

Abstract
Influenza viruses exhibit a fascinating level of antigenic heterogeneity that facilitates re-infection in the human population. The human antibody repertoire also manifests endless capability for variation in the genes that specify the portion of antibody molecules interacting with epitopes. A recent explosion of techniques for isolating human monoclonal antibodies to viruses has led to isolation of new antibodies that allow glimpses into the molecular basis for recognition and escape that underlies the constant antigenic drift in influenza surface proteins. These studies also reveal evidence for lifelong persistence of immunity to some influenza viruses.
Keywords: Influenza, Human, Antibodies, Immunity
1. Introduction
Vaccines against acute viruses principally work by inducing neutralizing antibodies. Understanding the molecular basis for virus neutralization by antibodies is important, but there has been remarkably little progress in the past in understanding the action of individual human antibodies because of the difficulty in generating human monoclonal antibodies with functional activity against viruses. Recent technical breakthroughs, however, have facilitated the isolation of new human monoclonal antibodies against viruses such as influenza. Careful studies of the molecular basis for specificity or cross-reactivity, and examination of the molecular mechanism of virus inhibition, shed new light on the important interface of viral antigens and the human immune system. As we learn more about the intricate surface interactions between these protein molecules, we gain insight into how we might rationally design (or re-design) viral antigens as vaccines that induce enhanced antibody responses with broader reactivity. The goal of understanding how to induce broadly cross-reactive neutralizing antibodies is particularly relevant for influenza studies because of yearly antigenic drift of field strains, but also is of interest to those designing vaccines to prevent chronic viral infections like HIV and hepatitis C.
2. Obstacles to generation of human monoclonal antibodies to viruses
Monoclonal antibodies (mAbs) have revolutionized the conduct of science since their first description in 1975 [1]. The use of these specific reagents also has made possible improved clinical diagnostics in the medical arena, and a large number of antibodies have found their way to clinical use as therapeutic agents, typically for therapy of cancer or autoimmunity. There is one antibody, palivizumab, which is licensed for prevention of a viral disease, RSV infection. Nevertheless, the potential of mAbs for therapy remains largely unfulfilled. The principal reason for the lack of a large number of mAb therapeutics is simply the difficulty in generating human mAbs of high affinity.
Virus-specific antibodies can prevent many important virus infections or diseases. Specific human antibodies have been shown to prevent disease caused by a wide variety of viruses belonging to diverse RNA or DNA virus families that include the orthomyxoviruses, paramyxoviruses, alphaviruses, flaviviruses, arenaviruses, lentiviruses, picornaviruses, hepadnaviruses, poxviruses, and herpesviruses. Examples from clinical medicine include hepatitis A virus (HAV), measles virus, poliovirus, and varicella zoster virus. The number of polyclonal immunoglobulin (Ig) products licensed for use in the U.S. is striking. Polyclonal Ig preparations with high titers to specific agents are used for a number of viruses including varicella zoster virus Ig, hepatitis B virus Ig, RSV Ig, rabies virus Ig, vaccinia virus Ig, and cytomegalovirus Ig. Difficulties with our ability to sustain these products include the need to find immune donors, the risk of adventitious agents in human-derived blood products, and significant lot-to-lot variability in donor pools. Human mAbs are desirable as replacements for these therapies, or as means for therapy of other conditions whether for infectious diseases or for noninfectious health problems such as autoimmunity or cancer.
A variety of techniques other than generation of human mAbs have been used to generate candidate mAb therapeutics. The first mAbs were murine mAbs developed from the fusion of murine splenocytes and non-secreting myelomas. Mice offer many advantages for the generation of mAbs, most notably the opportunity to hyper-immunize subjects and the ability to collect lymphoid tissues including spleen. Murine mAbs have been used in the past in patients, with moderate success. For example, in the early 1980s trials began for treating patients experiencing transplant rejection with the anti-thymocyte (anti-CD3) murine mAb OKT3. Although abrogation of rejection could be achieved in some cases, the patients were treated in the intensive care unit because a significant proportion of patients suffered systemic side effects including life-threatening anaphylaxis. Such severe reactions were due to human anti-mouse antibody (HAMA) reaction [2]. Besides anaphylaxis, HAMA also can cause a more subtle effect, the accelerated clearance of the therapeutic antibody with each infusion. Approximately 25% of the U.S. population has anti-mouse antibodies, and following infusion of mouse antibodies virtually all subjects exhibit a HAMA response.
