

Abstract
TM601 is a synthetic form of chlorotoxin, a 36-amino acid peptide derived from the venom of the Israeli scorpion, Leirius quinquestriatus, initially found to specifically bind and inhibit the migration of glioma cells in culture. Subsequent studies demonstrated specific in vitro binding to additional tumor cell lines. Recently, we demonstrated that proliferating human vascular endothelial cells are the only normal cell line tested that exhibits specific binding to TM601. Here, we identify annexin A2 as a novel binding partner for TM601 in multiple human tumor cell lines and human umbilical vein endothelial cell (HUVEC). We demonstrate that the surface binding of TM601 to the pancreatic tumor cell line Panc-1 is dependent on the expression of annexin A2. Identification of annexin A2 as a binding partner for TM601 is also consistent with the anti-angiogenic effects of TM601. Annexin A2 functions in angiogenesis by binding to tissue plasminogen activator and regulating plasminogen activation on vascular endothelial cells. We demonstrate that in HUVECs, TM601 inhibits both vascular endothelial growth factor- and basic fibroblast growth factor-induced tissue plasminogen activator activation, which is required for activation of plasminogen to plasmin. Consistent with inhibition of cell surface protease activity, TM601 also inhibits platelet-derived growth factor-C induced trans-well migration of both HUVEC and U373-MG glioma cells.
Keywords: Diseases/Cancer, Peptides, Protein/Protein-Protein Interactions, Tumor/Marker, Annexin, Annexin A2, Anti-angiogenesis, Invasion, Tissue Plasminogen Activator
Introduction
TM601 is a 36-amino acid synthetic peptide that is identical in structure and function to chlorotoxin that was isolated as an inhibitor of chloride ion channels from the venom of the giant Israeli scorpion, Leirius quinquestriatus (1, 2). Based on the specific tumor binding properties, TM601 has entered clinical evaluation as an iodinated radiopharmaceutical (131I-TM601) administered either locally or intravenously (3, 4). Early studies demonstrated that chlorotoxin can inhibit a potentially glioma-specific chloride ion channel (5). Chlorotoxin was shown to inhibit the migration and invasion of glioma cells possibly via the modulation of ion channels (6). Subsequent studies suggested that chlorotoxin modulates the chloride ion channel in glioma cells by facilitating the internalization and, hence, the down-regulation of the cell surface levels of the CLC-3 chloride channel (7). Chlorotoxin was shown to bind a macromolecular complex containing MMP-2,2 membrane type metalloprotease-1, tissue inhibitor of metalloprotease-2 (8), and the CLC-3 chloride channel at the surface of glioma cells and mediate the internalization and down-regulation of both MMP-2 and CLC-3 (7, 8). Chlorotoxin was also able to inhibit the in vitro activity of MMP-2 and the cell surface gelatinolytic activity in D54-MG cells, supporting an interaction between MMP-2 and chlorotoxin in glioma cells (8).
In addition to glioma cells, chlorotoxin has been shown to specifically bind other tumors of neuroectodermal origin (9). Recently, using mouse tumor models, a bio-conjugate of chlorotoxin with the nearly infrared dye Cy5.5 (CTX:Cy5.5) was shown to efficiently detect and monitor multiple tumor types, including glioma, medulloblastoma, prostate cancer, intestinal cancer, and sarcoma following intravenous injection (10). These studies also demonstrated that binding of CTX:Cy5.5 bio-conjugate to MCF-7 breast carcinoma cells in vitro is facilitated by the expression of exogenous MMP-2. However, these studies were unable to demonstrate a direct interaction between CTX:Cy5.5 and recombinant MMP-2, suggesting that the molecular target for chlorotoxin (TM601) is as yet unknown (10).
Recently we found that TM601 not only binds a wide range of tumor cell types but is also internalized by proliferating human vascular endothelial cells (11). These studies also demonstrated an anti-angiogenic effect of TM601 using both the chicken chorioallantoic membrane assays and the mouse Matrigel plug assays. Notably, TM601 inhibited angiogenesis induced by a wide range of stimuli including, VEGF, bFGF, hepatocyte growth factor, PDGF-AB, tumor necrosis factor-α, and interleukin-6. TM601 was also able to specifically inhibit angiogenesis stimulated by several different types of implanted tumor cells in a tumor chorioallantoic membrane assay, supporting a role for TM601 in inhibiting tumor angiogenesis (11). Finally, we demonstrated that TM601 inhibits both VEGF- and bFGF-stimulated trans-well migration of HUVECs, supporting a direct effect of TM601 on vascular endothelial cell types (11). Based on the interesting specificity for tumor and vascular endothelial cells, we sought to identify the molecular target for TM601 present on both the surface of tumor and vascular endothelial cell types.
In this paper we identify annexin A2 as a novel molecular target for TM601 that is expressed on the surface of multiple human tumor cell lines and vascular endothelial cells in culture. We show that surface binding of TM601 to the pancreatic tumor cell line Panc-1 is dependent on the expression of annexin A2 using siRNA-mediated specific knockdown of annexin A2 levels. We also demonstrate that treatment of HUVECs in culture with TM601 inhibits the activity of tissue plasminogen activator (tPA) present in the cell culture supernatants. Consistent with the inhibition of cell surface protease activity, we show that TM601 inhibits trans-well migration of both HUVEC and U373-MG glioma cells in response to PDGF-CC stimulation.
MATERIALS AND METHODS
Peptide Synthesis
TM601 is a chemically synthesized 36-amino acid peptide containing the same amino acid sequence and four intramolecular disulfide bonds as chlorotoxin (P45639). TM601 was synthesized by solid phase chemical synthesis, refolded, and lyophilized. TM602 is a synthetic TM601 peptide that is biotinylated at the N terminus at the time of chemical synthesis.
Cell Culture and Antibodies
U87-MG and U373-MG glioma cells were cultured in Eagle's minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 2 mm l-glutamine. U373-MG glioma cells (kindly provided by Dr. Darell D. Bigner, Duke University), A549 lung carcinoma cells, Panc-1 pancreatic carcinoma cells, and normal human dermal fibroblasts (Cambrex) were all cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated FBS. HUVECs were cultured in vascular endothelial cell medium supplemented with vascular endothelial cell growth supplement, 5% FBS, penicillin, and streptomycin (ScienCell). Normal human astrocytes were cultured in astrocyte medium supplemented with astrocyte growth supplement, 5% FBS, penicillin, and streptomycin (ScienCell). All of the cell lines were cultured in 37 °C humidified 5% CO2 incubators and unless otherwise noted obtained from ATCC.
