

Abstract
A growing body of evidence suggests that reactive oxygen species are critical components of cell signaling pathways, in particular regulating protein phosphorylation events. Here, we show that oxidative stress in response to hydrogen peroxide treatment of human epithelial cells induces robust tyrosine phosphorylation on multiple proteins. Using an anti-phosphotyrosine purification and liquid chromatography-tandem mass spectrometry approach, we have identified many of these H2O2-induced tyrosine-phosphorylated proteins. Importantly, we show that epidermal growth factor receptor (EGFR) and Src are the primary upstream kinases mediating these events through their redox activation. The finding that many of the identified proteins have functions in cell adhesion, cell-cell junctions, and the actin cytoskeleton prompted us to examine stress-induced changes in adhesion. Immunofluorescence analysis showed that H2O2 alters cell adhesion structures and the actin cytoskeleton causing loss of adhesion and apoptosis. Remarkably, these cellular changes could be attenuated by inhibition of EGFR and Src, identifying these kinases as targets to block oxidative damage. In summary, our data demonstrate that EGFR and Src together play a central role in oxidative stress-induced phosphorylation, which in turn results in loss of adhesion, morphological changes, and cell damage in epithelial cells. These data also provide a general model for redox signaling in other cell systems.
Keywords: Cell/Adhesion, Cytoskeleton/Actin, Methods/Mass Spectrometry, Proteomics, Receptors/Tyrosine Kinase, Signal Transduction/Protein Kinases/Tyrosine, Phosphotyrosine Signaling, Reactive Oxygen Species (ROS), Src
Introduction
Protein phosphorylation is a key post-translational modification event that plays a critical role in regulating diverse cellular processes such as signal transduction, metabolism, differentiation, cell division, movement, and cell survival (1). In mammalian cells, one-third of expressed proteins are thought to be phosphorylated on serine and threonine and less commonly on tyrosine residues (2). Tyrosine phosphorylation in particular plays a critical role in growth factor signal transduction, and unregulated tyrosine phosphorylation by oncogenic tyrosine kinases has been implicated in cancer through the generation of inappropriate proliferative and survival signals (3). Reactive oxygen species (ROS)2 are known to be produced by and regulate a multitude of cellular and physiological processes, including cell signaling. They include several species such as hydrogen peroxide (H2O2), the hydroxyl radical (•OH), superoxide (O2˙̄), and singlet oxygen and are interconvertible. For example, O2˙̄ is generated by the action of enzymes such as NADPH oxidase, xanthine oxidase, lipoxygenase, cyclooxygenase, cytochrome P450, or through UV irradiation and can be converted into H2O2 and O2 by the action of superoxide dismutases. ROS were originally recognized as active products of the mammalian host defense mechanism involved in the respiratory burst in phagocytic neutrophils. ROS have also been reported to promote tumorigenesis. For example, ROS-induced tumorigenesis has been linked to oxidative DNA damage through generation of the DNA oxidative product 8-oxo-deoxyguanosine, a highly mutagenic agent. In addition, ROS may stabilize and activate HIF-1α, a transcription factor controlling angiogenesis under normoxic or hypoxic conditions (4). ROS are also thought to contribute to cancer through interference with multiple signaling systems, including pathways involving nuclear transcription factor κB, activated protein-1, phospholipase A2, mitogen-activated protein kinases (MAPKs), Akt, and Jun kinase.
Among the ROS, H2O2 is the major cellular form because of its stability. It has been reported to play a key role in reversible protein phosphorylation through inactivation of protein-tyrosine phosphatases and the lipid phosphatase and tumor suppressor PTEN; this is by direct oxidation of their catalytic cysteine residues (5–11). As such, H2O2 has been recognized as an important intracellular messenger in growth factor and cytokine signal transduction (12–16). Indeed, the H2O2 generated transiently in response to peptide growth factor stimulation appears to be permissive for transmission of tyrosine phosphorylation-based signals and is thought to be generated through a mechanism requiring sequential activation of phosphatidylinositol 3-kinase, βPix, Rac1, and NADPH oxidase, with the resulting O2˙̄ converted to H2O2 by superoxide dismutase (17).
In this study, we identify and characterize proteins that are phosphorylated in response to treatment with H2O2 and relate these protein changes to the biological effects of oxidative stress. An SV40 large T antigen-immortalized human mammary luminal epithelial cell line, HB4a, was chosen as a cellular model for these studies (18). HB4a cells display several markers of the luminal epithelial cell type, have a characteristic luminal epithelial cellular morphology in culture, and are the cell type from which most breast cancers arise. In these cells, H2O2 treatment induced the activation of several signaling pathways and generated a robust but transient tyrosine phosphorylation of multiple proteins. An immunoprecipitation (IP) strategy was used to purify tyrosine-phosphorylated proteins that were subsequently identified by LC-MS/MS. Our work shows that many cytoskeletal and adhesion-related proteins are targets of H2O2-induced tyrosine phosphorylation in these cells and that the Src and EGFR kinases are critical upstream regulators of these events. Importantly, we link the modification of these proteins to reduced cell adhesion, altered cytoskeletal organization, and increased apoptosis in response to oxidative stress.
EXPERIMENTAL PROCEDURES
Cell Culture, Treatment, and Lysis
The luminal epithelial cell line HB4a was derived from reduction mammoplasty tissue of a normal subject by flow sorting and immortalization with a temperature-sensitive mutant of SV40 large T cells (18) and was obtained from M. O'Hare (Ludwig Institute for Cancer Research, London). Cells were maintained as described previously (19). Wild-type murine embryonic fibroblasts were a gift from A. Di Cristofano (Memorial Sloan-Kettering Cancer Center, New York) and were maintained in Iscove's modified Dulbecco's media supplemented with 15% (v/v) fetal calf serum, l-glutamine (2 mm), streptomycin (100 μg/ml), and penicillin (100 IU/ml) at 37 °C in a 5% CO2-humidified incubator. Cells were passaged at 70–80% confluence by trypsinization according to standard procedures. Cells at ∼80% confluence were treated with H2O2 (30% v/v; Sigma) at 0.5 mm for the indicated times. For Src kinase and EGFR inhibition, cells were pretreated with PP1 (2.5 μm; Alexis Biochemicals, San Diego) and AG1478 (5 μm; Calbiochem), respectively, for 1 h prior to treatment with 0.5 mm H2O2 or vehicle (double deionized water). For lysis, cells were washed in chilled PBS and scraped in lysis buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 2 mm sodium orthovanadate, 100 μg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, 17 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 5 μm fenvalerate, 5 μm bis-peroxovanadium 1,10-phenanthroline, and 1 μm okadaic acid). Lysates were clarified by centrifugation (14,000 × g, 10 min, 4 °C), and protein concentration was determined using Coomassie Protein Assay Reagent (Pierce). Protein samples were then separated by one-dimensional SDS-PAGE according to standard procedures.