Therefore, efforts were made to reduce the immunogenicity of murine mAbs by replacing murine sequences outside the antigen-combining site with human antibody sequences. Antibody gene cloning techniques made this possible by recombinant means. Initially, human antibody Fc regions were cloned onto mouse Fab regions, yielding so-called “chimeric antibodies”. These antibodies could still induce HAMA however, suggesting that a significant proportion of HAMA response is directed to the constant region 1 (CH1 and CL) domains of the antibodies. Subsequent heroic efforts have centered on “antibody humanization”, a process also known as “CDR-grafting.” In this method the variable loops of the mouse mAb antibody combining site (the complementarity determining region [CDR] loops) are cloned onto a human Fab framework that is predicted by three-dimensional modeling to have a similar overall conformation to the mouse Fab. Six CDRs (CDR 1–3 of the heavy chain, and CDRs 1–3 of the light chain) must be cloned correctly, and often the affinity and functional activity of the humanized antibody are significantly reduced or lost compared with the parental murine mAb. Further modeling can suggest particular framework residues to revert back to mouse sequences in order to restore some of the original conformation [3], [4]. Sometimes the variable domain amino acids are also found to be immunogenic, and further humanization by “variable domain resurfacing” requires that surface residues not normally found in a human variable fragment (Fv) be mutated to the expected human amino acid, thereby eliminating potentially immunogenic sites [5]. These engineering processes are laborious and time consuming, without guarantee of success.
Combinatorial antibody phage display libraries have yielded human antibodies. The principal drawback of this approach is that many antibody fragments (Fabs or single chain Fv) bind antigen but do not mediate the function desired in the antibody, such as virus neutralization. Therefore rapid screening procedures of phage-Fabs or expressed antibody fragments often miss the antibodies of interest. Converting candidate Fabs to full-length Ig molecules can be a laborious process and is challenging for high-throughput screening purposes, although some companies have begun to use high-throughput techniques based on the small molecule library screening model.
Some investigators have attempted to combine the advantages of the mouse model with the use of human B cells or antibody genes. Researchers have transplanted human peripheral blood lymphocytes (PBL) from vaccinated healthy blood donors into mice with severe combined immunodeficiency (”human PBL-SCID mice”) and demonstrated an antigen-dependent humoral immune response [6], [7]. Engraftment rates can be low however, and SCID mice do not provide an environment in which somatic mutation of Ig variable genes takes place efficiently. This approach also was combined with phage display antibody repertoire cloning to yield human Fabs [8]. A similar approach is the Trimera system (human/mouse radiation chimera). In this model, functional human lymphocytes are engrafted in normal strains of mice that are rendered immuno-incompetent by lethal total body irradiation followed by radioprotection with severe combined immunodeficient (SCID) mouse bone marrow [9]. High affinity secondary antibody responses generally are difficult to achieve with these methods.
Transgenic mice that contain both inactivated mouse Ig genes and large regions of the human heavy and light chain loci cloned on yeast artificial chromosomes (YACs) were developed in the 1990s [10], [11]. Mice possessing a human heavy chain YAC in germline configuration with core variable and constant sequences (D, J H, Cμ, C), and 66 VH genes, and a human kappa chain YAC containing in germline configuration the J and C regions and 32 VL genes, supported production of high-affinity human antibodies to some antigens [10]. These mice have been used in commercial settings to develop new antibodies.