Anti-TM601 antibodies were raised in rabbits immunized to a conjugate between TM701 and keyhole limpet hemocyanin. Tyrosine 29 in TM601 is replaced by a phenylalanine residue in TM701. The rabbit serum from the immunized mice was purified over a protein A column, followed by an affinity purification step using a TM601-Affi-gel 10 affinity column. The specificity of the TM601 antibody was tested with synthetic TM601 using a Western blot. Monoclonal anti-annexin A2 antibodies were purchased from Zymed Laboratories Inc. All other chemicals were purchased from Sigma.
99mTc-TM601 Surface Binding Affinity Assay
The N-terminal hydroxysuccinaimide (NHS) ester of [99mTcMAS3] with high specific activity and high radiolabel purity (>99%) was prepared in Me2SO using a 20-min protocol as described previously (12). Radiolabeled TM601 monomer was prepared at 25 μm by resuspending lyophilized TM601 in Me2SO followed by conjugation of 50 μl of TM601 with 500 μl of [99mTcMAS3]NHS (4mCi, 0.36nmol) and 5 μl of triethylamine (4 m). Constant stirring was performed at room temperature for 1 h. The radiolabeled [99mTcMAS3]TM601 was purified by reverse phase high pressure liquid chromatography and concentrated to 1mCi/ml using 3,000 molecular weight cut-off Vivaspin cartridges (Sartorius Stedim Biotech).
For the surface binding assay, the cell lines cultured as described above were split into 96-well filter plates (Millipore). Each well in the 96-well plate was plated with 4 × 104 cells and grown for 48 h. At the time of the binding assay, the cell number/well was measured by staining one well/cell line. To determine absolute affinity, homologous competition assay was performed using radiolabeled TM601 as tracer and TM601 monomer as test compound. Live cell binding was performed at 4 °C to prevent internalization because of constitutive endocytosis. The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and incubated with 0.5 mCi of the radiolabeled TM601 in the presence or absence of unlabeled TM601 for 20 min at 4 °C. The cells were washed three times with PBS, and the well contents were transferred directly into plastic tubes for counting using a modified 96-well puncher with disposable punch tips. The data are expressed as the means ± S.D. percent uptake ratio of triplicate measurements at each concentration, and curve fit was performed using Prism version 4.0 software.
Affinity Pulldowns and Mass Spectrometry
Cells in culture were harvested with trypsin that was inactivated using trypsin-neutralizing solution (Invitrogen). The cell pellets were washed once with ice-cold PBS and resuspended in 5 ml ice-cold PBS. The cells were incubated with or without either TM601 or TM602 (for affinity pulldowns) by adding 50 μl of 2 mg/ml of stock/ml of cells at 4 °C for 1 h with constant rotation. The cells were pelleted, washed once with ice-cold BuPHTM PBS (Pierce), and incubated with bis[sulfosuccinimidyl] suberate (BS3) (Pierce) cross-linking agent for 30 min at 4 °C. Cross-linker was neutralized by adding quenching solution (Pierce) and incubating for 10 min on ice. The cells were collected, and the lysates were prepared using lysis buffer containing 10 mm Tris (pH 7.5), 100 mm NaCl, 1 mm NaF, 20 mm Na2P4O7, 2 mm Na3VO4, 10% glycerol, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, and protease inhibitor mixture (Sigma). The lysates were freeze-thawed and cleared by centrifuging at 14,000 rpm for 20 min at 4 °C. The supernatants were assayed for protein concentration using the Bradford assay. For the pulldown assay, equal amounts of protein from lysates of treated or untreated (±TM602) samples were incubated with neutrAvidinTM (Invitrogen) beads overnight at 4 °C with continuous rotation. The beads were washed with lysis buffer and boiled in Laemmli buffer (Bio-Rad), and the samples were separated by SDS-PAGE. The gels were processed for either silver staining using a SilverQuestTM staining kit (Invitrogen) using the manufacturer's instructions or for Western blotting as described below. Silver-stained bands were cut from the gels, washed twice with 50% acetonitrile, and processed for liquid chromatography-tandem mass spectrometry analysis at the mass spectrometry core facility at Beth Israel Deaconess Medical Center (Boston, MA).
Surface Biotinylation
The cells were cultured in T75 tissue culture flasks and grown to confluence. The cells were washed with ice-cold BuPHTM PBS (Pierce), and surface proteins were biotinylated using EZ-link-sulfo-NHS-SS-Biotin (part of a Pierce cell surface protein isolation kit) by incubation at 4 °C for 30 min. After biotinylation, the cell pellets were lysed in modified radioimmune precipitation assay buffer containing 50 mm Tris (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mm Na3VO4, 10 mm NaF, 10% glycerol, and protease inhibitor mixture (Sigma). The lysates were frozen, thawed on ice, and cleared by centrifuging at 14,000 rpm for 20 min at 4 °C. The cleared lysates were quantified using the Bradford assay. Equal protein quantities of each lysate were added to neutrAvidinTM beads and incubated for 1 h at room temperature. The beads were washed with lysis buffer, boiled in Laemmli buffer, and separated by SDS-PAGE. The level of cell surface annexin A2 was determined by Western blot as described below.
Western Blotting
The gels were transferred using a semi-dry method using the Trans-blot SD semi-dry electrophoretic transfer cell (Bio-Rad) in transfer buffer. The blots were washed once in TBST containing 1× Tris-buffered saline (Bio-Rad) and 0.1% Tween 20 (Sigma) and incubated for 1 h at room temperature in 5% (w/v) nonfat dry milk in TBST. The blots were washed in TBST and immunoblotted in 5% (w/v) bovine serum albumin (Sigma) with either affinity-purified anti-TM601 (1:3000, 1 h at room temperature) or anti-annexin A2 (1:200, overnight 4 °C) antibody. The blots were washed with TBST and incubated with appropriate secondary antibody. After washes, the blots were developed using LumiGLO® chemiluminescent reagent (Cell Signaling Technology).
