in the RIKEN database (Kabushiki Kaisha DNAForm) as Mouse Fantom clone ID B230107D19. The identity of the clone was confirmed by sequencing. The full-length open reading frame of 1164 residues was cloned under the control of a galactose inducible promoter (Gal1) with hexahistidine and V5 epitope tags at the C-terminus, into the yeast expression vector pYES2/CT (Invitrogen™, Carlsbad, CA, USA), which also carries the selectable marker URA3. A PCR fragment of 3,495 bp was amplified using the cDNA clone as template with the primer pYes2.Hd (5′-CATTAAGCTTGCCACCATGGCGACCATGCGGAGGACAG-3′), which introduced a unique restriction site for Hind III (underlined) and a consensus Kozak sequence (double-underlined) and the primer pYes2.Xho (5′-GCATGCTCGAGCCACTCATCGGGCCTCTG-3′), which introduced a unique restriction site for Xho I (underlined) and remove the natural stop codon of the cDNA. Amplifications were performed with Expand High-Fidelity Taq Polymerase (Roche Applied Science, Indianapolis, IN, USA) as follows: one cycle at 94 °C for 100 s, 35 cycles at 94 °C for 30 s, 48 °C for 30 s, 72 °C for 150s, and one cycle at 68 °C for 10 min. The PCR fragment was cloned with the Zero-Blunt® PCR cloning kit (Invitrogen), according to the manufacturer’s instructions, to yield plasmid pFK178. After confirmation by sequencing, the ≈3.5-kbp Hind III-Xho I fragment was ligated to pYES2/CT, opened by Hind III and Xho I, to yield plasmid pFK184. The plasmid was then introduced into the yeast strain SY1 (see below). Human ACSL6 was cloned in the same manner in pYES2/CT to yield plasmid pFK195.
Culture and manipulation of S. cerevisiae
The yeast mutant strain SY1 (MATα ura3-52 leu2-3,112, his4-619, sec6-4ts, GAL2), a generous gift of Dr. C.W. Slayman (Nakamoto et al 1991), that carries a thermo-sensitive sec6 mutation and grows on galactose was used in this study. The strain was routinely maintained on YPD plates at 25 °C. The pYES2/CT, pYES2/CT/LacZ (Invitrogen) and pFK184 (Atp8a1) plasmids were introduced into the SY1 strain using the S.c. EasyComp kit (Invitrogen), according to the manufacturer’s instructions, to yield strains FK196, FK197 and FK204, respectively. Transformants were selected at 25 °C without uracil on YNB-Leucine medium (Yeast Nitrogen Base-with amino acids medium [BD Bioscience, San Jose, CA, USA] supplemented 180 μg/ml of L-leucine)with 2% glucose. After a week, single colonies were picked and purified twice on the same medium. The presence of the plasmids in the clones was confirmed by PCR analysis and by DNA isolation.
β-galactosidase and acyl-CoA synthetase assays
Yeast strains carrying the plasmid pYES2/CT/LacZ or pYES2/CT/ACSL6 were grown in YNB-Leucine medium, at 25 °C with 2% glucose or galactose. Samples of 1.5 ml were removed from the culture, centrifuged at 10,000 × g for 30s, and stored frozen until assayed for β-galactosidase activitity (Clontech manual, protocol PT3024-1) or for acyl-CoA synthetase activity in presence of [1-14C]-C18:1-OH (Soupene and Kuypers 2006b).
Protein expression in S. cerevisiae
Yeast strains, inoculated from fresh plates, were grown in YNB-Leucine medium and 2% glucose at 25 °C to an OD600 of ≈2.0 (≈24 hrs). Cells were collected by centrifugation at 2,000 g for 5 min, washed once in medium without glucose, and suspended to an OD600 of ≈0.1 into 250 ml of YNB-Leucine medium with 2% galactose. Cells were grown at 25 °C for 3 generations to allow induction of expression of proteins under the control of the Gal1 promoter. The culture was then shifted to the nonpermissive temperature of 37 °C to inactivate the Sec6ts protein, resulting in the accumulation of post-Golgi vesicles in the cytosol of the cells. After 6 hrs at 37 °C, cells were collected by centrifugation at 6,000 g for 10 min and stored frozen at −80 °C.