Another approach to the development of therapeutic mAbs for use in humans is the generation of nonhuman primate mAbs. Chimpanzees and humans are highly related genetically and therefore the development of human anti-chimpanzee immunoglobulin antibodies in human patients is expected to be rare. Sequence analysis indicates that human antibody genes are highly homologous to the corresponding chimpanzee genes. These differences are comparable to differences between human allotypes. In our studies and those of others, human antibodies have the same half-life as that of chimpanzee antibodies following infusion into chimpanzees [12], [13]. The utility of this approach is severely limited however by the scarcity of these primates for such studies.
3. Previous efforts to generate human mAbs have been inefficient
Human mAbs derived from the peripheral blood lymphocytes (PBLs) of research volunteers or patients are desirable. However many technical difficulties have resulted in a limited yield from the efforts to develop human mAbs in the past. Several methods for the generation of human mAbs have been described. Most methods primarily rely on transformation of B cells with Epstein-Barr virus (EBV) +/− the fusion of these transformed B cells with non-secreting myeloma cells of murine, human, or murine–human (heteromyeloma) origin. EBV transformation historically yielded antibodies at a low efficiency that are of low affinity and predominantly of the IgM class. Such cell lines are often unstable in culture. These limitations have been especially important for those who seek to use limited amounts of donor lymphocytes in which the number of antigen-specific B cells is usually very low even following active immunization or infection.
A recent innovation, the combined use of EBV transformation with simultaneous CpG treatment to stimulate Toll-like receptor 9 has resulted in marked enhancement of the efficiency of the transformation process. The technique has led to the isolation of interesting antibodies to SARS coronavirus and H5N1 avian influenza, among others [14], [15]. The technique also has been combined with electrofusion to isolate rare antibodies to 1918 influenza [16].
Conventional fusion techniques, principally polyethylene glycol [PEG] mediated fusion, were hampered by a very low frequency of fusion and immortalization. When fusing murine splenocytes from hyper-immunized animals, an extremely low inefficiency can be tolerated if one simply aims to find one or several mAbs that bind to an antigen. However, when using human samples such as PBLs or ultimately even more precious samples such as tumor infiltrating lymphocytes, such inefficiencies make success unlikely. We have developed high efficiency electrofusion methods that make generation of human mAbs from limited numbers of donor cells possible [16], [17].
4. Principles of electroporation
Application of high intensity electric field pulses to cells causes transient membrane permeabilization. The extent of permeabilization depends on several physical parameters associated with the technique such as pulse intensity, number, duration, shape, and interval. Electric field intensity is the most critical parameter in the induction of permeabilization. The intensity must exceed a critical threshold in order to induce membrane permeabilization. The electric field intensity delivered depends on the cell size. Extent of permeabilization (which correlates experimentally with the flow rate across the membrane) is controlled by both pulse number and duration [18]. Increasing electric field intensity above the critical threshold needed for permeabilization results in an increase in the area of the membrane that is involved. Permeabilization is transient and disappears with time after delivery of the electric field pulses. The rate of resealing of the pore in the membrane may be influenced by both pulse duration and number, but is independent of the electric field intensity that creates the permeabilization [18].
5. Principles of electrofusion
The membrane effects of applied electrical fields in electrofusion are similar to those in electroporation, except that membranes in close contact can fuse together during the process of pore formation. Therefore, electric field intensities used in electrofusion are similar to those used in electroporation. Electrofusion is performed in a sequence of steps. First, cells are brought into contact with other cells. Cell–cell contact can be achieved by several methods. Chemical methods such as avidin–biotin bridging have been used to specifically bring together two cell types [19], [20], [21], [22]. Chemical methods require more manipulation than other methods but can be useful in certain circumstances. Physical methods such as centrifugation can bring cells into contact prior to (or after) the fusion pulse [23]. Electroacoustic fusion of cells in sugar solutions and of cells brought into contact in an ultrasonic standing wave field has been described [24]. We have used a method for bringing cells into contact prior to electrofusion that is termed dielectrophoresis.