Annexin A2 siRNA Knockdown
2 × 105 Panc-1 cells were plated in 6-well plates such that they were 30–50% confluent at the time of transfection. The transfection was done using LipofectamineTM RNAiMAX and Opti-MEM® reduced serum medium as per the manufacturer's instructions (Invitrogen). The cultures were incubated for 4 h with 30 pmol of each siRNA (set of three human annexin A2-specific stealth siRNA; Invitrogen). The transfection medium was removed after the 4-h incubation and replaced with fresh medium. The cultures were incubated overnight, then trypsinized, and plated in 96-well plates for the 99mTc-TM601 surface binding assay or used to prepare lysates for annexin A2 Western blot analysis as described above.
tPA Activity Assay
HUVECs were plated in 60-mm dishes at 0.6 × 106 cells/dish and cultured overnight. The cells were washed once with serum-free basal medium and serum-starved overnight in basal medium. The serum-free medium was then replaced by 760 μl of serum-free medium with or without TM601 at 50 μm. The cells were incubated for 22 h at 37 °C in humidified CO2 incubator. The supernatants were collected and centrifuged at 3000 rpm at 4 °C to remove any cell debris using a tabletop centrifuge. The supernatants were transferred to fresh tubes, snap frozen in liquid nitrogen, and stored at −80 °C. tPA levels and activity were measured using colorimetric enzyme-linked immunosorbent assay kits purchased from AssayPro as per the manufacturer's instructions. The activity is measured as the ability of tPA present in the supernatants to activate plasminogen in an indirect assay that measures the amount of plasmin generated using a highly specific plasmin substrate that releases a yellow para-nitroaniline chromophore. The chromophore was quantified at 415 nm using an enzyme-linked immunosorbent assay plate reader.
Trans-well Migration Assay
HUVEC and U373-MG cells were cultured as described above. The cells were washed once in serum-free basal medium and serum-starved overnight. The cells were harvested with trypsin, and trypsinization was stopped with trypsin-neutralizing solution supplemented with 0.2% methylcellulose (Sigma). The cells were then washed twice with Hanks' balanced salt solution containing 0.2% methylcellulose. The cells were resuspended at 2 × 106/ml in serum-free basal medium containing 0.2% methylcellulose. The cells were then incubated with or without TM601 (final concentration, 50 μm) for 30 min at room temperature. 5 × 104 cells ± TM601 were then transferred to the upper chamber of the trans-well (COSTAR; 0.8-micron pore size). Trans-wells were placed in 24-well plates containing either basal medium with 0.4% FBS or basal medium with 0.4% FBS plus 50 ng/ml PDGF-CC (R & D Systems). Trans-well chambers were incubated at 37 °C in humidified CO2 incubators for 22 h. The nonmigrated cells remaining on the upper side of the filter were removed using a cotton swab. The migrated cells on the lower side were fixed and stained using the Diff-Quik kit (Dade-Behring). Migrated cells were then quantified by counting five random fields using a hemocytometer.
RESULTS
TM601 Binds the Cell Surface of Tumor and Normal Human Vascular Endothelial Cell Lines with Varying Affinities
TM601 has been shown to specifically bind several different tumor cell lines in vitro. Here we quantified the relative binding of TM601 to the cell surface of different tumor cell lines to determine the number of binding sites/cell and the affinity of TM601 for each cell line. For this purpose the Technetium-99m labeled TM601 (99mTc-TM601) was incubated with cells in the presence or absence of unlabeled TM601 competitor. The tumor cell lines tested had variable affinity and could be categorized as having higher (U87-MG, A549) or lower (Panc-1) affinity for TM601 (Fig. 1, A–C). The tumor cell lines also showed variable Bmax levels, indicating that the number of cell surface binding sites for TM601 varies between the different cell lines (Fig. 1, A–C, and data not shown). Consistent with previous results (11), in this assay proliferating HUVECs were the only normal cell line tested to show cell surface binding of 99mTc-TM601, whereas both dermal fibroblast and astrocytes showed little binding (Fig. 1, D–F). TM601 binds the cell surface of HUVECs with an affinity comparable with the binding of TM601 to Panc-1 pancreatic carcinoma cells (Fig. 1, C and D).
FIGURE 1.

Specific binding of TM601 to the cell surface of tumor and vascular endothelial cells. Cell surface binding of TM601 was quantified by measuring the amount of technetium-99m-labeled TM601 bound to the cell surface at 4 °C in the presence of unlabeled TM601 monomer used as competitor. TM601 binds the surface of multiple tumor cells with a high affinity for U87-MG glioma (A) and A549 lung carcinoma (B) and lower affinity for Panc-1 pancreatic carcinoma (C) cell lines. TM601 binds HUVECs (D) in culture with an affinity comparable with Panc-1 cells and shows little surface binding to either normal human astrocytes (E) or normal human dermal fibroblasts (F).
TM601 Associates with a Specific Subset of Cell Surface Proteins on Both Tumor and HUVEC Cells in Culture
Initial studies from the Sontheimer laboratory identified the MMP-2·membrane type metalloprotease-1 macromolecular complex and the CLC-3 chloride ion channel as targets for chlorotoxin on the surface of human glioma cells (7, 8). Subsequent studies by the Olson laboratory demonstrated that MMP-2 also facilitates the binding of chlorotoxin to MCF-7 breast cancer cells (10). However, the studies from the Olson laboratory could not demonstrate binding between chlorotoxin and MMP-2 (10). Here we have used a biochemical approach to identify the target for TM601 on the surface of tumor and endothelial cells. TM601 was incubated with U87-MG glioma cells followed by chemical cross-linking with a membrane-impermeable cross-linking agent. The lysates from treated and untreated cells (±TM601) were separated by SDS-PAGE and immunoblotted with anti-TM601 antibodies to reveal a specific subset of cross-linked gel-shifted proteins migrating at a higher molecular mass than the native TM601 peptide, which has a mass of 3950 Da (Fig. 2A). Furthermore, with the exception of a 60-kDa band, the gel-shifted proteins were specific to the presence of TM601 followed by the cross-linking agent (Fig. 2A, lane 4) and were absent when either TM601 or cross-linker alone was added to the cells (Fig. 2A, lanes 2 and 3, respectively). To isolate these proteins for subsequent identification, a pulldown assay was performed using an N-terminally biotinylated form of TM601 (TM602). An anti-TM601 Western blot of the TM602 cross-linked samples from U87-MG cells following neutrAvidinTM pulldown and separation by SDS-PAGE revealed a pattern of bands identical to that associating with TM601 (data not shown). Additionally, the pattern of bands associating with TM602 was similar in multiple tumor cell types (including U87-MG, A549, and Panc-1) and HUVECs (Fig. 2, B–E). The results support the presence of common molecular target(s) for TM601 on the surface of these different cell lines.