Yeast vesicles isolation
The frozen cell pellet was thawed, washed with 50 ml of 10 mM NaN3, and centrifuged for 10 min at 2,000 × g. The pellet was weighed and suspended with 10 ml of lyticase solution per gram of cells. Lyticase (360 units/mg) was dissolved at 5 mg/ml in sorbitol buffer (1.4 M D-sorbitol, 50 mM K2HPO4 pH 7.5, 10 mM NaN3, 40 mM ϐ-mercaptoethanol and 1 mM PMSF). Digestion of the cell wall was carried out in a water bath at 37 °C with gentle agitation for 1 to 2 hrs. The formation of spheroplasts was monitored by the decreased of the absorbance of the suspension diluted 50 times in water at OD800 nm. Spheroplasts were collected by centrifugation at 500 g for 10 min and gently suspended with cold sorbitol buffer containing 10 mM MgCl2 and 0.8 mg/ml of concanavalin A at an equivalent of 50 OD600 per ml, and were incubated for 15 min on ice. Cross-linking of the plasma membranes was monitored under a light microscope. After centrifugation at 500 g for 5 min, spheroplasts were suspended in cold lysis buffer (0.8 M D-sorbitol, 10 mM triethanolamine pH 7.2 and 1 mM EDTA) at an equivalent of 60 OD600 per ml. They were mechanically disrupted by applying 20 to 30 strokes with a Dounce homogenizer, which was verified under a light microscope, and cleared by centrifugation at 10,000 × g for 10 min at 4 °C. The vesicles contained in the supernatant were then collected by ultra-centrifugation at 100,000 g for 1 hr at 4 °C. The pellet containing the vesicles was gently washed with 4 ml of cold 50 mM Tris-HCl pH 7.4 with 1 mM EGTA and collected as above. Finally, vesicles were slowly and gently suspended in about 200 μl of cold 50 mM Tris-HCl pH 7.4, 1 mM EGTA, and 10% glycerol using a stir-bar. The vesicles were stored frozen at −80 °C.
Protein quantification and detection
The concentration of proteins in vesicles was determined with the Bio-Rad DC Protein Assay, according to the manufacturer’s instructions. Vesicles were brought to a volume of 25 μl with water and mixed with 175 μl of 10% SDS. Then, 100 μl of reagent A and 800 μl of reagent B were added. After 15 minutes incubation at room temperature, the absorbance at 750 nm was read. BSA was used as the standard under the same conditions. Proteins extracts from whole yeast cells were obtained by breakage with vigorous vortexing in the presence of glass beads. Total protein extracts and vesicles samples were dissolved in the SDS-PAGE loading buffer and denatured for 30 min at 37 °C. Five μg of proteins were loaded in each lane of a 7.5%-gel SDS-PAGE. After migration, the proteins were electro-transferred to a nitrocellulose membrane, and immuno-detection was performed in TBS (Tris buffer saline pH 7.4) with 0.1% Tween-20 and 1% milk with a commercial monoclonal anti-V5 antibody (Invitrogen) at a 1/5,000 dilution for 1 hr at room temperature. The membrane was washed 3 times 10 min with TBS-0.1% Tween-20, and incubated with a HRP-conjugated goat anti-rabbit IgG antibody (Pierce) at a 1/1,000 dilution for 1 hr. HRP detection was performed with ImmonoPure DAB (Pierce, Rockford, IL, USA) according to the manufacturer instructions.
ATPase activity assays
ATPase assays were performed at 37 °C in 40 mM HEPES-KOH pH 7.2, 10 mM MgCl2, 1 mM EGTA, and 10 mM ATP with an amount of vesicles equivalent to 2 μg of proteins for each time point. The reaction was carried out for up to 2 hours and, at least, 4 samples of 80 μl were removed at various times during the incubation, and were mixed with 20 μl of Malachite Green dye (BioAssay Systems, Hayward, CA, USA) to terminate the reaction. After 20 min of incubation at room temperature with the dye, absorbance at 650 nm was determined with a microplate reader, and the amount of phosphate released was calculated using a standard curve obtained with a phosphate standard (KH2PO4) (BioAssay Systems). All the ATPase assays performed with vesicles containing Atp8a1 protein were corrected for the amount of Pi released with vesicles isolated from a yeast strain carrying the vector, pYES2/CT. Atp8a1-vesicles and control vesicles (vesicles obtained from the yeast strain transformed with the vector only) were always assayed in parallel under all the different conditions tested as shown in Figure 3. The amount of Pi released in the reaction containing the vector-vesicles was subtracted from the values obtained with the Atp8a1-vesicles for each time point. The rates of hydrolysis of ATP (pmoles of Pi released per mg of proteins per minute) was calculated by taking the slope of the linear regression line through the data points, amount of Pi released (y-axis) plotted as function of time (x-axis). Phospholipids (PS, PE, PC), dissolved in chloroform/methanol (2/1, v/v), were dried under nitrogen, suspended in 0.01% (w/v) of C12E9, and added to the ATPase buffer at a 1/150 dilution before addition of the vesicles. Vanadate, oubain, CaCl2, MgCl2, and EDTA were also added to the ATPase buffer prior to incubation with the vesicles. N-ethylmaleimide (NEM) was either added in the ATPase buffer with the vesicles (in-reaction condition) or to the vesicles before dilution in the ATPase buffer (pre-reaction condition). Pre-treatments of vesicles were performed with increasing concentrations of NEM in a volume of 20 μl for 30 min on ice. Vesicles were then diluted to 360 μl with the ATPase buffer. The pretreatment of vesicles led to the carry over of some NEM during the ATPase reaction. The highest concentration of NEM during the reaction phase of the incubation was 0.55 mM, which is not inhibitory as established by the reactions performed in presence of NEM without pre-treatment (see Results).