6. Dielectrophoresis
In any fusion method, sufficient force must be applied to each cell to overcome the negative surface charge. If electricity is used, merely applying a uniform electric field will not move a cell because the net charge of the cell is zero. However, a non-uniform field moves the positive ions, inside each cell, to one side and the negative ions to the opposite side producing a dipole. Once the dipole is induced, a net force is exerted on the cell because the intensity of the field is greater on one side than the other. The movement of cells in one direction causes the cells to concentrate in an area. Since the cells are now dipoles the negative side of one cell will attract the positive side of another cell over coming the negative surface charge. Fig. 1 illustrates the principles. In Fig. 1(A) A low-voltage alternating current (AC) is delivered to mixed cell suspensions of transformed B cells and myeloma cells, (B) cell mixtures in the field result in myeloma/B cell contact, and (C) the resulting fields cause the formation of “pearl chains” of mixed cells. After establishing close contact using AC current, a large electroporating direct current (DC) pulse is delivered (Fig. 1(A)), followed by maintenance of contact with delivery of AC pulses again for a time. After membrane resealing, some myeloma cells will have fused with an antigen-specific B cell.
Fig. 1.
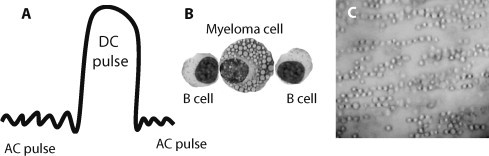
Principles of electrofusion to generate human hybridomas. (A) A low-voltage alternating current (AC) is delivered to suspensions of EBV/CpG transformed B cells and myeloma cells, a large electroporating direct current (DC) pulse is delivered to cause defects in the cell membranes, then cell–cell contact is maintained with delivery of AC pulses again. (B) Treatment of cell mixtures in the AC pulse field results in myeloma/B cell contact. (C) Photomicrograph of the formation of “pearl chains” of mixed cells in the AC pulse (photo courtesy of David Blum).
7. Plasmablast sorting
A recently developed technique for sorting circulating human plasmablasts has been developed with excellent efficiency of isolation of human influenza-specific antibodies [25]. Investigators noted that plasmablasts, normally not found in peripheral blood in substantial numbers, occurred in high frequency in the blood of subjects who had been vaccinated with influenza vaccine about a week prior. The timing of the circulation of these cells suggested that they likely possessed genes specific for the vaccine antigens, and molecular cloning of the expressed antibody genes subsequently proved that influenza-specific antibody genes were common in these cells. This technique promises to yield a large number of antibodies to influenzas for which conventional or experimental vaccines can be used as immunogens in humans. It is not clear at this time whether the circulating pool of plasmablasts in patients with chronic infections (such as HIV or hepatitis C) is enriched in antigen-specific clones. It is also not established that these cells can be transformed efficiently; therefore direct cloning of antibody genes followed by expression of antibodies is required for screening.
8. New human mAbs directed to the influenza HA protein
The recent explosion of human antibody techniques has yielded interesting mAbs to influenza virus in recent years. Phage display techniques yielded very interesting cross-reactive antibodies that recognize a large number of diverse influenza strains [26], [27]. These antibodies are of great interest, although it seems doubtful that knowledge about the epitope will immediately be useful for inducing cross-reactive antibodies. The epitope is present in most circulating influenza strains, but most individuals do not possess significant levels of such antibodies despite previous infections.
Our own studies have focused on the occurrence of highly potent human neutralizing antibodies to the 1918 pandemic influenza strain isolated from elderly survivors of that pandemic [16]. Several interesting features of these studies are highlighted below.