FIGURE 2.

TM601 associates with a specific subset of cell surface proteins in both tumor and HUVECs. A, lysates from treated and untreated (±TM601 at 4 °C) U87-MG glioma cells chemically cross-linked with a membrane-impermeable cross-linking agent (BS3) were separated by SDS-PAGE and immunoblotted with anti-TM601 antibodies (A). Comparison of lane 2 (TM601 without cross-linking) and lane 3 (BS3 cross-linking without TM601) with lane 4 (TM601 followed by BS3 cross-linking) reveals a specific subset of cross-linked gel-shifted cell surface proteins that associate with TM601. B–E, the associating proteins can be isolated using TM602 (biotinylated TM601) in a pulldown assay with neutrAvidinTM beads. Anti-TM601 Western blot of neutrAvidinTM pulldowns from lysates of TM602 treated (+) and untreated cells (−) chemically cross-linked with BS3 shows a similar subset of surface proteins specifically associating with TM602 on A549 (B), Panc-1 (C), and U87-MG (D) tumor cell lines and HUVECs (E).
Mass Spectrometric Identification of TM602 Binding Partner Expressed on Human Tumor and Vascular Endothelial Cells
To identify the cell surface proteins binding the TM601 peptide, U87-MG cells were incubated with or without TM602 at 4 °C, followed by BS3 cross-linking. The lysates were prepared as described, and equal protein amounts were used for neutrAvidinTM pulldown. TM602 cross-linked proteins were separated by SDS-PAGE and visualized by silver staining (Fig. 3). The indicated bands (Fig. 3, filled triangles) were selected on account of co-migration with TM601 containing a complex detected by anti-TM601 immunoblot (Fig. 2). The identified bands were isolated and analyzed by mass spectrometry. The protocol was repeated for additional tumor cell lines, A549 and Panc-1 cells, and for HUVECs (data not shown). Annexin A2 was selected as a potential target for surface binding of TM601 on the basis of high abundance (high average total ion current value) and high specificity because annexin A2 was present only in the test sample containing TM602, but not in protein bands of similar molecular weight in the control reaction lacking TM602 (Fig. 3). Annexin A2 protein was identified in mass spectrometric results from all of the tested cell lines cross-linked with TM602. Additionally, annexin A2-derived peptides were detected in higher molecular mass bands of (>40 kDa, which is the combined molecular mass of TM601 and annexin A2) indicated in Fig. 3, implying the formation of a macromolecular complex.
FIGURE 3.
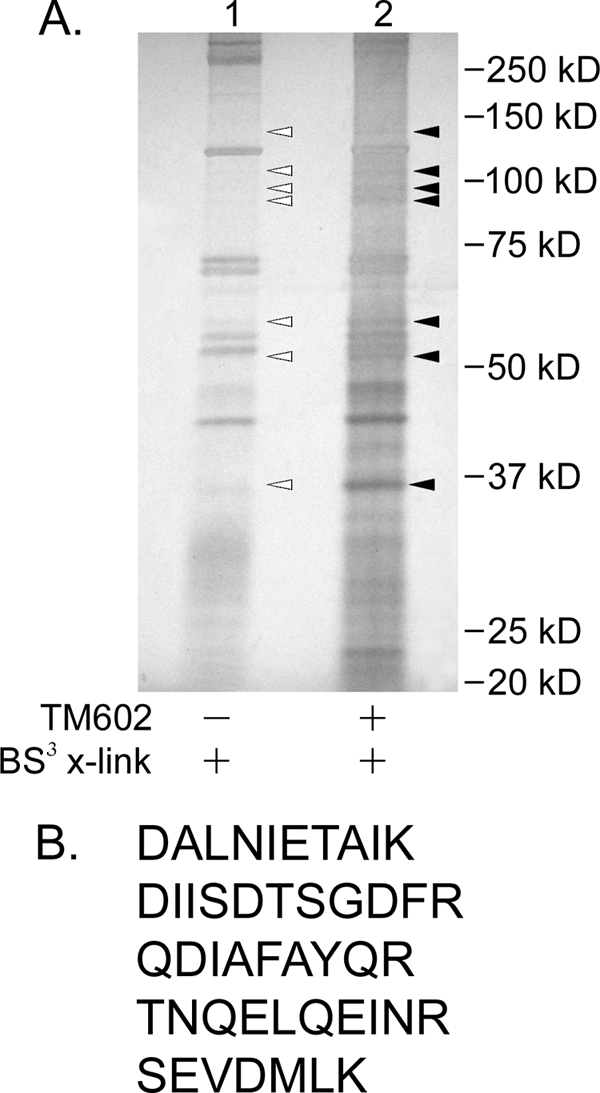
Mass spectrometric identification of TM602 binding partner expressed on tumor cell surface. A, lysates from treated and untreated (±TM602 at 4 °C) U87-MG glioma cells chemically cross-linked with membrane-impermeable cross-linking agent (BS3) and isolated by neutrAvidinTM pulldown were separated by SDS-PAGE and silver-stained using the SilverQuestTM staining kit. The indicated bands were extracted from control (lane 1, open triangles) and TM602 pulldown samples (lane 2, filled triangles) and analyzed by mass spectrometry to identify isolated protein sequences. As indicated, comparisons were made between mass spectrometric results of gel slices of identical molecular mass (kDa) between the two lanes to identify proteins specifically enriched in TM602-containing lane. B, peptides identified exclusively in lane 2 that have sequence identity to human annexin A2.