Immunoblotting and Immunoprecipitation
Immunoblotting was carried out essentially as described (20). Primary antibodies used with dilutions and sources were as follows: α-Ser(P)-473-Akt (1:2000), α-Akt (1:2000), anti-p38 (1:1000), α-Thr(P)-180/Tyr(P)-182-p38 (1:1000), α-JNK (1:1000), α- Thr(P)-183/Tyr(P)-185-JNK (1:1000), α-β-catenin (1:1000), α-E-cadherin (1:1000), α-Tyr(P)-416-Src (1:2000), and α-Tyr(P)-527-Src (1:2000) (all from Cell Signaling Technology); α-activated-ERK1/2 (1:5000) and α-ERK1/2(1:5000) (both from Promega); α-p130Cas (1:1000), α-cortactin (1:1000), and α-FAK (1:2000) (all from BD Transduction Laboratories); α-EGFR (1:1000) and α-pY (pY-99; 1:5000) (both from Santa Cruz Biotechnology); α-β-actin (1:1000; Sigma); α-Prdx3 (1:2000; Lab Frontier); and α-pY1197-EGFR (1:500; Millipore), α-pY421-cortactin (1:1000; Novus Biologicals) and α-pY118-paxillin (1:1000; BD Transduction Laboratories). For small scale IPs, proteins were immunoprecipitated from 500 μg of cell lysate using 5 μg of antibody and 40 μl of a 50% slurry of protein A-Sepharose for 16 h at 4 °C. Immune complexes were then washed three times in lysis buffer and boiled in Laemmli buffer prior to immunoblotting.
Large Scale Tyrosine-phosphorylated Protein Enrichment
Exponentially growing HB4a cells (40 × 15-cm dishes; ∼2 × 108 cells) were either left untreated or treated with 0.5 mm H2O2 for 20 min followed by washing in cold PBS and lysis in chilled lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.1% sodium deoxycholate, 1 mm EDTA plus protease inhibitors, and 1 mm sodium orthovanadate). Lysates were precleared by incubation with 500 μl of protein A-agarose for 4 h at 4 °C on a rotator and then incubated overnight with 300 μl of agarose-conjugated 4G10 α-Tyr(P) monoclonal antibody beads (Santa Cruz Biotechnology). IPs were washed extensively with lysis buffer, and proteins were eluted with a 1-bed volume of 100 mm phenyl phosphate. The elution step was repeated, and the two eluates were pooled. Samples were precipitated by adding 1 volume of 100% trichloroacetic acid (at −20 °C) to 4 volumes of sample with incubation for 10 min at 4 °C. Precipitated protein was recovered by centrifugation at 14,000 × g for 5 min, and the pellet washed twice with ice-cold acetone. Air-dried pellets were resuspended in 100 μl of Laemmli buffer and boiled; proteins were resolved by 12% one-dimensional SDS-PAGE, and gels were stained with colloidal Coomassie Blue G-250, as described (20).
Protein Identification by LC-MS/MS
Excised gel pieces were subjected to trypsin digestion as described previously (20). Peptide extracts were vacuum-dried and resuspended in 6 μl of double deionized water containing 0.1% formic acid. LC-MS/MS was performed by injecting 5 μl of digested peptides onto a reversed phase capillary column (PepMap 75 μm × 150 mm, LC Packings) using a nanoflow high pressure liquid chromatography system (Ultimate, Dionex) connected on-line to an electrospray ionization Q-TOF I mass spectrometer (Waters). The flow rate was 300 nl/min, and separation was performed by gradient elution from 5 to 50% solution B (80% (v/v) acetonitrile, 0.1% formic acid) for 60 min followed by an isocratic step at 100% solution B for 10 min. Balance solution A was 0.1% formic acid. Data-dependent acquisition was used with mass spectrometry scans set every second (m/z 350–1500), and MS/MS performed on automatically selected peptide ions, also for 1 s (m/z 50–2000, continuum mode), using the function switching in MassLynx version 4.0 software. Raw MS/MS data were smoothed (Savitzky Golay, two channels twice) and centroided (at 80%) and peaks lists generated using MassLynx software. Peak lists were submitted for data base searching using Mascot (version 2.2.04). Searches were performed against the IPI Human Database (release version 3.44; May 2008; 72,346 sequence entries). Parameters for protein searches were as follows: enzyme (trypsin and porcine); miscleavages (2); charge of ions (+2 and +3); mass tolerance of precursor peptide ion (100 ppm); and mass tolerance for MS/MS fragment ions (0.8 mmu). Carbamidomethylation of cysteines was considered as a fixed modification, whereas oxidation of methionine and pyroglutamic acid, N-acetylation, and phosphorylation of tyrosine, serine, and threonine were considered as variable modifications. Positive protein identifications were called when at least two peptide sequences matched an entry and Mascot scores were above the significance threshold value at p = 0.05. When proteins of overlapping sequence identity (e.g. splice isoforms, processed precursors) matched the same set of peptides, all protein isoforms returned from the search were reported. To evaluate the false discovery rate, data were researched against a randomized “decoy” IPI human data base using Mascot, employing identical search parameters and validation criteria. A false discovery rate of <1% was determined for the combined dataset. Phosphorylation sites were evaluated using Scaffold software version 2.0. Phosphopeptide identifications were accepted when the Mascot score was above the significance threshold value at p = 0.05, and major fragment ions could be clearly assigned to MS/MS spectra. The sequences of phosphorylated peptides with Mascot/X tandem scores, precursor m/z values, and charge are provided as supplemental material, which also includes spectra and y and b ion lists for each of the identified phosphopeptides.
Immunofluorescence Staining and Confocal Microscopy
For immunofluorescence staining, HB4a cells seeded onto coverslips were pretreated with 2.5 μm PP1 or vehicle for 1 h prior to treatment with 0.5 mm H2O2 or vehicle for 20 min. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton-X-100, and washed with PBS. Localization of selected proteins was assessed using the following antibodies and dilutions: α-E-cadherin (1:50), α-pY118-paxillin (1:100), α-pY416-Src (1:100), α-cleaved caspase-3-(Asp-175) fluorescein-conjugated (1:100) (all from Cell Signaling Technology); α-ZO-1 (1:100; Invitrogen); α-pY (1:500; pY99; Santa Cruz Biotechnology); and α-cytochrome c (1:100) and α-BCL2 (1:100) (both from Pharmingen). Antibodies were diluted in 2.5% bovine serum albumin/PBS and incubated with cells at room temperature for 1 h. After three PBS washes, samples were incubated with fluorescently labeled secondary antibodies diluted in 2.5% bovine serum albumin/PBS. For localization of filamentous actin, cells were incubated with 0.1 μg/ml Alexa568-phalloidin (Sigma) for 45 min at 37 °C. Coverslips were washed three times with PBS and at least twice with double deionized water before mounting in Vectashield mounting medium (Vector Laboratories). Coverslip edges were sealed with nail polish onto glass slides (BDH) and dried in the dark at 4 °C. Cells were imaged using a Zeiss LSM510 laser scanning confocal microscope equipped with an inverted Axiovert 200 M stand (Carl Zeiss Inc., Germany). Emission wavelengths of 488 nm from an argon laser, 543 nm from an HeNe1 laser, and 633 nm from an HeNe2 laser were used. All laser intensities used to detect the same immunostained markers from different cell treatments were the same, and all laser intensities used for capturing images were not saturated. Images were obtained from four horizontal sections (x-y plane) taken at equal intervals through the whole cell volume and were displayed as a projection along the z axis using maxima fluorescence values. Images were exported as .tif files using the Zeiss LSM image browser and processed using Adobe Photoshop version 7.0 software.
Adhesion Assays
96-Well tissue culture plates were coated with sterile 3% bovine serum albumin for 1 h at room temperature. HB4a cells pretreated for 1 h with 2.5 μm PP1, 5 μm AG1478, or vehicle followed by 0.5 mm H2O2 for 20 min were added to wells at 5000 cells/well in RPMI 1640 medium containing 10% fetal calf serum. Cells were left to adhere for the indicated times at 37 °C in a 10% CO2-humidified incubator. Unattached cells were aspirated, and the remaining cells were gently washed three times with PBS to remove unbound or loosely attached cells. Substratum bound cells were counted using a hemocytometer. All conditions were repeated four times.