9. Human antibodies to the 1918 pandemic virus
It is remarkable that extremely elderly subjects maintain circulating B cells encoding antibodies to the 1918 influenza virus over 90 years after that pandemic. This virus, and antigenically similar viruses, circulated in humans for the first several decades of the 20th century. Therefore it is expected that 1918 pandemic survivors would have been exposed and probably infected by similar viruses on multiple occasions during childhood and the early adult years. However, it has been over a half century since antigenically related viruses have circulated in humans. Surprisingly, nearly all of the extreme elderly subjects we examined (but not younger subjects) possessed 1918-specific B cells in the peripheral blood. The frequency was low, with an average of about 1 in 5 million circulating cells that were 1918 HA-specific. To some extent, these studies raise more questions than they answer. What signals maintain the B cells in circulation? Do early 20th century influenza antigens persist in these subjects? Are these subjects genetically unusual in some way? Isolation of antibodies and their sequences only provide a glimpse into the nature of immune memory, but not into the mechanisms that maintain it.
10. Molecular features of 1918 HA-specific human monoclonal antibodies
The antibodies we have isolated to the 1918 HA from pandemic survivors exhibit some distinctive molecular features. Most interestingly, the antibody gene sequences possess a high number of somatic mutations, nearly three times the average number of mutations found in the sequences of human memory B cells. Intuitively one might speculate that a high number of somatic mutations would facilitate a high affinity of binding, and we found this to be true. Biosensor studies reveal that these antibodies interact with extraordinary affinity with recombinant HA molecules. Finally, we tested whether the high affinity of interaction correlated with a high functional activity. Indeed, some of these antibodies exhibited ultra-potent neutralizing activity against 1918 virus and other early 20th century H1N1 viruses, when measured by HAI or neutralizing antibody assays in vitro or protection against lethal challenge in mice.
11. Epitope specificity
Typically, influenza neutralizing antibodies recognize variable loops on the HA protein head that surround the sialic acid receptor binding site. Five major antigenic sites have been previously defined using murine monoclonal antibodies and designated sites Sa, Sb, Ca1, Ca2, and Cb (Fig. 2 ). Using generation of antibody escape mutants coupled with HA gene sequence analysis, and other techniques, we have identified that the 1918 virus-specific human antibodies bind principally to the Sa and Sb antigenic sites. These data suggest that these potent inhibiting antibodies bind to conventional sites located in highly accessible but also variable loops. Interestingly, some of the binding appears to involve residues that flank the classical sites, suggesting human antibodies may interface with the HA with a somewhat broader interface and more articulated surface than murine antibodies. Ongoing structural studies will reveal the exact nature of interactions and the extent of shape complementarity that the human antibodies are able to achieve. One of the surprising features of the epitope studies is the fact that one of the antibodies, designated 1F1 exhibits a low level of cross-reactivity for a number of H1N1 viruses over the 20th century. This antibody, unexpectedly, binds and inhibits virus as far apart as 1918 and 1977 H1N1 viruses, although the activity is highest against the 1918 strain. A better understanding of the structural basis for this cross-reactivity could suggest conserved elements of the H1 HA molecule on which we could focus vaccine development for a vaccine that induces broadly cross-reactive antibodies. Isolation of additional cross-reactive antibodies to the head domain of influenza is a high priority if we are to understand the structural basis for conservation of these otherwise cryptic conserved epitopes.
Fig. 2.
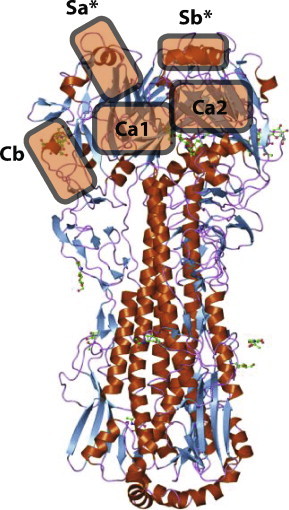
Major antigenic sites in the 1918 influenza hemagglutinin molecule. The ribbon diagram is based on PDB entry 1rd8 [28]. The designation of antigenic sites was described previously [29]. Asterisks indicate sites for which human neutralizing antibodies have been isolated.