Figure 3.

ATPase activity of Atp8a1 in yeast vesicles. Vesicles isolated from cells carrying either the vector (open square) or the Atp8a1 construct (filled square) were assayed for release of phosphate from added ATP as function of time at 37 °C. Two μg of proteins were used for each time point. The rate of hydrolysis of ATP (pmoles of Pi released per mg of proteins per minute) was calculated by taking the slope of the linear regression line. In the experiment shown, the 120 min data point was no longer in the linear range and it was not included in the calculation.
Transport activity assays
Assays were performed at 25 °C in 200 μl of 20 mM HEPES-KOH pH 7.2, 15 mM KCl, 5 mM NaCl, 10 mM MgCl2, 2 mM EGTA, 5 mM hydroxylamine, 120 mM K-glutamate with or without 10 mM ATP (see below), with an amount of vesicles equivalent to 25 μg of protein and 4.25 pmoles of NBD-PS for each time point. Fluorescence intensity was recorded at 535 nm (excitation = 468 nm, slit width = 10 nm) in a Perkin-Elmer LS5B luminescence spectrometer at 25 °C using a quartz micro fluorometer cell (3 × 3 × 40 mm). Vesicles were first mixed with NBD-PS in absence of ATP. Incorporation of NBD-PS into the outer layer of the membrane of the vesicles was achieved in the first minute after the addition. The vesicles were first incubated in absence of ATP for 2 hrs, then 10 mM ATP was added and the vesicles were further incubated for 1 hr. Presence of NBD-PS in the outer layer was detected by reducing the NBD fluorophore to a nonfluorescent entity by the nonmembrane permeant reagent dithionite. After recording fluorescence in absence of dithionite (F−), 5 μl of 1 M dithionite was added to the 200 μl vesicles mixtures (25 mM dithionite final concentration) and the fluorescence was recorded after 1 min (F+). Percent of quenched NBD-PS was calculated as follows: (1-F+/F−) × 100. To determine the total amount of NBD-PS incorporated in the different conditions and to assess efficiency of the quenching, control reactions were performed by the addition of 1/10 volume of 5% SDS in the cuvettes after the F− and F+ values had been recorded.
Results
Production of Atp8a1 protein in vesicles
We first established that mammalian membrane proteins could be expressed in the yeast mutant strain used in our study, and that their activity could be measured in cells as well as in preparation of intra-cellular vesicles obtained with a simplified isolation procedure (see Methods). Genes were cloned under the control of a promoter induced during growth of the cells on galactose, as the carbon source, and the accumulation of the post-Golgi vesicles in the cytosol was obtained by shifting galactose-grown cells to the nonpermissive temperature of 37 °C (see Materials and methods) (Walworth and Novick 1987; Nakamoto et al 1991; Alder-Baerens et al 2006). The product of the lacZ gene, encoding the β-galactosidase enzyme, and of the ACSL6 gene, encoding a human membrane protein of known activity (acyl-CoA synthetase) (Soupene and Kuypers 2006b), were used to verify that the yeast strain correctly expressed recombinant proteins during growth on galactose and was suitable for the study of the activity of mammalian membrane-bound enzymes, such as Atp8a1. Expression of the cytosolic β-galactosidase and of the membrane protein ACSL6 did not have a deleterious effect on the growth of the strain (Figure 1, panel A). As expected, the activities of both enzymes were absent in cells grown on glucose but were detected during growth on galactose (Figure 1, panel B and C). In addition, the activity of the ACSL6 protein was also detected in transit vesicles obtained from cells grown on galactose and shifted at 37 °C (data not shown).
Figure 1.

Protein expression in the Saccharomyces cerevisiae SY1 strain. (A) Growth on galactose of cells transformed with either the vector (open square), the construct expressing lacZ, encoding ϐ-galactosidase enzyme (filled square), or the construct expressing the human membrane protein acyl-CoA synthetase 6 (triangle). Growth was monitored by following the absorbance of the culture at 600 nm as function of time. (B) ϐ-Galactosidase activity of the SY1 strain carrying the lacZ construct during growth with either galactose (filled square) or glucose (open square). (C) Acyl-CoA synthetase activity of SY1 strain carrying the ACSL6 construct grown with either galactose (filled square) or glucose (open square). Cells were harvested at an OD of 1.0 and assayed for acyl-CoA synthetase activity as described in Materials and methods.