MTT Cell Viability Assay
Exponentially growing HB4a cells were trypsinized and counted using a hemocytometer, and 5000 cells/well were seeded into 96-well plates. The culture was then incubated for 24 h before pretreatment with 2.5 μm PP1 for 1 h and then 0.5 mm H2O2 for 20, 60, and 240 min or left untreated. After removal of medium, 50 μl of MTT solution (1 mg/ml) (Sigma) was added to the wells, followed by incubation at 37 °C for 4 h. The supernatant was carefully removed; 100 μl of DMSO was added to each well, and the plates were shaken for 20 min. The absorbance at 540 nm was then measured in a plate reader. Values were normalized against untreated samples and were the mean of four independent measurements.
RESULTS
Hydrogen Peroxide Induces Tyrosine Phosphorylation and Activation of Multiple Signaling Pathways in Human Mammary Luminal Epithelial Cells
Our goal was to examine targets of ROS-induced tyrosine phosphorylation in a relevant epithelial cell model. We chose the human mammary luminal epithelial cell line HB4a, as it is ideal for examining early molecular events associated with breast cancer formation because the majority of breast cancers arise from the luminal epithelial cell type. In previous work, we have used this model to examine how overexpression of the receptor tyrosine kinase ErbB2 results in oncogenic signaling (19, 21, 22) and how oxidative stress alters the human mammary luminal epithelial cell proteome in an ErbB2-dependent manner (20). Here, we expand on this work by studying in more detail the signaling events associated with oxidative stress.
Previous studies using H2O2 to treat different cell types have reported nonlethal cellular responses in the range 0.1–1 mm H2O2. Here, we tested a wider range of doses and treated HB4a cells for different times to find a maximal response in terms of protein tyrosine phosphorylation as assessed by immunoblotting. A concentration of 0.5 mm H2O2 gave a robust though transient increase in tyrosine phosphorylation, which peaked around 20 min after treatment (Fig. 1A, left panel). At 20 min, there was no noticeable detachment of cells from the substratum, although by 4 h around half of the cells had become detached (see below). At the highest concentration of H2O2 tested (10 mm), most cells became detached from the substratum by 20 min, although at the lowest concentration tested (10 μm), there was no detectable induction of tyrosine phosphorylation (data not shown). Notably, 0.5 mm H2O2 treatment for 20 min resulted in maximal activation of the signaling kinases Akt, ERK1/2, and p38 but not JNK (Fig. 1A, right panel). In agreement with other reports, these data show that oxidative stress induces global tyrosine phosphorylation and activates multiple signaling pathways involved in regulating cellular proliferation and survival.
FIGURE 1.

Effect of H2O2 on signaling in HB4a and purification of tyrosine-phosphorylated proteins. A, total cell lysates (TCL) were prepared from HB4a cells treated with 0.5 mm H2O2 for the indicated times. Protein (30 μg) was separated by one-dimensional SDS-PAGE, transferred to Immobilon P membrane, and probed with specific antibodies against the indicated proteins and phosphorylation motifs. B, HB4a cells were either left untreated or treated with 0.5 mm H2O2 for 20 min followed by preclearing with protein A and enrichment with α-Tyr(P) mouse monoclonal antibody (4G10). 10 μg of total cell lysates, 10 μg of protein A precleared lysate (PC), 1 μg of immunoprecipitated protein (IP) from untreated cells, and 5 μg of affinity-purified protein from H2O2-treated cells were resolved by one-dimensional SDS-PAGE followed by immunoblotting with α-Tyr(P) antibody pY99 (left panel) and visualization of proteins by colloidal Coomassie Blue G-250 (CCB) staining (right panel).
Identification of H2O2-induced Tyrosine-phosphorylated Proteins
To enrich the H2O2-induced tyrosine-phosphorylated proteins for mass spectrometry-based identification, a large scale IP approach was adopted using agarose-conjugated α-phosphotyrosine (Tyr(P)) antibody and a phenyl phosphate elution step. Starting with ∼1 × 108 cells (18 mg of total protein), the purification yielded 54 μg of protein (0.3% of total) from H2O2-treated cells (0.5 mm; 20 min) versus 4.5 μg (0.025% of total) from untreated cells. Purification and one-dimensional gel separation of tyrosine-phosphorylated proteins are shown in Fig. 1B. LC-MS/MS and data base searching identified 85 proteins and 8 previously reported phosphorylation sites (supplemental Table 1 and supplemental material). These sites were as follows: p120 catenin Tyr(P)-228 (Src kinase phosphorylation site); cortactin Ser(P)-405, Ser(P)-418, and Tyr(P)-421 (the latter is an Src kinase phosphorylation site); epidermal growth factor receptor (EGFR) Thr(P)-693 and Tyr(P)-1197 (the latter is an autophosphorylation site); paxillin Tyr(P)-118 (FAK phosphorylation site); and tight junction protein-2/ZO-2 Tyr(P)-1118. Importantly, nearly half of the identified proteins are reported to be tyrosine-phosphorylated in the SwissProt, PHOSIDA, and/or PhosphoSite Databases, confirming the effectiveness of the enrichment strategy. These included the following: receptor tyrosine kinases EGFR, ephrin type A receptor 2, and epithelial discoidin domain-containing receptor 1/DDR1; the nonreceptor tyrosine kinases FAK and PYK2; tyrosine phosphatase SHPTP2; and proteins involved in the regulation of the actin cytoskeleton and adhesion (Abl interactor 1 isoform 2, β-actin, actin-γ2, α-actinin 1 and 4, ARF GTPase-activating protein GIT1, β-catenin, p120 catenin, cortactin, p130CAS, CUB domain-containing protein 1, integrin β4, junction plakoglobin, Par3, paxillin, and ZO-2). Few proteins were identified from untreated cells where basal tyrosine phosphorylation levels were low. These were mainly abundant proteins not normally known to be tyrosine-phosphorylated (HSP70 family members, HSP90, cytokeratin 18, tubulins, and GRP78). We hypothesize that these proteins may be present as a result of nonspecific binding. Two forms of actin were also identified. Despite the high abundance of actin in cells and the possibility of nonspecific binding, previous work has shown that β-actin is tyrosine-phosphorylated in 3T3 fibroblasts expressing an activated form of Src kinase (23).
Several proteins were identified from gel bands that were above their predicted masses. These were S100A8, S100A9, calmodulin-like protein 5, epidermal fatty acid-binding protein, galectin 7, and ubiquitin and ribosomal protein S27a (supplemental Table 1, gray-shaded cells). Although the latter identification suggests that ubiquitin peptides from a ubiquitinylated protein of larger mass may have been identified, the presence of the other proteins from these higher mass bands is unclear. Conversely, peptides from desmoplakin isoforms 1 and 2 were found in a gel band of ∼140 kDa, well below their predicted masses of 332 and 260 kDa, respectively. It is tempting to speculate that this may be the result of caspase-dependent cleavage, known to occur during epithelial cell apoptosis (24), a response of HB4a cells to H2O2 treatment (see below).