12. Summary
A detailed understanding of the molecular basis for antibody-mediated inhibition of influenza by human antibodies has not been possible in the past, because of the paucity of human monoclonal antibodies. Recent technical developments in the field of human monoclonal antibody isolation, by diverse techniques including phage display, single B cell sorting and cloning, plasmablast sorting and cloning, EBV/CpG transformation of B cells, and human hybridoma techniques have begun to yield large numbers of interesting influenza-specific antibodies. Studies of the genetic and structural basis for neutralization and for cross-reactivity should lead the way to rational input on the design and selection of HA molecules for inclusion in vaccines, especially for the design of vaccines that induce broadly neutralizing antibodies.
Acknowledgements
I thank Xiaocong Yu, Patricia McGraw, Jens Krause, David Blum and Christopher Basler for helpful discussions on this topic. This work was supported by NIH U54 AI057157 and P01 AI-058113. JEC is supported by a Burroughs Wellcome Fund Clinical Scientist in Translational Research Award.
References
- 1.Kohler G., Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 2.Shawler D.L., Bartholomew R.M., Smith L.M., Dillman R.O. Human immune response to multiple injections of murine monoclonal IgG. J Immunol. 1985;135:1530–1535. [PubMed] [Google Scholar]
- 3.Taylor G., Furze J., Tempest P.R., Bremner P., Carr F.J., Harris W.J. Humanised monoclonal antibody to respiratory syncytial virus. Lancet. 1991;337:1411–1412. doi: 10.1016/0140-6736(91)93091-m. [DOI] [PubMed] [Google Scholar]
- 4.Tempest P.R., Bremner P., Lambert M., Taylor G., Furze J.M., Carr F.J. Reshaping a human monoclonal antibody to inhibit human respiratory syncytial virus infection in vivo. Biotechnology (NY) 1991;9:266–271. doi: 10.1038/nbt0391-266. [DOI] [PubMed] [Google Scholar]
- 5.Delagrave S., Catalan J., Sweet C., Drabik G., Henry A., Rees A. Effects of humanization by variable domain resurfacing on the antiviral activity of a single-chain antibody against respiratory syncytial virus. Protein Eng. 1999;12:357–362. doi: 10.1093/protein/12.4.357. [DOI] [PubMed] [Google Scholar]
- 6.Neil G.A., Sammons D.W. Immunization of SCID-Hu mice and generation of anti-hepatitis B surface antigen-specific hybridomas by electrofusion. Hum Antibodies Hybridomas. 1992;3:201–205. [PubMed] [Google Scholar]
- 7.Walker W., Gallagher G. The in vivo production of specific human antibodies by vaccination of human-PBL-SCID mice. Immunology. 1994;83:163–170. [PMC free article] [PubMed] [Google Scholar]
- 8.Duchosal M.A., Eming S.A., Fischer P., Leturcq D., Barbas C.F. 3rd, McConahey P.J. Immunization of hu-PBL-SCID mice and the rescue of human monoclonal Fab fragments through combinatorial libraries. Nature. 1992;355:258–262. doi: 10.1038/355258a0. [DOI] [PubMed] [Google Scholar]
- 9.Eren R., Lubin I., Terkieltaub D., Ben-Moshe O., Zauberman A., Uhlmann R. Human monoclonal antibodies specific to hepatitis B virus generated in a human/mouse radiation chimera: the Trimera system. Immunology. 1998;93:154–161. doi: 10.1046/j.1365-2567.1998.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mendez M.J., Green L.L., Corvalan J.R., Jia X.C., Maynard-Currie C.E., Yang X.D. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet. 1997;15:146–156. doi: 10.1038/ng0297-146. [DOI] [PubMed] [Google Scholar]
- 11.Green L.L., Hardy M.C., Maynard-Currie C.E., Tsuda H., Louie D.M., Mendez M.J. Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat Genet. 1994;7:13–21. doi: 10.1038/ng0594-13. [DOI] [PubMed] [Google Scholar]