A cDNA representing the full-length version of the Atp8a1 protein, annotated as isoform a, was cloned in the yeast mutant strain (see Materials and methods). Atp8a1 was detected in a total protein extract obtained from cells grown on galactose (Figure 2A) and was localized in the transit vesicles that accumulated in the cytosol at 37 °C (Figure 2B, lanes 4 and 5). Atp8a1 protein is very sensitive to proteolytic cleavage when expressed in various cells (Ding et al 2000) and several degradation products were detected in a protein extract made from a lysate obtained after enzymatic digestion of the yeast cell wall and mechanical disruption of the spheroplasts (Figure 2B, lane 2). However, the vesicles contained mainly the full-length protein and very little of the smaller products could be detected (Figure 2B, lanes 4 and 5). Atp8a1 protein was not detected in cells grown on glucose or in cells transformed with the vector used for cloning (Figure 2A and 2B, lanes 1 and 3).
Figure 2.

Expression and localization of Atp8a1 protein in vesicles. Western blotting detection of Atp8a1 protein in SY1 cells carrying the construct expressing Atp8a1 grown with either glucose or galactose. Proteins were extracted, separated by SDS-PAGE and transferred to a nitrocellulose membrane. Atp8a1 was detected with a monoclonal antibody against the V5 epitope fused at the C-terminus of the protein. (A) Total protein extracts obtained from cells carrying the Atp8a1 construct grown with either glucose (Glc) or galactose (Galac). (B) After enzymatic digestion of the cell wall, spheroplasts were mechanically disrupted and the lysate was cleared at 4,000 g. Proteins present in the supernatant were loaded on lanes 1 and 2. Protein extracts of post-Golgi vesicles, isolated as described in Materials and methods, are shown on lanes 3, 4, and 5. Protein extracts obtained from cells carrying the vector are shown on lanes 1 and 3 and from cells expressing Atp8a1 are shown on lanes 2, 4, and 5. Lane 5 shows the result of a different vesicle isolation’s experiment obtained from the cells expressing Atp8a1 as shown in lane 4.
ATPase activity in purified vesicles
We verified that the ATPase activity of the protein in yeast was similar to the activity established in other membranes. ATPase activity was determined in purified vesicles by the detection of release of Pi from added ATP as function of time, as described in the Method section. Although, endogenous ATP-hydrolyzing enzymes are present in the vesicles (Nakamoto et al 1991; Alder-Baerens et al 2006), Atp8a1-vesicles have a specific activity rate 5-fold higher than the vector-vesicles (Figure 3). Thus, Atp8a1 is active in this system and the low activity of the yeast ATPases in these vesicles does not preclude detection of Atp8a1. The ATPase activity of Atp8a1 was inhibited by the phosphate analog vanadate and by the presence of Ca2+ (Figure 4). Atp8a1 required the presence of Mg2+ and was insensitive to the drug ouabain. The inhibitory effect of calcium is also observed in RBCs but inhibition of the flippase by calcium is not specific as other cations such as Ni2+, Co2+, and Zn2+ have been shown to also be inhibitory (Bitbol et al 1987; Moriyama and Nelson 1988).
Figure 4.

Characterization of the P-type Mg2+-ATPase activity of Atp8a1. The amount of phosphate released during incubation of ATP and the control vesicles (vector vesicles) was determined as function of time in presence or absence of the indicated concentration of chemicals, and was subtracted to the amount of phosphate released in reactions containing the Atp8a1-vesicles assayed under the same conditions. The fold change of the ATPase activity rates was calculated as the ratio of the rate obtained in presence of the indicated chemical to the rate obtained in its absence.
N-ethylmaleimide inhibition
The effect of the sulfhydryl-modifying reagent NEM on the ATPase activity of the protein was tested using two conditions. The vesicles were pre-incubated with NEM for 30 min on ice and then, they were diluted 20-fold in the assay buffer and incubated with ATP. Under these conditions, low NEM concentrations (≤1 mM) were sufficient to completely inhibit the enzyme (Figure 5A and C, column Pre-Rxn), which is similar to the inhibitory effect observed on the flippase activity in the RBC membrane. However, when the vesicles were not pre-treated and NEM was added directly into the reaction mixture containing ATP, a much higher amount was needed to detect an inhibition. Under this condition, 100 mM NEM was required to inhibit the ATPase activity to a similar low level obtained with a pre-treatment of 1 mM (Figure 5B and C, column In Rxn). In addition, the activity of the protein in vesicles exposed to 100 or 150 mM NEM was not irreversibly inhibited and resumed after 90 min incubation (Figure 5B and C, column New Phase). At a lower concentration of 50 mM NEM, activity was only inhibited during the first 30 min of the reaction. The effect of NEM under these conditions was in strong contrast to the findings obtained when the vesicles were pre-incubated with NEM (Figure 5A) and to the complete and, apparently, permanent inhibition observed with vanadate (Figure 4A and not shown). Thus, inhibition by NEM, which is commonly used to inactivate the RBC flippase, appears to be a function of the treatment conditions and of the delay between exposure to the drug and measurement of the activity.