Validation and Functional Classification of Identified Proteins
Selected H2O2-induced phosphorylated proteins were further confirmed by immunoblotting of small scale α-Tyr(P) IPs with specific antibodies. The known Tyr(P) proteins p130CAS, β-catenin, cortactin, EGFR, E-cadherin, and FAK were shown to be present in IPs (Fig. 2A). The redox regulator Prdx3 (peroxiredoxin 3), a protein not been reported to be tyrosine-phosphorylated, was also enriched in IPs from H2O2-treated cells. Although we were unable to confirm whether Prdx3 was tyrosine-phosphorylated in response to oxidative stress, its enrichment in the IPs is intriguing given its role in protecting proteins from oxidative damage. Interestingly, a number of known Src kinase substrates (e.g. FAK, p130CAS, β-catenin, p120-catenin, cortactin, EGFR, and paxillin) were identified, prompting us to examine if Src itself was present in the IPs. Immunoblotting using antibodies against the activated form of c-Src (α-Tyr(P)-416-Src) confirmed that this was the case and that Src is activated in H2O2-treated HB4a cells (Fig. 2A).
FIGURE 2.

Validation and functional classification of immunoaffinity-purified Tyr(P)-containing proteins. A, lysates from untreated and H2O2-treated HB4a cells were immunoprecipitated with α-Tyr(P) (pTyr) antibody and immunoblotted with the antibodies shown. B, functional distribution of the identified H2O2-induced tyrosine-phosphorylated proteins.
Functional annotation of the identified proteins revealed that around 60% are reported to be associated with the actin cytoskeleton, focal adhesions, cell-cell junctions, and cell motility (Fig. 2B). This suggests that H2O2 has profound effects on the actin cytoskeleton, adhesion, and/or intercellular connections (see below), as suggested in previous work (25–27). We acknowledge that the presence of enriched proteins using this strategy does not directly prove that they are tyrosine-phosphorylated and that associated proteins will also be enriched. Indeed, an enrichment of complexes was evident from searching the STRING protein interaction data base using the identified proteins (including Src) as input (Fig. 3A). Using the “medium” confidence level defined in the software, there is evidence that 84% of these proteins can interact with one or more of the other proteins. Src kinase interacted with the highest number of input proteins (22 interactions) followed by β-actin/ENSP00000349960 (21 interactions), EGFR (19 interactions), tight junction protein 1/TJP1/ZO-1 (18 interactions), and E-cadherin/UVO (17 interactions). The Src kinase interactors and their “evidenced” connectivities are shown in Fig. 3B. Although these interactions may not all occur in H2O2-treated HB4a cells, these findings suggest that many of the identified proteins occur within signaling complexes and networks regulating the cytoskeleton and adhesion and that Src and EGFR play a major role in catalyzing tyrosine phosphorylation events that regulate these networks.
FIGURE 3.

Interaction networks of identified proteins. A, proteins identified in supplemental Table 1 and Src kinase were imported into the EMBL Search Tool for the Retrieval of Interacting Proteins (STRING) data base, and an interaction map was generated. Each node represents a protein entry. Interactions or edges were generated from experimental, text mining, and data base evidences using the medium confidence level. Thicker lines represent higher confidence interactions. B, schematic illustrating the 22 Src-interacting input proteins identified.
Roles of Src and EGFR Kinases in H2O2-dependent Tyrosine Phosphorylation
To address the possible roles of Src and EGFR in eliciting peroxide-induced tyrosine phosphorylation in these cells, we used inhibitors PP1 and AG1478 to examine the effects of reducing the Src and EGFR kinase activities, respectively. Pretreatment of cells with either PP1 or AG1478 significantly reduced H2O2-induced tyrosine phosphorylation, implicating both kinases as mediators of peroxide-induced phosphorylation (Fig. 4A). Two prominent phosphorylated bands of ∼41 and 45 kDa were retained, however, in the PP1- and AG1478-treated samples, respectively, suggesting some specificity in the response to the different inhibitors. As alluded to above, Src kinase was activated in response to H2O2 treatment as shown by its active site (Tyr-416) phosphorylation. Notably, AG1478 treatment, as well as PP1 treatment, resulted in ablation of the Tyr(P)-416 signal, with no effect on phosphorylation of the Src kinase Tyr-527 inhibitory site (Fig. 4A). Similar results were observed in MEFs, where the H2O2-induced tyrosine phosphorylation and Src kinase activation were appreciably inhibited by PP1 (Fig. 4A, right panel). However, AG1478 was less potent in these cells, and higher doses of H2O2 (2–10 mm) were required to induce robust tyrosine phosphorylation. PP1 treatment also reduced Akt, ERK1/2, and p38 activation in response to H2O2, albeit modestly (Fig. 4B), suggesting an upstream role for Src in mediating activation of these known survival, proliferative, and stress-activated pathways in response to oxidative stress. PP1 pretreatment also resulted in loss of the adhesion proteins and Src substrates β-catenin, cortactin, FAK, and p130CAS from α-Tyr(P) IPs from H2O2-treated cells, presumably as a result of dephosphorylation and disruption of complexes in the absence of Src activity (Fig. 4C). Finally, the effects of H2O2 and inhibitor treatments on identified phosphorylation sites EGFR-Tyr(P)-1197, cortactin-Tyr(P)-421, and paxillin-Tyr(P)-118 were examined by immunoblotting with phospho-specific antibodies (Fig. 4D). H2O2 treatment resulted in increased phosphorylation of all three sites, which was partially or completely blocked by either PP1 or AG1478 pretreatment. Paxillin-Tyr(P)-118 phosphorylation was only reduced by PP1 treatment. In addition, tyrosine phosphorylation of E-cadherin was shown by immunoprecipitating the protein and blotting for Tyr(P) (Fig. 4D, lower panel). Taken together, these data show that the Src and EGFR kinases are activated by H2O2 and suggest that they play a direct role in mediating the cellular response to oxidative stress through phosphorylation of the identified targets.
FIGURE 4.

Effect of EGFR and Src inhibition on H2O2-induced signaling events and tyrosine-phosphorylated protein recovery. A, total cell lysates (TCL) prepared from untreated and treated HB4a cells (left panel) and murine embryonic fibroblasts (MEF) (right panel) were immunoblotted with antibodies against the indicated proteins and their phosphorylated forms. Cells were left untreated or pretreated with 2.5 μm Src kinase inhibitor PP1 or 5 μm EGFR inhibitor AG1478 for 1 h, prior to treatment with 0.5 mm H2O2 for 20 min. B, activation of signaling kinases in response to H2O2 and the effect of PP1 pretreatment. C, effect of Src kinase inhibition on recovery of tyrosine-phosphorylated proteins. Small scale α-Tyr(P) IPs from untreated and PP1-pretreated and H2O2-treated cells were immunoblotted for the indicated proteins. D, effect of H2O2 treatment and Src and EGFR kinase inhibition on phosphorylation of EGFR at Tyr-1197, cortactin at Tyr-421, and paxillin at Tyr-118 and proof of E-cadherin tyrosine phosphorylation. Cells were treated as indicated, lysates prepared, and specific phosphorylation events monitored by immunoblotting with phospho-specific antibodies. E-cadherin was immunoprecipitated from lysates and IPs immunoblotted with anti-Tyr(P) (pTyr) antibody.