- 12.Crowe J.E., Jr., Bui P.T., Siber G.R., Elkins W.R., Chanock R.M., Murphy B.R. Cold-passaged, temperature-sensitive mutants of human respiratory syncytial virus (RSV) are highly attenuated, immunogenic, and protective in seronegative chimpanzees, even when RSV antibodies are infused shortly before immunization. Vaccine. 1995;13:847–855. doi: 10.1016/0264-410x(94)00074-w. [DOI] [PubMed] [Google Scholar]
- 13.Prince A.M., Horowitz B., Baker L., Shulman R.W., Ralph H., Valinsky J. Failure of a human immunodeficiency virus (HIV) immune globulin to protect chimpanzees against experimental challenge with HIV. Proc Natl Acad Sci USA. 1988;85:6944–6948. doi: 10.1073/pnas.85.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simmons C.P., Bernasconi N.L., Suguitan A.L., Mills K., Ward J.M., Chau N.V. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 2007;4:e178. doi: 10.1371/journal.pmed.0040178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Traggiai E., Becker S., Subbarao K., Kolesnikova L., Uematsu Y., Gismondo M.R. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X., Tsibane T., McGraw P.A., House F.S., Keefer C.J., Hicar M.D. Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature. 2008;455:532–536. doi: 10.1038/nature07231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu X., McGraw P.A., House F.S., Crowe J.E., Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J Immunol Methods. 2008;336:142–151. doi: 10.1016/j.jim.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rols M.P., Teissie J. Electropermeabilization of mammalian cells. Quantitative analysis of the phenomenon. Biophys J. 1990;58:1089–1098. doi: 10.1016/S0006-3495(90)82451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo M.M., Tsong T.Y., Conrad M.K., Strittmatter S.M., Hester L.D., Snyder S.H. Monoclonal antibody production by receptor-mediated electrically induced cell fusion. Nature. 1984;310:792–794. doi: 10.1038/310792a0. [DOI] [PubMed] [Google Scholar]
- 20.Wojchowski D.M., Sytkowski A.J. Hybridoma production by simplified avidin-mediated electrofusion. J Immunol Methods. 1986;90:173–177. doi: 10.1016/0022-1759(86)90073-6. [DOI] [PubMed] [Google Scholar]
- 21.Hewish D.R., Werkmeister J.A. The use of an electroporation apparatus for the production of murine hybridomas. J Immunol Methods. 1989;120:285–289. doi: 10.1016/0022-1759(89)90254-8. [DOI] [PubMed] [Google Scholar]
- 22.Bakker Schut T.C., Kraan Y.M., Barlag W., de Leij L., de Grooth B.G., Greve J. Selective electrofusion of conjugated cells in flow. Biophys J. 1993;65:568–572. doi: 10.1016/S0006-3495(93)81128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teissie J., Rols M.P. Fusion of mammalian cells in culture is obtained by creating the contact between cells after their electropermeabilization. Biochem Biophys Res Commun. 1986;140:258–266. doi: 10.1016/0006-291x(86)91084-3. [DOI] [PubMed] [Google Scholar]
- 24.Bardsley D.W., Liddell J.E., Coakley W.T., Clarke D.J. Electroacoustic production of murine hybridomas. J Immunol Methods. 1990;129:41–47. doi: 10.1016/0022-1759(90)90418-u. [DOI] [PubMed] [Google Scholar]
- 25.Wrammert J., Smith K., Miller J., Langley W.A., Kokko K., Larsen C. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ekiert D.C., Bhabha G., Elsliger M.A., Friesen R.H., Jongeneelen M., Throsby M. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sui J., Hwang W.C., Perez S., Wei G., Aird D., Chen L.M. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stevens J., Corper A.L., Basler C.F., Taubenberger J.K., Palese P., Wilson I.A. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science. 2004;303:1866–1870. doi: 10.1126/science.1093373. [DOI] [PubMed] [Google Scholar]
- 29.Caton A.J., Brownlee G.G., Yewdell J.W., Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype) Cell. 1982;31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]