TM602 Binds Annexin A2 Expressed on the Surface of Multiple Tumor Cell Lines and Vascular Endothelial Cells
As a first step toward the validation of annexin A2 as a cell surface target for TM601, we evaluated the levels of annexin A2 expressed on the surface of multiple tumor cell lines and vascular endothelial cell using the surface biotinylation assay. The results confirmed the presence of annexin A2 at the cell surface of A549, Panc-1, multiple human glioma cell lines, and vascular endothelial cells (Fig. 4, A and B, and data not shown). The results also revealed sensitivity of surface annexin A2 in endothelial cells to trypsinization (Fig. 4B, lanes 3 and 4). To validate the association of surface annexin A2 with TM602, trypsinized A549 and Panc-1 cells were incubated with increasing concentrations of TM602 at 4 °C, followed by BS3 cross-linking and neutrAvidinTM pulldown from lysates that were then immunoblotted with anti-annexin A2 antibodies. The results show that surface annexin A2 from both higher affinity A549 and lower affinity Panc-1 cells associates with TM602 (Fig. 4C, lanes 1–8). Similarly, TM602 was incubated with and cross-linked to the surface of plate-bound HUVECs at 4 °C followed by neutrAvidinTM affinity pulldown from lysates that were then Western blotted for detection of annexin A2. The result confirms the interaction between surface annexin A2 and TM602 on vascular endothelial cells (Fig. 4D, lanes 1 and 2). In the case of HUVECs, affinity binding and cross-linking of TM602 was done using plate-bound cells, because the sensitivity of annexin A2 to trypsin hampered detection using Western blotting. This finding supports the aforementioned mass spectrometric result that was sufficiently sensitive to detect annexin A2 as a specific binding partner for TM602 in pulldown assays using trypsinized HUVECs. The blots in Fig. 4 (C and D) were stripped and reprobed with anti-TM601 antibodies to demonstrate that the annexin A2 protein co-migrates with the cross-linked TM602 (data not shown).
FIGURE 4.

TM602 binds annexin A2 expressed on the surface of multiple tumor and vascular endothelial cells. Confluent monolayers of A549, Panc-1, U87-MG, and HUVECs treated with or without trypsin were surface-biotinylated, and biotinylated proteins were isolated using neutrAvidinTM beads. The levels of annexin A2 expressed on the surface of tumor cell lines and HUVECs and the sensitivity to trypsin were determined using an anti-annexin A2 Western blot. A, annexin A2 levels at the cell surface and in total cell lysates for plate-bound (lanes 1, 3, 5, and 7) and trypsinized (lanes 2, 4, 6, and 8) A549 (lanes 1, 2, 5, and 6) and Panc-1 (lanes 3, 4, 7, and 8) cells. B, annexin A2 levels at the cell surface and in total cell lysates for plate-bound (lanes 1, 3, 5, and 7) and trypsinized (lanes 2, 4, 6, and 8) U87-MG (lanes 1, 2, 5, and 6) glioma and HUVECs (lanes 3, 4, 7, and 8). Annexin A2 is expressed at the cell surface in all the aforementioned cell lines. In HUVECs, surface annexin A2 is sensitive to trypsinization (B, lanes 3 and 4). C and D, the association of TM602 with cell surface annexin A2 was validated using an anti-annexin A2 Western blot of neutrAvidinTM pulldowns of lysates from A549, Panc-1, and HUVECs following TM602 surface cross-linking. Annexin A2 specifically associates with TM602 at the cell surface of A549 and Panc-1 tumor cells (C) as well as HUVECs (D).
Annexin A2 Mediates Cell Surface Binding of TM601 to Tumor Cells in Vitro
The functional importance of the interaction between surface annexin A2 and TM602 was tested using siRNA knockdown and surface binding assays. The pancreatic carcinoma cell line Panc-1 was selected on account of efficient knockdown of annexin A2 levels. Panc-1 cells were either mock transfected or transfected with annexin A2-specific siRNA. The specific down-regulation of annexin A2 levels was confirmed by annexin A2 Western blot (Fig. 5A). The surface binding of TM601 to mock transfected or annexin A2 knockdown Panc-1 cells was then tested using the 99mTc-TM601 surface binding assay. The knockdown of annexin A2 resulted in a complete loss of cell surface binding of 99mTc-TM601 to Panc-1 cells (Fig. 5, B and C). These results demonstrate that annexin A2 is required for surface binding of TM601 to Panc-1 cells.
FIGURE 5.

Annexin A2 mediates cell surface binding of TM601 to Panc-1 pancreatic carcinoma cell line. Functional importance of the interaction between annexin A2 and TM601 was tested using a siRNA knockdown approach in Panc-1 carcinoma cells. A, Western blot using anti-annexin A2 antibody shows that the level of annexin A2 in untransfected Panc-1 cells was comparable with that of mock-transfected cells (lanes 1 and 2). Annexin A2 siRNA transfected cells showed a significant reduction of annexin A2 levels at 48 h post-transfection by annexin A2 Western blot of total cell lysates. As shown in A, the level of annexin A2 is similar in untransfected and mock-transfected cells (lanes 1 and 2) and is significantly decreased in annexin A2 siRNA treated cells (lane 3). Control and annexin A2 siRNA-treated Panc-1 cells were also tested for surface 99mTc-TM601 binding assays. B, competitive binding of 99mTc-TM601 in untransfected Panc-1 cells. C, competitive binding of 99mTc-TM601 to surface of Panc-1 cells transfected with annexin A2 siRNA. Annexin A2 siRNA-mediated knockdown in Panc-1 cells abolished the surface binding of TM601.
TM601 Inhibits VEGF Stimulated tPA Activity in Human Endothelial Cell Supernatants
Recently, we demonstrated that TM601 can inhibit neovascularization stimulated by both VEGF and bFGF in mouse Matrigel plug assays (11). Similarly, previous studies using annexin A2 knock-out mice demonstrated a requirement of annexin A2 for neovascularization of Matrigel plugs containing either VEGF-A or bFGF (13). Early studies also identified annexin A2 as a receptor for plasminogen and tPA on vascular endothelial cells, resulting in the accelerated conversion of plasminogen to plasmin (14). Finally, studies with knock-out mice revealed that annexin A2 deficiency severely inhibits tPA-dependent vascular endothelial cell plasminogen activation (13). Hence, we tested the effect of TM601 on HUVEC tPA activity in response to VEGF or bFGF stimulation. The results shown in Fig. 6 demonstrate that TM601 treatment of HUVECs significantly inhibits both VEGF and bFGF-stimulated tPA activity.
FIGURE 6.