Figure 5.

Effect of N-ethylmaleimide treatment on Atp8a1 activity. The amount of phosphate released during incubation of ATP and the control vesicles (vector only) was determined as function of time in presence or absence of the indicated concentration of NEM, and was subtracted to the amount of phosphate released in the reactions containing the Atp8a1-vesicles assayed under the same conditions. (A) ATPase activity of the Atp8a1-vesicles pre-incubated (pre-reaction, Pre-Rxn) with 0 (square), 0.1 mM (cross), 1 mM (triangle), 5 mM (inverted triangle) or 10 mM (circle) NEM. Note that the concentrations of NEM carried-over into the reaction are indicated in the column In-Rxn of panel C. (B) ATPase activity of the Atp8a1-vesicles added in reaction buffer (in-reaction, In-Rxn) containing 0 (square), 50 mM (cross), 100 mM (triangle), and 150 mM (circle) NEM. (C) The rates of ATP hydrolysis were calculated from 0 to 2 hrs, as described in legend of Figure 4, and presented in the column entitled 0 to 2 hrs. When appropriate, activity rates were calculated from 90 to 120 min and presented in the column entitled New Phase, as described in Results.
Abbreviation: n/a, not applicable.
Stimulation by amino-phospholipids
The ATPase activity was stimulated by low amounts of phosphatidyl-serine (PS) added in the reaction (Figure 6), reaching an optimum at 0.3 mM. Phosphatidyl-ethanolamine, PE, stimulated the activity to a lesser extend, whereas a lipid with a nonamino-head group, phosphatidyl-choline (PC), was neither stimulatory nor inhibitory.
Figure 6.
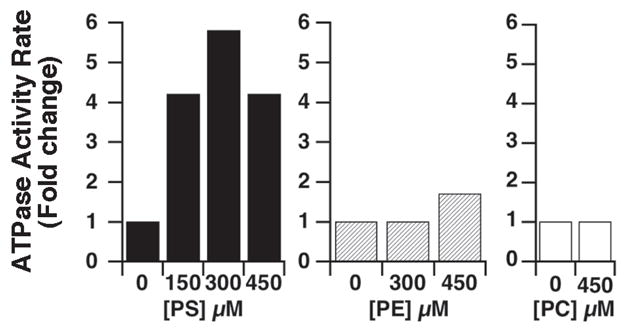
Stimulation by amino-phospholipids. The different lipids were added in the reaction buffer at the indicated concentrations before addition of the vesicles. The amount of phosphate released during incubation of ATP and the control vesicles (vector only) was determined as function of time in presence or absence of the indicated lipids, and was subtracted to the amount of phosphate released in the reactions containing the Atp8a1-vesicles assayed under the same conditions. Assays were performed and ATPase activity rates were calculated as described in legend of Figure 4.
Transport activity of Atp8a1
The movement of PS in Atp8a1-vesicles was determined with a fluorescently-labeled species, NBD-PS, which rapidly incorporated in the outer leaflet of the membrane of the vesicles. Incorporation of NBD-PS was followed by addition of the nonmembrane permeant reductant dithionite. Dithionite reduces the fluorochrome of the NBD-PS molecules exposed on the surface and eliminates their fluorescence. The difference in the fluorescence of the vesicles before and after addition of the quencher is a measure of the distribution of NBD-PS in the two leaflets: high quenching when exposed in the outer leaflet and low quenching when protected in the inner leaflet (Figure 7A). Yeast post-Golgi vesicles contain several endogenous lipids transporters that maintain the asymmetry of the vesicle membrane. They are not specific for aminophospholipids and slowly move lipids from the inner to outer layer (Alder-Baerens et al 2006). Control-vesicles were assayed to assess their activity in our system. In absence of ATP, about 80% of the NBD-PS fluorescence was quenched after incubation for 2 hrs (Figure 7B). As anticipated, a similar slow movement was obtained with the Atp8a1-vesicles when no ATP was present. After addition of ATP, the distribution of the NBD-PS molecules of the Atp8a1-vesicles was noticeably different from the control-vesicles and different from the condition without ATP (Figure 7B). In presence of ATP, about 70% of the fluorescence was still quenched in the control-vesicles, whereas all of the NDB-PS was protected in the Atp8a1-vesicles after 1 hr incubation. Thus, after addition of ATP, rapid movement of PS from the outer to inner leaflet had occurred in the vesicles containing Atp8a1.
Figure 7.