Role of Src Kinase in H2O2-induced Loss of Cell Adhesion, Morphology, and Cell Viability
The high proportion of proteins identified with roles in cellular adhesion and cytoskeletal regulation prompted us to examine the effect of H2O2 on cell-cell and cell-substratum adhesion events and cell morphology. We hypothesized that H2O2-induced tyrosine phosphorylation through Src may affect the function of proteins involved in cytoskeletal regulation and adhesion, particularly because some of these are known Src substrates and because Src is a regulator of cell adhesion and motility (28). Immunofluorescence staining confirmed the robust increase in tyrosine phosphorylation in response to H2O2, which was potently inhibited by PP1 pretreatment (Fig. 5, A–C). The tyrosine phosphorylation signal was primarily localized to the cell cortex and regions of cell-cell contacts in untreated cells, whereas H2O2-treated cells were heavily stained in all areas but the nuclei. H2O2 treatment resulted in changes to the actin cytoskeleton; in control cells most actin filaments were localized close to cell-cell adhesions, whereas in H2O2-treated cells, disorganized actin filaments were observed throughout the cytoplasm of most cells, where there was a loss of uniform cell shape (Fig. 5, D–F). Staining for Tyr(P)-416-Src was increased as expected in H2O2-treated cells, reduced by PP1 pretreatment, and was uniformly localized with some discrete puncta (Fig. 5, G–I). H2O2-induced and PP1-sensitive phosphorylation of the focal adhesion and integrin-regulated adaptor protein paxillin at its Tyr-118 FAK phosphorylation site was also confirmed by staining (Fig. 5, J–L), validating the MS/MS-based identification of this site (supplemental Table 1). In untreated cells, punctate Tyr(P)-118-paxillin staining was more evident at cell edges and indicated sites of focal adhesions. H2O2 treatment appeared to increase diffuse cytoplasmic staining of Tyr(P)-118-paxillin, whereas PP1 pretreatment resulted in staining only at the periphery of cell clusters. As expected, expression of the cell-cell adhesion protein E-cadherin was localized predominantly to regions of cell-cell contact in untreated cells (Fig. 5m). This localized staining was reduced in response to H2O2, but it was rescued by pretreatment with the PP1 inhibitor (Fig. 5, N and O). Similarly, staining for the tight junction protein ZO-1 revealed more intense punctate staining in areas of cell-cell contact, representing tight junctions, and this staining was significantly reduced in H2O2-treated cells and rescued by PP1 pretreatment (Fig. 5, P–R). Together, these data indicate that oxidative stress of epithelial cells results in a moderate alteration of the actin cytoskeleton and cell morphology and a substantial loss of normal cell-cell contacts. It must be noted that these immunofluorescence experiments required cells to be attached to the substratum, although H2O2 causes significant loss of adhesion. We therefore propose that the observed events occur prior to de-adhesion as part of the response to oxidative stress.
FIGURE 5.
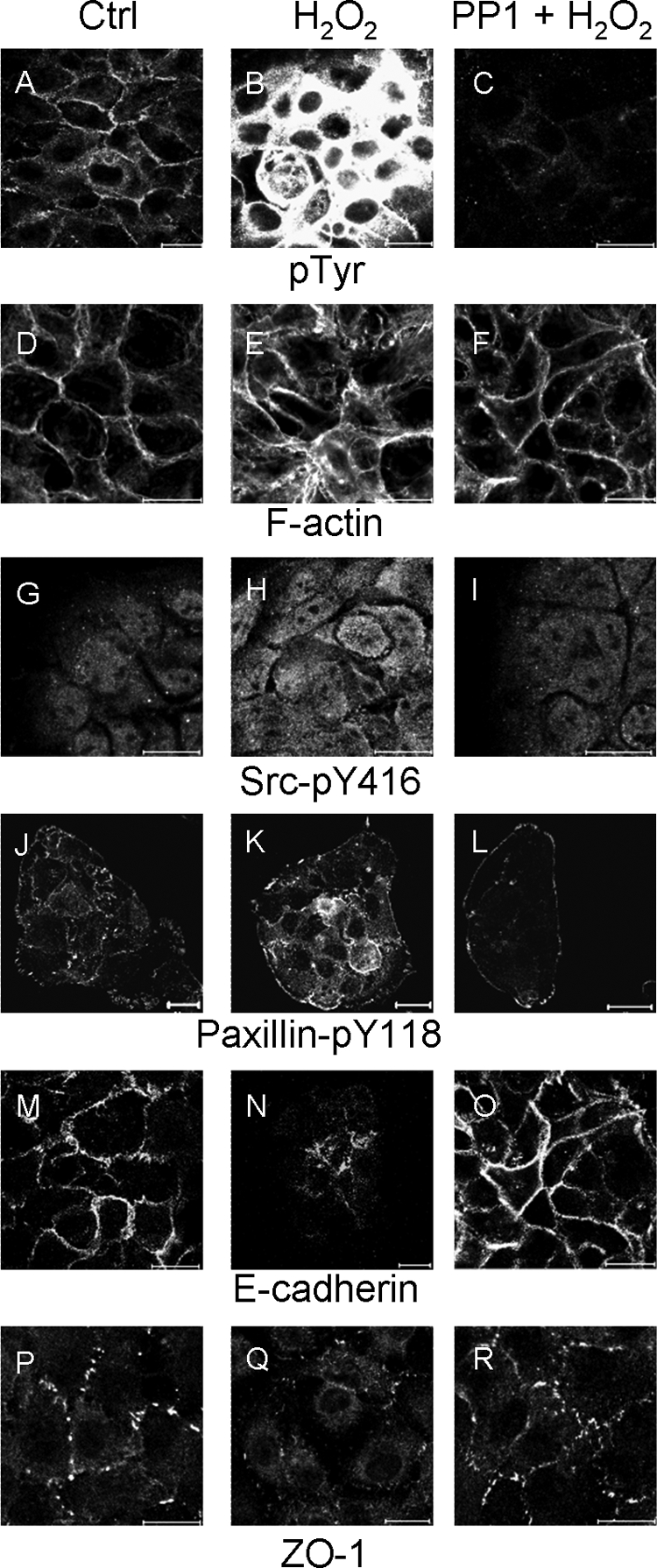
H2O2 induces morphological and protein localization changes in HB4a cells. HB4a cells seeded onto coverslips were either left untreated or treated with 0.5 mm H2O2 for 20 min or pretreated with 2.5 μm PP1 for 1 h and then with H2O2 for 20 min before fixation and staining for Tyr(P) (pTyr), F-actin, pY416-Src, pY118-paxillin, E-cadherin, and ZO-1. Images for each set of fields for each protein were taken using the same exposure times. Images are representative of at least five different fields in at least three independent experiments. Scale bar, 20 μm. Ctrl, control.
We further examined the effect of peroxide and Src or EGFR inhibition on cell adhesion itself. HB4a cells were pretreated with PP1 or AG1478 and then treated with peroxide or left untreated and plated onto substratum for 1 and 4 h followed by gently washing to remove unbound cells. The remaining adherent cells were counted. The results showed that H2O2 treatment significantly reduced the adhesive capability of cells and that this effect was partially dependent upon Src and EGFR kinase activity (Fig. 6A). This suggests that peroxide-induced Src and EGFR activation decrease adhesive capability. Further experiments to determine the effect of peroxide on cell viability were performed using an MTT assay that takes into account the loss of cells due to de-adhesion. Cell viability/cell adhesion was decreased to about 50% after 20 min, 25% after 1 h, and 20% after 4 h of H2O2 treatment, whereas PP1 pretreatment resulted in the maintenance of ∼100, ∼85, and ∼50% of viability/adhesion after 20 min and 1 and 4 h of treatment, respectively (Fig. 6B). This suggests that Src-dependent signaling may also play a role in mediating peroxide-induced cell death, as well as adhesion.
FIGURE 6.