TM601 inhibits VEGF-stimulated tPA activity in human vascular endothelial cell supernatants. The effect of TM601 on VEGF- and bFGF-stimulated activation of secreted tPA in HUVECs was measured. Both VEGF- and bFGF-stimulated tPA activity in serum-starved HUVECs and TM601 treatment significantly decreased the amount of active tPA present in the supernatants of vascular endothelial cells treated with either VEGF or bFGF. *, p < 0.05. Bar, S.D.
TM601 Inhibits PDGF-CC-induced U373-MG Glioma and HUVEC Cell Migration in Trans-well Assays
Recent studies identified PDGF-CC, a novel member of the PDGF family of growth factors, as a specific substrate for proteolytic processing and activation by tPA (15). PDGF-CC was also identified as a mediator of tumor-associated fibroblast-induced angiogenesis (16). We tested the effects of TM601 on PDGF-CC-induced migration of vascular endothelial cells using a trans-well assay. As shown in Fig. 7A, recombinant PDGF-CC strongly stimulates the migration of HUVEC and U373-MG glioma cells, and TM601 treatment of both cell types significantly inhibits PDGF-CC-stimulated migration. We also demonstrate that knockdown of annexin A2 in U373-MG glioma cells has an effect comparable with TM601 treatment of wild-type cells on PDGF-CC-stimulated migration (Fig. 7B). Furthermore, treatment of annexin knockdown cells with a lower dose of TM601 (25 μm) as compared with wild-type cells completely abolished migration, most likely because of inhibition of cells still expressing annexin A2 protein. These results were obtained using recombinant PDGF-CC corresponding to the tPA-activated truncated form of full-length PDGF-CC. The results support the hypothesis that TM601 can inhibit both the activated form of growth factors such as PDGF-CC and the further activation of full-length PDGF-CC, a key player in VEGF inhibitor-resistant cancers.
FIGURE 7.

TM601 inhibits PDGF-CC-induced U373-MG glioma and HUVEC migration in trans-well chamber assays. A, in a trans-well assay using HUVEC or U373-MG cells, PDGF-CC strongly stimulates cell migration, and TM601 significantly inhibits the PDGF-CC-stimulated migration of both vascular endothelial cells and U373-MG glioma cells. *, p < 0.05. Bar, S.D. B, functional importance of annexin A2 in PDGF-CC-stimulated glioma migration was tested using an annexin A2 siRNA knockdown approach. Mock-transfected wild-type U373-MG and annexin A2 knockdown U373-MG glioma cells were used in a trans-well assay testing migration stimulated by PDGF-CC. Wild-type cells demonstrated significant migration stimulated by PDGF-CC that was inhibited by TM601 treatment. Annexin A2 knockdown cells demonstrated very weak migration stimulated by PDGF-CC that was completely abolished by a lower dose of TM601 (25 μm) as compared with wild-type cells. C, Western blot using anti-annexin A2 antibody demonstrates the levels of annexin A2 after annexin A2 siRNA treatment.
DISCUSSION
TM601 is a synthetic chlorotoxin peptide that preferentially binds several tumor cell types both in vitro and in ongoing clinical trials (2). In the current study, we identify annexin A2 as a novel target for TM601 expressed on the cell surface of multiple human tumor cell types and vascular endothelial cells in culture. Annexin A2 was validated as a specific binding partner of TM601 in diverse tumor cell lines, including U87-MG glioma, A549 lung carcinoma, and Panc-1 pancreatic carcinoma cells using Western blot analysis. A knockdown approach using siRNA demonstrated that annexin A2 is required for surface binding of TM601 to Panc-1 cells. Identification of annexin A2 as a specific target for TM601 is consistent with the tumor-targeting effects of TM601 and the corresponding overexpression of annexin A2 in multiple human cancers including high grade glioma (17–20), colorectal cancer (21), pancreatic cancer (22), hepatocellular carcinoma (23, 24), renal cell carcinoma (25), squamous cell carcinoma (26), prostate cancer (27), and lung cancer (28). Annexin A2 overexpression has been correlated with poor prognosis in colorectal cancer (21) and recurrence after surgery in pancreatic cancer patients (29). Following intravenous administration, tumor-specific uptake of 131I-TM601 has been observed in many of these tumor types tested in clinical trials, including glioma, metastatic melanoma, colorectal, pancreatic, prostate, and non-small cell lung carcinoma (4).
At a functional level, studies have demonstrated both the requirement of annexin A2 for glioma migration and the inhibitory effect of chlorotoxin/TM601 on glioma migration. Specifically, siRNA knockdown of annexin A2 in glioma cells has been shown to decrease the in vitro migration of both U87-MG and U373-MG cells (30). Conversely, chlorotoxin treatment of D54-MG, U251-MG (6, 8),and TM601 treatment of U373-MG glioma cells (Fig. 7) inhibits trans-well migration of the glioma cells. In U373-MG cells we have also demonstrated that annexin A2 knockdown inhibits trans-well migration to levels comparable with that of TM601 treatment of U373-MG cells at concentrations between 25 and 50 μm. Taken together these studies are consistent with the role of annexin A2 as a target for TM601 in glioma cells that might explain the inhibitory effect of TM601 on glioma cell migration.
The TM601 peptide was also shown to inhibit vascular endothelial cell functions in vitro and have wide ranging anti-angiogenic effects (11). Annexin A2 is also expressed on the surface of vascular endothelial cells and has been shown to play a significant role in the regulation of tPA activity, plasminogen activation, and angiogenic functions (13, 14). Consistent with these reports, annexin A2 was specifically detected by Western blotting of neutrAvidinTM pulldowns of biotinylated TM601 cross-linked to the surface of growing HUVECs (Fig. 4D, lanes 1 and 2). The relevance of the interaction between the vascular endothelial cell annexin A2 and TM601 is further supported by the significant inhibitory affect of TM601 on tPA activity in HUVECs stimulated by both VEGF and bFGF (Fig. 6).