NBD-PS movement in Atp8a1-vesicles. (A) Cartoon representation of the transport assay. Fluorescent phosphatidyl-serine (NBD-PS) molecules were mixed with vesicles and incubated at 25 °C in absence or presence of ATP. The movement of NBD-PS from the outer (exposed) to the inner (protected) layer was monitored by measuring the fluorescence of the vesicles before and after addition of the nonmembrane permeant reductant dithionite, which eliminates the fluorescence of the exposed NBD-PS molecules. (B) Quenching of the NBD-PS fluorescence in control and Atp8a1-vesicles. Quenching values were calculated as described in Methods and were expressed relative to the value obtained when all of the NBD-PS molecules were exposed (t = 0 min), which was set at 100%. ATP was added in both control- and Atp8a1-vesicles 5 minutes after determination of the 2 hrs time point. Bars represent the SE of 5 and 3 different experiments performed with the control- and Atp8a1-vesicles, respectively.
Discussion
The presence of the ATP8A1 protein in erythrocytes has been established both in human and mouse RBCs (Pasini et al 2006; Soupene and Kuypers 2006a; Pasini et al 2008). The mRNA of the murine form was also reported in erythrocyte precursors (Soupene and Kuypers 2006a). The RBC flippase was estimated at a molecular mass of 120 kDa on denaturing acrylamide gel (Morrot et al 1990), which is similar to the migration pattern of Atp8a1 detected in the membrane of RBCs (Soupene and Kuypers 2006a) and in yeast vesicles. The activities and the enzymatic studies of ATP8A1 account for, and explain the observed biochemical characteristics of the flippase activity of RBCs, as summarized in Table 1: requirement for Mg2+ and ATP, specificity for amino-glycerophospholipids, inhibition by vanadate, Ca2+ and sulfhydryl-modifying reagents (Seigneuret and Devaux 1984; Bitbol et al 1987; Bitbol and Devaux 1988; Connor et al 1990). The formation of the phospho-aspartate intermediate of Atp8a1 is the step in the catalytic cycle of this P-type ATPase that is inhibited by vanadate, by some divalent cations (eg, Ca2+) and by N-ethylmaleimide (Moriyama and Nelson 1988; Ding et al 2000). The next step, the de-phosphorylation of the phospho-enzyme, requires the presence of aminophospholipids, either PS or PE. In absence of these specific lipids, the enzyme cannot end the ATP-cycle (Ding et al 2000). In addition, this PS/PE-stimulated ATPase protein can transport PS uni-directionally in the yeast vesicles, as observed in RBCs and other cells (Tang et al 1996; Chin et al 2003). Although, members of the CDC50 family are essential for the localization and the activity of the human ATP8B1 protein (Paulusma et al 2008), the ATPase and transport activity of Atp8a1 established that no other mammalian protein is essential for its function in post-Golgi vesicles of yeast.
Table 1.
Characteristics of the Atp8a1 protein in the membrane of RBC and yeast vesicles
| RBC flipase | Atp8a1 (isoform a) | |
|---|---|---|
| Molecular Mass apparent, kDa | ≈120 | ≈120 (130, calculated) |
| Activity | Mg2+-ATPase | + |
| PS transport | + NBD-PS | |
| Stimulation | PS | + |
| PE | + weak | |
| Inhibition | vanadate | + |
| Ca2+ | + | |
| NEM | + |
Abbreviations: NBD-PS, nitroBenzoxaDiazol-phosphatidyl-serine; NEM, N-ethylmaleimide; PE, phosphatidyl-ethanolamine; PS, phosphatidyl-serine; RBC, red blood cells.
Treatment of RBCs with NEM and its effect on the flippase activity are believed to mimic the inactivation of the enzyme that occurs in an oxidative environment that affects the transbilayer movement in hemoglobinopathies. The inhibition of the flippase activity by sulfhydryl-modifying reagents like PDA can be reversed by treatment with membrane-permeant reductants (eg, DTT), which supports our observation that under some condition the inactivation of the enzyme is not irreversible (Balasubramanian et al 2007). In addition, NEM can bind to the un-phosphorylated protein but not to the phosphoenzyme intermediate. Thus, it has been suggested that the sulfhydryl-sensitive residue(s), one or several of the 23 cysteines of the protein, is close to the phosphorylation site and on the cytosolic side (Connor and Schroit 1990, 1991; Chang et al 2004; Balasubramanian et al 2007; Tyurina et al 2007). Similar to the protective effect of ATP on KATP channels against NEM (Trapp et al 1998), phosphorylation of the flippase could protect the critical cysteine(s), important for activity, from attack by NEM. This would account for the relative insensitivity of the enzyme when exposed to the reagent in presence of ATP. At very high concentration (≥100 mM) of NEM, the ATPase activity of Atp8a1 was initially inhibited but resumed after prolonged incubation. NEM certainly nonspecifically reacted with -SH groups of other proteins present in the vesicles during the reaction and was, thus, removed from the buffer in a time and concentration-dependent fashion, as observed. Perhaps, in presence of ATP, the initial inhibitory effect of NEM was the result of a competition to access the aspartate residue rather than the result of the inactivation of the enzyme through sulfhydryl-modification of the cysteine(s) residue.