Effect of H2O2, Src, and EGFR inhibition on HB4a cell adhesion and viability. A, adhesion assays were performed where 5000 cells pretreated with PP1, AG1478, and/or H2O2 or left untreated were plated into 96-well plates in medium containing 10% fetal calf serum at 37 °C. After 1 and 4 h, cells were gently washed three times, and the number of adherent cells was counted using a hemocytometer. The mean cell number of four independent assays is shown ± S.D. An unpaired Student's t test was used to assess the significance of difference between treatments. *, p < 0.05; **, p < 0.01. B, MTT-based viability assays were performed where 5000 HB4a cells were plated into 96-well plates in medium containing 10% fetal bovine serum. After 24 h, the cells were pretreated with 2.5 μm PP1 for 1 h followed by treatment with 0.5 mm H2O2 for 20 min and 1 and 4 h or left untreated. Cells were incubated with MTT, and then DMSO was added and the plates were shaken for 20 min followed by measurement of the absorbance at 540 nm. Values were normalized against the untreated samples and are the average of four independent measurements ± S.D. C, MTT assays were performed essentially as described above, except that H2O2-containing media were replaced after 20 min with fresh medium containing 10% fetal bovine serum (washout) and cells cultured for different time points up to 24 h prior to MTT assay.
To test whether the observed effects on cell viability and morphology were a specific response to ROS-dependent signaling or a consequence of nonspecific toxicity, “washout” experiments were performed where H2O2-containing media were replaced after 20 min with fresh media without H2O2, and cell viability and morphology were assessed over 24 h. Cell viability was partially rescued (20–40% viability after 24 h) when H2O2 was removed and completely rescued (50–100%) in the cells pretreated with PP1 (Fig. 6C). Recovery of cell morphology after H2O2 washout was monitored by phase-contrast microscopy and F-actin staining (Fig. 7). The ordered morphology of cells was lost in response to H2O2 treatment, and this was restored 16 h after washout. Restoration of F-actin staining appeared to be more rapid, and cortical staining was restored by 8 h. These data support the conclusion that the observed effects are due to ROS-specific signaling events and not due to irreversible toxicity.
FIGURE 7.

Recovery of H2O2-induced morphological defects after H2O2 washout. Confluent HB4a cells on coated coverslips were either left untreated or treated with 0.5 mm H2O2 for 20 min followed by replacement with fresh medium containing 10% fetal bovine serum for the indicated time periods before fixation and staining for F-actin. Images were captured using both phase contrast and fluorescence with the same exposure. Images are representative of six different fields. Scale bar, 20 μm. Ctrl, control.
Role of Src Kinase in H2O2-induced Apoptosis
To explore further a possible role of Src activation in peroxide-induced cell death, several markers of apoptosis (cytochrome c release from mitochondria, activation of caspase-3, and decreased Bcl-2 expression) were examined by immunofluorescence. In response to peroxide, there was a decrease in fluorescence intensity for cytochrome c resulting from its release from mitochondria and indicative of apoptosis (Fig. 8). Cleaved caspase-3, a mediator of apoptosis, accumulated in the cytosol of some cells following H2O2 treatment, although staining of the anti-apoptotic protein Bcl-2, which governs the permeability of the mitochondrial outer membrane, displayed decreased fluorescence intensity. These observations indicated that a certain level of apoptosis occurs in cells exposed to H2O2. In contrast, in cells pretreated with PP1 prior to H2O2 treatment, cytochrome c, cleaved caspase-3, and Bcl-2 staining were similar to that found in untreated cells (Fig. 8, right panels). These data suggest that blocking Src kinase activity suppresses oxidant-induced apoptosis in these cells, which may be a consequence of tyrosine hyper-phosphorylation.
FIGURE 8.

Effect of Src inhibition on H2O2-induced apoptosis in HB4a cells. Confluent HB4a cells were either left untreated or treated with 0.5 mm H2O2 for 20 min or pretreated with PP1 for 1 h and then treated with H2O2 for 20 min before fixation and staining for cytochrome c, cleaved activated caspase-3, and BCL2. Images were taken using the same exposures for the three different treatment conditions and are representative of three independent experiments. Scale bar, 20 μm. Ctrl, control.
DISCUSSION
ROS play critical roles in regulating cell behavior through modification of proteins. However, compared with knowledge accumulated from other cell regulatory systems, relatively little is known about the extent of redox modifications of signaling proteins. In this study, we have shown that oxidative stress of human mammary luminal epithelial cells induces a robust and transient increase in protein tyrosine phosphorylation, and we have identified a subset of these proteins. Almost 60% of them are known to be involved in modulating the cytoskeleton and cell adhesion, and we provide evidence for the existence of multiple signaling complexes controlling these processes. We further showed that oxidative stress alters the actin cytoskeleton, causing disruption of cell-cell and cell-matrix contacts. H2O2 treatment also elicited pro-apoptotic changes in cells that were still attached to the substratum and that may contribute to detachment. Our most important finding was that the Src and EGFR kinases (present within these complexes) play a central role in eliciting H2O2-induced tyrosine phosphorylation, which we link directly to changes in cell morphology, loss of cell-cell adhesions, cell detachment, and apoptosis.
Our data suggest that loss of adhesion involves tyrosine phosphorylation-dependent disruption of several types of epithelial adhesion complexes as follows: E-cadherin-mediated cell-cell adherens junctions (AJs) in which catenins, α-actinin, and afadin link these junctions to the actin cytoskeleton; cell-cell tight junctions (TJs), where claudins and occludin are stabilized by proteins such as ZO-1/TJP1, ZO-2, and Par3, which help to maintain epithelial polarity and integrity; cell-matrix focal adhesions (FAs), where integrin heterodimers link extracellular matrix proteins with FAK, p130CAS, paxillin, and other proteins to regulate attachment-dependent survival, proliferation, and motility signaling; and desmosome complexes where plakoglobins link cadherin dimers to intermediate filaments. Multiple components of these adhesion complexes were identified using the enrichment strategy described.
Src kinase family members and receptor tyrosine kinases such as EGFR have been implicating in regulating the assembly of and signaling events from epithelial AJs, TJs, and FAs. For example, Src and EGFR have been reported to phosphorylate and dissociate β-catenin and p120 catenin in AJs (29–31), and expression of oncogenic v-Src results in redistribution of E-cadherin and AJ disassembly (32). The re-organization and/or disassembly of AJs in our system is likely to occur through the same mechanism, i.e. oxidant-induced activation of Src and EGFR leading to hyper-phosphorylation of AJ proteins (E-cadherin, catenins, cortactin, and afadin), junctional disruption, and cell de-adhesion. A previous study also showed that overexpression of ephrin type A2 receptor (also identified herein) can destabilize AJs via a Src- and RhoA-dependent mechanism (33). Thus, hyper-activation of receptor tyrosine kinases other than EGFR may also promote loss of adhesion.
In tight junctions, the roles of Src and EGFR are less clear, although it has been shown that kinase-inactive Src delays oxidative stress-induced disruption of TJs (34), that EGF-stimulated, Src-dependent phosphorylation of Par3 at Tyr-1127 regulates TJ assembly (35), and that Src-stimulated tyrosine phosphorylation of ZO-1 and ZO-2 leads to disruption of TJ-protein interactions (36). Thus, it appears that H2O2 may cause the TJ disassembly observed here through Src activation and phosphorylation of junction proteins such as Par3 and ZO-1/2, which were identified here. In FAs, the interplay between extracellular matrix-integrin engagement and FAK and Src phosphorylation in coupling adhesion with survival and motility signaling has been intensely studied. However, little is known about the effects of ROS on FA complexes. ROS have been shown to promote FAK, paxillin, and p130CAS tyrosine phosphorylation in endothelial cells (37), supporting our findings in epithelial cells. Herein, paxillin phosphorylated at the Tyr-118 FAK site was delocalized from FAs in response to H2O2 and would be expected to promote de-adhesion, supporting previous work showing that lysyl oxidase facilitates migration of invasive breast cancer cells through an H2O2-mediated mechanism involving the FAK/Src signaling pathway (38). Oxidative stress is also known to modify the actin cytoskeleton in many cell types, and this has been characterized by fragmentation, patching, and oxidative modification of F-actin itself (reviewed in Ref. 39). Here, we observed subtle changes in F-actin and irregular cell morphology. Although the observed changes may be consistent with previous findings, we suggest that the outcome is dependent upon cell type and the effective ROS dose.