In contrast to plasmin, which is a broad spectrum protease (31), tPA is a secreted protease with highly restricted substrate specificity that to date includes plasminogen, N-methyl-d-aspartate receptor, and the most recently identified PDGF-CC growth factor (15). tPA cleaves and activates the latent PDGF-CC growth factor that is a novel member of the PDGF family of growth factors (15, 32). Expression of PDGF-CC has been demonstrated in multiple glioblastoma cell lines and primary glioblastoma multiforme (33). Several recent studies have demonstrated variable effects of PDGF-CC, including the recruitment of tumor-associated fibroblasts by melanoma cells (34), recruitment of vascular endothelial cells by tumor-associated fibroblasts (16), and recruitment of pericytes by tumor cells (35). Interestingly, PDGF-CC signaling and migration has been implicated as a mechanism for tumors becoming refractory to anti-VEGF treatment (16, 35). PDGF-CC-stimulated angiogenic response is indistinguishable from PDGF-AB-induced angiogenesis and functions via the activation of both PDGF-αα and PDGF-αβ receptor (36). TM601 can inhibit PDGF-AB-stimulated angiogenesis in the chicken chorioallantoic membrane assay (11). Consistent with these reports we found that TM601 inhibits both recombinant PDGF-CC- and PDGF-AB-stimulated trans-well migration of both HUVEC and glioma cells (Fig. 7 and data not shown). Hence, our results support the hypothesis that TM601 treatment can inhibit both the activation of full-length PDGF-CC by inhibiting tPA-induced proteolytic cleavage of PDGF-CC and inhibit migration stimulated by activated PDGF-CC. It is likely that the inhibitory effect of TM601 on growth factor-stimulated invasion is due to the inhibition of activation the broad spectrum protease, plasminogen functioning downstream of tPA activation. Taken together, data presented in this paper further support a role for TM601 as an anti-angiogenic therapeutic agent potentially via mechanisms involving binding to annexin A2. The results are consistent with previous studies that suggest a role for annexin A2 as a therapeutic target in angiogenesis and tumor progression (37).
In conclusion, the results presented in this report identify annexin A2 as a novel target for TM601 on tumor and vascular endothelial cells. It is possible that annexin A2 binding to TM601 might mediate the mechanistic effects of TM601 on tumor and vascular endothelial cell migration and angiogenesis via modulation of tPA and plasmin activity. Activated plasmin can mediate the subsequent cleavage and activation of pro-MMP-2 (38, 39), a key protease involved in migration and invasion. Indeed, the importance of annexin A2 to tumor-specific binding was clearly demonstrated using siRNA knockdown of annexin A2 in Panc-1 tumor cells, which resulted in the elimination of tumor cell binding (Fig. 5). However, it is possible that TM601 binds additional proteins in a macromolecular cell surface complex that may also contribute to the tumor and endothelial effects of TM601. For instance, although we and others (10) have been unable to detect an interaction between MMP-2 and TM601/chlorotoxin, surface cross-linking followed by affinity pulldown assays in our laboratory do support an interaction between membrane type metalloprotease-1 and biotinylated TM601 on HUVECs (data not shown). Future studies are being directed toward better understanding the additional components of the macromolecular target of TM601 on tumor and vascular endothelial cells and their role in mediating TM601 functions as a tumor-targeting and anti-angiogenic agent. Because TM601 has specific tumor-targeting properties, these studies will also further the identification of tumor biomarkers and their use in clinical applications.
Acknowledgments
We thank Dr. Alison O'Neill, Robert Radie, and Susan Stewart for thoughtful comments and suggestions on the manuscript. We also thank Dr. Lewis Cantley for many helpful discussions during the course of this work. Lastly, we acknowledge Michael Egan in memoriam for enthusiastic support for the project.
Footnotes
- MMP
- matrix metalloprotease
- VEGF
- vascular endothelial growth factor
- tPA
- tissue plasminogen activator
- PDGF-CC
- platelet-derived growth factor form C
- HUVEC
- human umbilical vein endothelial cell
- bFGF
- basic fibroblast growth factor
- siRNA
- small interfering RNA
- FBS
- fetal bovine serum
- NHS
- N-terminal hydroxysuccinaimide
- PBS
- phosphate-buffered saline
- BS3
- bis[sulfosuccinimidyl] suberate.
REFERENCES
- 1.DeBin J. A., Maggio J. E., Strichartz G. R. (1993) Am. J. Physiol. 264, C361–C369 [DOI] [PubMed] [Google Scholar]
- 2.Mamelak A. N., Jacoby D. B. (2007) Expert Opin. Drug Deliv. 4, 175–186 [DOI] [PubMed] [Google Scholar]
- 3.Mamelak A. N., Rosenfeld S., Bucholz R., Raubitschek A., Nabors L. B., Fiveash J. B., Shen S., Khazaeli M. B., Colcher D., Liu A., Osman M., Guthrie B., Schade-Bijur S., Hablitz D. M., Alvarez V. L., Gonda M. A. (2006) J. Clin. Oncol. 24, 3644–3650 [DOI] [PubMed] [Google Scholar]
- 4.Gibbin T. E., Senzer N., Raizer J. J., Shen J., Nabors L. B., Wiranowska M., Fiveash J. B. (2009) J. Clin. Oncol. 27, e14507 [Google Scholar]
- 5.Ullrich N., Gillespie G. Y., Sontheimer H. (1995) Neuroreport 7, 343–347 [PubMed] [Google Scholar]