Regardless of the mechanism(s) of the inhibition of the flippase by NEM in vitro, the effect of this chemical gives a better understanding of how PS can become exposed on the surface of damaged cells such as sickle RBC. In the cell, ATP is continuously produced and given that the only other requirement is Mg2+, it would be anticipated that the flippase is always phosphorylated and in a transport-activated state. Because the phospho-intermediate form is not, or is at least less, sensitive to attack of -SH groups, damage to the flippase should only occur during, or shortly after transport. Thus, in sickle RBCs the completion of each cycle would result in a progressive accumulation of damages during the life span of the cell. Unwarranted activation of the scramblase activity, resulting in exposure of PS, will further increase the frequency during which the flippase is susceptible to ROS.
How the enzyme specifically interacts with PS and PE, and flips them from one leaflet of the membrane to the other is not known. In normal RBC, all of the PS molecules are on the cytosolic side of the membrane but only PS from the outer layer is transported by the enzyme. Thus, the binding site(s) for PS and PE should be inaccessible to the species residing in the inner leaflet. In addition, the binding site is probably not exposed on the surface of the protein as the substrates themselves are in the membrane. The two Atp8a1 isoforms present in the membrane of RBC are the result of splicing events affecting two alternative motifs that are eight residues upstream of a domain, conserved among P-type ATPases, implicated in coupling hydrolysis of ATP to transport. PS stimulates both isoforms but PE can only induce the de-phosphorylation of one of the two (Ding et al 2000). This suggests that the alternative motifs are involved in binding or in the specificity of interaction to PS and PE.
We show that Atp8a1 isoform a, the longest of the known spliced products, is active when expressed in yeast vesicles. In this system, the study of the activity of this and the other isoforms present in the RBC membrane represents an opportunity to determine the molecular mechanisms that maintain PS asymmetry in normal RBC and to define the alteration of the flippase that leads to the loss of PS-asymmetry in RBC pathology.
Acknowledgments
We thank Dr C.W. Slayman for providing the yeast mutant used in this work. This work was supported by NIH grant HL070583. The authors report no conflicts of interest in this work.
References
- Alder-Baerens N, Lisman Q, Luong L, et al. Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol Biol Cell. 2006;17:1632–42. doi: 10.1091/mbc.E05-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–64. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- Beleznay Z, Zachowski A, Devaux PF, et al. ATP-dependent aminophospholipid translocation in erythrocyte vesicles: stoichiometry of transport. Biochemistry. 1993;32:3146–52. doi: 10.1021/bi00063a029. [DOI] [PubMed] [Google Scholar]
- Bitbol M, Devaux PF. Measurement of outward translocation of phospholipids across human erythrocyte membrane. Proc Natl Acad Sci U S A. 1988;85:6783–7. doi: 10.1073/pnas.85.18.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitbol M, Fellmann P, Zachowski A, et al. Ion regulation of phosphatidylserine and phosphatidylethanolamine outside-inside translocation in human erythrocytes. Biochim Biophys Acta. 1987;904:268–82. doi: 10.1016/0005-2736(87)90376-2. [DOI] [PubMed] [Google Scholar]
- Chang QL, Gummadi SN, Menon AK. Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry. 2004;43:10710–18. doi: 10.1021/bi049063a. [DOI] [PubMed] [Google Scholar]
- Chin G, El-Sherif Y, Jayman F, et al. Appearance of voltage-gated calcium channels following overexpression of ATPase II cDNA in neuronal HN2 cells. Mol Brain Res. 2003;117:109–15. doi: 10.1016/s0169-328x(03)00210-9. [DOI] [PubMed] [Google Scholar]
- Connor J, Gillum K, Schroit AJ. Maintenance of lipid asymmetry in red blood cells and ghosts: effect of divalent cations and serum albumin on the transbilayer distribution of phosphatidylserine. Biochim Biophys Acta. 1990;1025:82–6. doi: 10.1016/0005-2736(90)90193-r. [DOI] [PubMed] [Google Scholar]
- Connor J, Schroit AJ. Aminophospholipid translocation in erythrocytes: evidence for the involvement of a specific transporter and an endofacial protein. Biochemistry. 1990;29:37–43. doi: 10.1021/bi00453a005. [DOI] [PubMed] [Google Scholar]
- Connor J, Schroit AJ. Transbilayer movement of phosphatidylserine in erythrocytes. Inhibitors of aminophospholipid transport block the association of photolabeled lipid to its transporter. Biochim Biophys Acta. 1991;1066:37–42. doi: 10.1016/0005-2736(91)90247-6. [DOI] [PubMed] [Google Scholar]