The interplay between Src family kinases and receptor tyrosine kinases is well established, and it is known that EGFR activity stimulates Src and vice versa (40, 41). Indeed, Src has been shown to associate with and phosphorylate EGFR at Tyr-845 (40). In our study, we observed that blockade of Src or EGFR kinase activity with PP1 and AG1478, respectively, could dramatically inhibit H2O2-induced tyrosine phosphorylation and alleviate H2O2-induced loss of cell adhesion. The fact that either inhibitor substantially reduced the signal suggests that these kinases do indeed cooperate with one another and/or share the same substrates. One key finding was that AG1478 treatment effectively blocked Src kinase activity induced by H2O2 treatment. This would place EGFR receptor activation upstream of Src, although the identification of multiple Src kinase substrates and interactors suggests that Src is the key regulator of multiple downstream effectors.
In terms of a mechanism by which H2O2 activates the EGFR and Src kinases, we propose that protein-tyrosine phosphatases with EGFR and/or Src as their substrates are targets of direct H2O2-mediated oxidative modification and inhibition (see Introduction). For EGFR, these may include PTP1B, SHPTP1, SHPTP2, Cdc25A, TC-PTP, and the receptor-type protein-tyrosine phosphatase κ, which have all been reported to dephosphorylate the receptor. Although SHPTP2 was identified in our pulldowns, this protein-tyrosine phosphatase appears to play a positive role in growth factor signaling, possibly acting through dephosphorylation of receptor recruitment sites for RasGAP or the Tyr(P)-517 Src inhibitory site. Although this makes SHPTP2 a less likely target of oxidation and mediator of the observed effects, it may well regulate downstream AJ components directly, as shown for VE-cadherin complex components in endothelial cells (42). In the case of c-Src, our study supports previous work linking its activity with oxidative stress-induced signal transduction (43–47). Our data suggest a mechanism whereby a phosphatase for Tyr(P)-416 of Src is inhibited by H2O2, because we found no evidence of the altered phosphorylation of the Tyr-517 inhibitory site. However, engagement of the Src homology 2 domain of Src through binding to phosphorylated EGFR would also promote autophosphorylation at Tyr-416 by relieving an auto-inhibited conformation (48). This again implicates the hyper-phosphorylation of EGFR as the initiator of oxidant-dependent signaling, which is diversified through downstream Src activation (Fig. 9). Alternatively, a sulfhydryl-based structural modification has been proposed for the direct activation of Src in response to oxidants (49).
FIGURE 9.

Model of H2O2-induced tyrosine phosphorylation signaling. H2O2 inhibits protein-tyrosine phosphatases (PTPs) resulting in hyper-phosphorylation of the EGFR and recruitment and activation of Src kinase. Src kinase (and EGFR) phosphorylates multiple protein targets in FA, AJ, TJ, and the actin cytoskeleton, resulting in disruption of cell-matrix and cell-cell contacts and possibly changes to the actin cytoskeleton leading to loss of adhesion. Loss of adhesion itself or direct mechanisms involving protein-tyrosine phosphatases, Src, or H2O2 promote apoptosis.
Finally, the findings of this study may have clinical implications in that Src or EGFR kinase inhibition may be useful in pathological conditions where oxidative stress is implicated. One such condition is myocardial ischemia-reperfusion injury, where a shortage of blood supply to a region of tissue is followed by resumption of blood flow and tissue damage due to generation of ROS. Although the detailed mechanisms for ROS generation and induced tissue damage are unclear, Src has been implicated in this process (50). From our study, the roles of receptor tyrosine kinases and Src family kinases in ischemia-reperfusion injury warrant further investigation, and small molecule inhibitors of these kinases should be tested in animal models as potential therapeutic interventions. In epithelial cancers, the overexpression and constitutive activation of ErbB receptors acting in concert with Src may promote transformation and invasion through loss of cell-cell and cell-matrix contacts in a similar manner to that elicited by oxidative stress. However, in this setting, mechanisms that block apoptosis in response to loss of adhesion must come into play in order for cells to survive.
Supplementary Material
This work was supported by grants from the EMF Biological Research Trust Grant BRT 06/28, the Association for International Cancer Research Grant 05-426, European Union Grants FP6 LSHC-CT-2003-502935, National Science Council of Taiwan Grant 97-2311-B-007-005, and from the Eve Appeal. This work was undertaken at University College London Hospital/University College London that received a portion of funding from the Department of Health National Institute for Health Research Biomedical Research Centres funding scheme.

The on-line version of this article (available at http://www.jbc.org) contains supplemental tables.
- ROS
- reactive oxygen species
- AJ
- adherens junction
- FA
- focal adhesion
- H2O2
- hydrogen peroxide
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- Tyr(P)
- phosphotyrosine
- TJ
- tight junctions
- EGFR
- epidermal growth factor receptor
- FAK
- focal adhesion kinase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS
- phosphate-buffered saline
- JNK
- c-Jun N-terminal kinase
- IP
- immunoprecipitation
- ERK
- extracellular signal-regulated kinase.