- 6.Soroceanu L., Manning T. J., Jr., Sontheimer H. (1999) J. Neurosci. 19, 5942–5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McFerrin M. B., Sontheimer H. (2006) Neuron Glia Biol. 2, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deshane J., Garner C. C., Sontheimer H. (2003) J. Biol. Chem. 278, 4135–4144 [DOI] [PubMed] [Google Scholar]
- 9.Lyons S. A., O'Neal J., Sontheimer H. (2002) Glia 39, 162–173 [DOI] [PubMed] [Google Scholar]
- 10.Veiseh M., Gabikian P., Bahrami S. B., Veiseh O., Zhang M., Hackman R. C., Ravanpay A. C., Stroud M. R., Kusuma Y., Hansen S. J., Kwok D., Munoz N. M., Sze R. W., Grady W. M., Greenberg N. M., Ellenbogen R. G., Olson J. M. (2007) Cancer Res. 67, 6882–6888 [DOI] [PubMed] [Google Scholar]
- 11.Jacoby D. B., Dyskin E., Yalcin M., Kesavan K., Dahlberg W., Ratliff J., Johnson E. W., Mousa S. A. (2010) Anticancer Res. 30, in press [PubMed] [Google Scholar]
- 12.Misra P., Humblet V., Pannier N., Maison W., Frangioni J. V. (2007) J. Nucl. Med. 48, 1379–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ling Q., Jacovina A. T., Deora A., Febbraio M., Simantov R., Silverstein R. L., Hempstead B., Mark W. H., Hajjar K. A. (2004) J. Clin. Invest. 113, 38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hajjar K. A., Jacovina A. T., Chacko J. (1994) J. Biol. Chem. 269, 21191–21197 [PubMed] [Google Scholar]
- 15.Fredriksson L., Li H., Fieber C., Li X., Eriksson U. (2004) EMBO J. 23, 3793–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford Y., Kasman I., Yu L., Zhong C., Wu X., Modrusan Z., Kaminker J., Ferrara N. (2009) Cancer Cell 15, 21–34 [DOI] [PubMed] [Google Scholar]
- 17.Reeves S. A., Chavez-Kappel C., Davis R., Rosenblum M., Israel M. A. (1992) Cancer Res. 52, 6871–6876 [PubMed] [Google Scholar]
- 18.Roseman B. J., Bollen A., Hsu J., Lamborn K., Israel M. A. (1994) Oncol. Res. 6, 561–567 [PubMed] [Google Scholar]
- 19.Nygaard S. J., Haugland H. K., Kristoffersen E. K., Lund-Johansen M., Laerum O. D., Tysnes O. B. (1998) J. Neurooncol. 38, 11–18 [DOI] [PubMed] [Google Scholar]
- 20.Iwadate Y., Sakaida T., Hiwasa T., Nagai Y., Ishikura H., Takiguchi M., Yamaura A. (2004) Cancer Res. 64, 2496–2501 [DOI] [PubMed] [Google Scholar]
- 21.Emoto K., Yamada Y., Sawada H., Fujimoto H., Ueno M., Takayama T., Kamada K., Naito A., Hirao S., Nakajima Y. (2001) Cancer 92, 1419–1426 [DOI] [PubMed] [Google Scholar]
- 22.Díaz V. M., Hurtado M., Thomson T. M., Reventós J., Paciucci R. (2004) Gut 53, 993–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu G. R., Kim S. H., Park S. H., Cui X. D., Xu D. Y., Yu H. C., Cho B. H., Yeom Y. I., Kim S. S., Kim S. B., Chu I. S., Kim D. G. (2007) Exp. Mol. Med. 39, 641–652 [DOI] [PubMed] [Google Scholar]
- 24.Mohammad H. S., Kurokohchi K., Yoneyama H., Tokuda M., Morishita A., Jian G., Shi L., Murota M., Tani J., Kato K., Miyoshi H., Deguchi A., Himoto T., Usuki H., Wakabayashi H., Izuishi K., Suzuki Y., Iwama H., Deguchi K., Uchida N., Sabet E. A., Arafa U. A., Hassan A. T., El-Sayed A. A., Masaki T. (2008) Int. J. Oncol. 33, 1157–1163 [PubMed] [Google Scholar]
- 25.Zimmermann U., Woenckhaus C., Pietschmann S., Junker H., Maile S., Schultz K., Protzel C., Giebel J. (2004) Virchows Arch. 445, 368–374 [DOI] [PubMed] [Google Scholar]
- 26.Zhong L. P., Wei K. J., Yang X., Zhang L., Zhou X. J., Pan H. Y., Li J., Chen W. T., Zhang Z. Y. (2009) Arch. Oral Biol. 54, 17–25 [DOI] [PubMed] [Google Scholar]
- 27.Inokuchi J., Narula N., Yee D. S., Skarecky D. W., Lau A., Ornstein D. K., Tyson D. R. (2009) Int. J. Cancer 124, 68–74 [DOI] [PubMed] [Google Scholar]
- 28.Brichory F. M., Misek D. E., Yim A. M., Krause M. C., Giordano T. J., Beer D. G., Hanash S. M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9824–9829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takano S., Togawa A., Yoshitomi H., Shida T., Kimura F., Shimizu H., Yoshidome H., Ohtsuka M., Kato A., Tomonaga T., Nomura F., Miyazaki M. (2008) Ann. Surg. Oncol. 15, 3157–3168 [DOI] [PubMed] [Google Scholar]
- 30.Tatenhorst L., Rescher U., Gerke V., Paulus W. (2006) Neuropathol. Appl. Neurobiol. 32, 271–277 [DOI] [PubMed] [Google Scholar]
- 31.Pepper M. S. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 1104–1117 [DOI] [PubMed] [Google Scholar]
- 32.Li X., Pontén A., Aase K., Karlsson L., Abramsson A., Uutela M., Bäckström G., Hellström M., Boström H., Li H., Soriano P., Betsholtz C., Heldin C. H., Alitalo K., Ostman A., Eriksson U. (2000) Nat. Cell Biol. 2, 302–309 [DOI] [PubMed] [Google Scholar]
- 33.Lokker N. A., Sullivan C. M., Hollenbach S. J., Israel M. A., Giese N. A. (2002) Cancer Res. 62, 3729–3735 [PubMed] [Google Scholar]
- 34.Anderberg C., Li H., Fredriksson L., Andrae J., Betsholtz C., Li X., Eriksson U., Pietras K. (2009) Cancer Res. 69, 369–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.di Tomaso E., London N., Fuja D., Logie J., Tyrrell J. A., Kamoun W., Munn L. L., Jain R. K. (2009) PLoS One 4, e5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao R., Bråkenhielm E., Li X., Pietras K., Widenfalk J., Ostman A., Eriksson U., Cao Y. (2002) FASEB J. 16, 1575–1583 [DOI] [PubMed] [Google Scholar]
- 37.Sharma M. C., Sharma M. (2007) Curr. Pharm. Des. 13, 3568–3575 [DOI] [PubMed] [Google Scholar]
- 38.He Y., Liu X. D., Chen Z. Y., Zhu J., Xiong Y., Li K., Dong J. H., Li X. (2007) Clin. Cancer Res. 13, 3115–3124 [DOI] [PubMed] [Google Scholar]
- 39.Le D. M., Besson A., Fogg D. K., Choi K. S., Waisman D. M., Goodyer C. G., Rewcastle B., Yong V. W. (2003) J. Neurosci. 23, 4034–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]