- Ding J, Wu Z, Crider BP, et al. Identification and functional expression of four isoforms of ATPase II, the putative aminophospholipid translocase. J Biol Chem. 2000;275:23378–86. doi: 10.1074/jbc.M910319199. [DOI] [PubMed] [Google Scholar]
- Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol. 2005;6:209–20. doi: 10.1038/nrm1591. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Kihara A, Igarashi Y. Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Pharm Bull. 2006;29:1542–6. doi: 10.1248/bpb.29.1542. [DOI] [PubMed] [Google Scholar]
- Kuypers FA, De Jong K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol Biol. 2004;50:147–58. [PubMed] [Google Scholar]
- Moriyama Y, Nelson N. Purification and properties of a vanadate- and N-ethylmaleimide-sensitive ATPase from chromaffin granule membranes. J Biol Chem. 1988;263:8521–7. [PubMed] [Google Scholar]
- Morrot G, Zachowski A, Devaux PF. Partial purification and characterization of the human erythrocyte Mg2(+)-ATPase. A candidate aminophospholipid translocase. FEBS Lett. 1990;266:29–32. doi: 10.1016/0014-5793(90)81498-d. [DOI] [PubMed] [Google Scholar]
- Nakamoto RK, Rao R, Slayman CW. Expression of the yeast plasma membrane [H+]ATPase in secretory vesicles. A new strategy for directed mutagenesis. J Biol Chem. 1991;266:7940–9. [PubMed] [Google Scholar]
- Neidlinger NA, Larkin SK, Bhagat A, et al. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006;281:775–81. doi: 10.1074/jbc.M505790200. [DOI] [PubMed] [Google Scholar]
- Pasini EM, Kirkegaard M, Mortensen P, et al. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood. 2006;108:791–801. doi: 10.1182/blood-2005-11-007799. [DOI] [PubMed] [Google Scholar]
- Pasini EM, Kirkegaard M, Salerno D, et al. Deep-coverage mouse red blood cell proteome: a first comparison with the human red blood cell. Mol Cell Proteomics. 2008;7:1317–30. doi: 10.1074/mcp.M700458-MCP200. [DOI] [PubMed] [Google Scholar]
- Paulusma CC, Folmer DE, Ho-Mok KS, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 2008;47:268–78. doi: 10.1002/hep.21950. [DOI] [PubMed] [Google Scholar]
- Paulusma CC, Oude Elferink RP. The type 4 subfamily of P-type ATPases, putative aminophospholipid translocases with a role in human disease. Biochim Biophys Acta. 2005;1741:11–24. doi: 10.1016/j.bbadis.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Holthuis JC, Herrmann A, et al. Tracking down lipid flippases and their biological functions. J Cell Sci. 2004;117:805–13. doi: 10.1242/jcs.01055. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Menon AK. Lipid flippases and their biological functions. Cell MolLife Sci. 2006;63:2908–21. doi: 10.1007/s00018-006-6167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter R, Brugger B, Sandhoff R, et al. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol. 1999;146:741–54. doi: 10.1083/jcb.146.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneuret M, Devaux PF. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc Natl Acad Sci U S A. 1984;81:3751–5. doi: 10.1073/pnas.81.12.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupene E, Kuypers FA. Identification of an erythroid ATP-dependent aminophospholipid transporter. Br J Haematol. 2006a;133:436–8. doi: 10.1111/j.1365-2141.2006.06051.x. [DOI] [PubMed] [Google Scholar]
- Soupene E, Kuypers FA. Multiple erythroid isoforms of human long-chain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. BMC Mol Biol. 2006b;7:21. doi: 10.1186/1471-2199-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Halleck MS, Schlegel RA, et al. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996;272:1495–7. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Mechanism of ATP-sensitive K channel inhibition by sulfhydryl modification. J Gen Physiol. 1998;112:325–32. doi: 10.1085/jgp.112.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyurina YY, Basova LV, Konduru NV, et al. Nitrosative stress inhibits the aminophospholipid translocase resulting in phosphatidylserine externalization and macrophage engulfment: implications for the resolution of inflammation. J Biol Chem. 2007;282:8498–509. doi: 10.1074/jbc.M606950200. [DOI] [PubMed] [Google Scholar]
- Walworth NC, Novick PJ. Purification and characterization of constitutive secretory vesicles from yeast. J Cell Biol. 1987;105:163–74. doi: 10.1083/jcb.105.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Beserra C, Garbers DL. A novel aminophospholipid transporter exclusively expressed in spermatozoa is required for membrane lipid asymmetry and normal fertilization. Dev Biol. 2004;267:203–15. doi: 10.1016/j.ydbio.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Zinser E, Sperka-Gottlieb CD, Fasch EV, et al. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–34. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]