REFERENCES
- 1.Pawson T., Nash P. (2003) Science 300, 445–452 [DOI] [PubMed] [Google Scholar]
- 2.Krebs E. G. (1994) Trends Biochem. Sci. 19, 439. [DOI] [PubMed] [Google Scholar]
- 3.Blume-Jensen P., Hunter T. (2001) Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 4.Harris A. L. (2002) Nat. Rev. Cancer 2, 38–47 [DOI] [PubMed] [Google Scholar]
- 5.Sullivan S. G., Chiu D. T., Errasfa M., Wang J. M., Qi J. S., Stern A. (1994) Free Radic. Biol. Med. 16, 399–403 [DOI] [PubMed] [Google Scholar]
- 6.Denu J. M., Tanner K. G. (1998) Biochemistry 37, 5633–5642 [DOI] [PubMed] [Google Scholar]
- 7.Lee S. R., Kwon K. S., Kim S. R., Rhee S. G. (1998) J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 8.Rhee S. G., Bae Y. S., Lee S. R., Kwon J. (2000) Sci. STKE 2000, E1. [DOI] [PubMed] [Google Scholar]
- 9.Kwon J., Lee S. R., Yang K. S., Ahn Y., Kim Y. J., Stadtman E. R., Rhee S. G. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16419–16424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leslie N. R., Lindsay Y., Ross S. H., Downes C. P. (2004) Biochem. Soc. Trans. 32, 1018–1020 [DOI] [PubMed] [Google Scholar]
- 11.Ross S. H., Lindsay Y., Safrany S. T., Lorenzo O., Villa F., Toth R., Clague M. J., Downes C. P., Leslie N. R. (2007) Cell. Signal. 19, 1521–1530 [DOI] [PubMed] [Google Scholar]
- 12.Sundaresan M., Yu Z. X., Ferrans V. J., Irani K., Finkel T. (1995) Science 270, 296–299 [DOI] [PubMed] [Google Scholar]
- 13.Bae Y. S., Kang S. W., Seo M. S., Baines I. C., Tekle E., Chock P. B., Rhee S. G. (1997) J. Biol. Chem. 272, 217–221 [PubMed] [Google Scholar]
- 14.Finkel T. (2000) FEBS Lett. 476, 52–54 [DOI] [PubMed] [Google Scholar]
- 15.Bae Y. S., Sung J. Y., Kim O. S., Kim Y. J., Hur K. C., Kazlauskas A., Rhee S. G. (2000) J. Biol. Chem. 275, 10527–10531 [DOI] [PubMed] [Google Scholar]
- 16.Rhee S. G., Kang S. W., Jeong W., Chang T. S., Yang K. S., Woo H. A. (2005) Curr. Opin. Cell Biol. 17, 183–189 [DOI] [PubMed] [Google Scholar]
- 17.Park H. S., Lee S. H., Park D., Lee J. S., Ryu S. H., Lee W. J., Rhee S. G., Bae Y. S. (2004) Mol. Cell. Biol. 24, 4384–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stamps A. C., Davies S. C., Burman J., O'Hare M. J. (1994) Int. J. Cancer 57, 865–874 [DOI] [PubMed] [Google Scholar]
- 19.Gharbi S., Gaffney P., Yang A., Zvelebil M. J., Cramer R., Waterfield M. D., Timms J. F. (2002) Mol. Cell. Proteomics 1, 91–98 [DOI] [PubMed] [Google Scholar]
- 20.Chan H. L., Gharbi S., Gaffney P. R., Cramer R., Waterfield M. D., Timms J. F. (2005) Proteomics 5, 2908–2926 [DOI] [PubMed] [Google Scholar]
- 21.Timms J. F., White S. L., O'Hare M. J., Waterfield M. D. (2002) Oncogene 21, 6573–6586 [DOI] [PubMed] [Google Scholar]
- 22.White S. L., Gharbi S., Bertani M. F., Chan H. L., Waterfield M. D., Timms J. F. (2004) Br. J. Cancer 90, 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 24.Weiske J., Schöneberg T., Schröder W., Hatzfeld M., Tauber R., Huber O. (2001) J. Biol. Chem. 276, 41175–41181 [DOI] [PubMed] [Google Scholar]
- 25.van Wetering S., van Buul J. D., Quik S., Mul F. P., Anthony E. C., ten Klooster J. P., Collard J. G., Hordijk P. L. (2002) J. Cell Sci. 115, 1837–1846 [DOI] [PubMed] [Google Scholar]
- 26.Alexandrova A. Y., Kopnin P. B., Vasiliev J. M., Kopnin B. P. (2006) Exp. Cell Res. 312, 2066–2073 [DOI] [PubMed] [Google Scholar]
- 27.Boardman K. C., Aryal A. M., Miller W. M., Waters C. M. (2004) J. Cell. Physiol. 199, 57–66 [DOI] [PubMed] [Google Scholar]
- 28.Frame M. C., Fincham V. J., Carragher N. O., Wyke J. A. (2002) Nat. Rev. Mol. Cell Biol. 3, 233–245 [DOI] [PubMed] [Google Scholar]
- 29.Roura S., Miravet S., Piedra J., García de Herreros A., Duñach M. (1999) J. Biol. Chem. 274, 36734–36740 [DOI] [PubMed] [Google Scholar]
- 30.Irby R. B., Yeatman T. J. (2002) Cancer Res. 62, 2669–2674 [PubMed] [Google Scholar]
- 31.Mariner D. J., Davis M. A., Reynolds A. B. (2004) J. Cell Sci. 117, 1339–1350 [DOI] [PubMed] [Google Scholar]
- 32.Gómez S., del Mont Llosas M., Verdú J., Roura S., Lloreta J., Fabre M., García de Herreros A. (1999) Biochim. Biophys. Acta 1452, 121–132 [DOI] [PubMed] [Google Scholar]
- 33.Fang W. B., Ireton R. C., Zhuang G., Takahashi T., Reynolds A., Chen J. (2008) J. Cell Sci. 121, 358–368 [DOI] [PubMed] [Google Scholar]
- 34.Basuroy S., Sheth P., Kuppuswamy D., Balasubramanian S., Ray R. M., Rao R. K. (2003) J. Biol. Chem. 278, 11916–11924 [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Du D., Fang L., Yang G., Zhang C., Zeng R., Ullrich A., Lottspeich F., Chen Z. (2006) EMBO J. 25, 5058–5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabath E., Negoro H., Beaudry S., Paniagua M., Angelow S., Shah J., Grammatikakis N., Yu A. S., Denker B. M. (2008) J. Cell Sci. 121, 814–824 [DOI] [PubMed] [Google Scholar]
- 37.Gozin A., Franzini E., Andrieu V., Da Costa L., Rollet-Labelle E., Pasquier C. (1998) Free Radic. Biol. Med. 25, 1021–1032 [DOI] [PubMed] [Google Scholar]
- 38.Payne S. L., Fogelgren B., Hess A. R., Seftor E. A., Wiley E. L., Fong S. F., Csiszar K., Hendrix M. J., Kirschmann D. A. (2005) Cancer Res. 65, 11429–11436 [DOI] [PubMed] [Google Scholar]
- 39.Dalle-Donne I., Rossi R., Milzani A., Di Simplicio P., Colombo R. (2001) Free Radic. Biol. Med. 31, 1624–1632 [DOI] [PubMed] [Google Scholar]
- 40.Tice D. A., Biscardi J. S., Nickles A. L., Parsons S. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1415–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bromann P. A., Korkaya H., Courtneidge S. A. (2004) Oncogene 23, 7957–7968 [DOI] [PubMed] [Google Scholar]
- 42.Ukropec J. A., Hollinger M. K., Salva S. M., Woolkalis M. J. (2000) J. Biol. Chem. 275, 5983–5986 [DOI] [PubMed] [Google Scholar]
- 43.Abe J., Takahashi M., Ishida M., Lee J. D., Berk B. C. (1997) J. Biol. Chem. 272, 20389–20394 [DOI] [PubMed] [Google Scholar]
- 44.Chen K., Vita J. A., Berk B. C., Keaney J. F., Jr. (2001) J. Biol. Chem. 276, 16045–16050 [DOI] [PubMed] [Google Scholar]
- 45.Khadaroo R. G., Parodo J., Powers K. A., Papia G., Marshall J. C., Kapus A., Rotstein O. D. (2003) Surgery 134, 242–246 [DOI] [PubMed] [Google Scholar]
- 46.Zhuang S., Schnellmann R. G., Zhougang S. (2004) Am. J. Physiol. Renal. Physiol. 286, F858–F865 [DOI] [PubMed] [Google Scholar]
- 47.Mehdi M. Z., Pandey N. R., Pandey S. K., Srivastava A. K. (2005) Antioxid. Redox. Signal. 7, 1014–1020 [DOI] [PubMed] [Google Scholar]
- 48.Boggon T. J., Eck M. J. (2004) Oncogene 23, 7918–7927 [DOI] [PubMed] [Google Scholar]
- 49.Pu M., Akhand A. A., Kato M., Hamaguchi M., Koike T., Iwata H., Sabe H., Suzuki H., Nakashima I. (1996) Oncogene 13, 2615–2622 [PubMed] [Google Scholar]
- 50.Guo J., Meng F., Zhang G., Zhang Q. (2003) Neurosci. Lett. 345, 101–104 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.