

Abstract
Venus is a yellow fluorescent protein that has been developed for its fast chromophore maturation rate and bright yellow fluorescence that is relatively insensitive to changes in pH and ion concentrations. Here, we present a detailed study of the stability and folding of Venus in the pH range from 6.0 to 8.0 using chemical denaturants and a variety of spectroscopic probes. By following hydrogen-deuterium exchange of 15N-labeled Venus using NMR spectroscopy over 13 months, residue-specific free energies of unfolding of some highly protected amide groups have been determined. Exchange rates of less than one per year are observed for some amide groups. A super-stable core is identified for Venus and compared with that previously reported for green fluorescent protein. These results are discussed in terms of the stability and folding of fluorescent proteins. Under mildly acidic conditions, we show that Venus undergoes a drastic decrease in yellow fluorescence at relatively low concentrations of guanidinium chloride. A detailed study of this effect establishes that it is due to pH-dependent, nonspecific interactions of ions with the protein. In contrast to previous studies on enhanced green fluorescence protein variant S65T/T203Y, which showed a specific halide ion-binding site, NMR chemical shift mapping shows no evidence for specific ion binding. Instead, chemical shift perturbations are observed for many residues primarily located in both lids of the β-barrel structure, which suggests that small scale structural rearrangements occur on increasing ionic strength under mildly acidic conditions and that these are propagated to the chromophore resulting in fluorescence quenching.
Keywords: Biophysics, Methods/Circular Dichroism CD, Methods/Fluorescence, Methods/NMR, Protein/Conformation, Protein/Denaturation, Protein/Folding, Protein/Thermodynamics
Introduction
Since the identification of the GFP7 from the jellyfish Aequorea victoria in the early 1990s, a large number of fluorescent proteins (FPs) have been isolated from natural sources, primarily from marine animals and corals (1, 2). These proteins have been developed further using protein engineering techniques to obtain a variety of FPs with broad fluorescence emission spectra, ranging from blue to far-red (3). The FP family of proteins is now arguably the most versatile and commonly used biomarker employed in a myriad of applications in the Life Sciences (1, 4–6). The impact that GFP and FPs have had is clearly illustrated by the award of the 2008 Nobel Prize in Chemistry to M. Chalfie, R. Y. Tsien, and O. Shimomura.
The maturation of GFP and its variants involves the initial folding of the polypeptide chain into a native-like conformation that then undergoes an autocatalytic cyclization and dehydration of the tripeptide encompassing residues 65–67. This is then followed by a rate-limiting oxidation step to form the chromophore whose color is determined by the side-chain composition of the tripeptide as well as its local environment (2, 7, 8). In the case of yellow fluorescent protein (YFP), a yellow variant of GFP, a number of mutations give rise to the red-shifted excitation and emission spectra (1). These include the following: (i) S65G, which prevents the formation of a hydrogen bond between Ser-65 and Glu-222, and (ii) T203Y, which produces additional polarizability around the chromophore, whereas the π-π stacking between Tyr-203 and the chromophore phenol ring reduces the excited state energy. Together with cyan fluorescent protein (a variant of GFP that contains a critical Y66W mutation that results in a chromophore with an indole rather than a phenol ring), YFP is often used as a fluorescence acceptor-donor pair in Förster resonance energy transfer measurements (1, 4, 5, 9). This technique is commonly used both in vitro and in vivo to monitor changes in the intermolecular distances between cyan fluorescent protein- and YFP-tagged interacting partners by measuring the relative intensities of both cyan and yellow fluorescence (9–11).
The spectroscopic properties of many GFP variants are known to be sensitive to pH (12) and halide ions (13). This property has been exploited in a number of applications where FPs have been developed to report on changes in the cellular environment (14, 15). However, for Förster resonance energy transfer-based studies where changes in fluorescence serve as the basis for monitoring changes in distance, the sensitivity of GFP and its variants to the chemical environment is highly undesirable. For this reason, a number of GFP variants have been engineered with reduced pH and halide ion sensitivity. Citrine is one such example; it contains a Q69M mutation in addition to the four mutations that were originally introduced to derive a yellow variant, enhanced YFP (S65G/V68L/S72A/T203Y). The Q69M mutation reduces the pH and halide sensitivity and has an increased yield in terms of protein expression (1). Recently, a new variant of YFP called Venus has been reported to have further reduced sensitivity with respect to proton and halide ion concentrations; additionally, it has a significantly accelerated rate of chromophore maturation (within minutes rather than hours) and the highest quantum yield among all FP variants at the time that it was first reported (16). Instead of the Q69M mutation used in Citrine, Venus contains the mutation F46L, which was found to accelerate the oxidation step during chromophore maturation, in addition to four others, F64L/M153T/V163A/S175G, that were found to facilitate folding at 37 °C.
Despite the apparent robustness and high stability of many FPs under native conditions, emerging data suggest that the folding of GFP and its variants is highly complex, with intermediate states populated under equilibrium and nonequilibrium conditions, and parallel pathways existing on the unfolding and refolding energy landscape (17–25). In addition, hysteresis observed in the unfolding/refolding curves of one particular variant, superfolder GFP, has been associated with the post-translational modification of chromophore formation (26, 27). However, there remains much that is not yet known on how this important class of proteins fold.
Here, we report a study on the stability and unfolding of Venus in which solution-state NMR spectroscopy was employed to extract the thermodynamic parameters of unfolding under native conditions by using hydrogen-deuterium exchange (HDX) experiments. In addition, fluorescence and far-UV CD spectroscopies were used to monitor the chemical denaturant-induced unfolding process under different conditions.
As a result of using the chemical denaturant guanidinium chloride (GdmCl) to study the stability and unfolding of Venus as a function of pH, we have also investigated the chloride ion sensitivity of the protein. In contrast to earlier reports (16), our results show that the fluorescence of Venus is sensitive to chloride ions at lower (pH 6.0) but not higher (pH 8.0) pH values. We report here a series of experiments using fluorescence and NMR spectroscopy to probe the nature of the chloride ion sensitivity, and we use both chloride and nitrate salts to establish that the effects are not due to the specific binding of halide ions to Venus, as has been reported for GFP (13), but can be attributed instead to many small scale structural rearrangements in Venus that occur on increasing the ionic strength of the solution.
Our results contribute to the general knowledge and understanding of the stability and folding of FPs, in addition to providing important information on how these large β-barrel structures react to changes in environmental conditions. We prove that Venus is not as insensitive to chloride ions as originally thought (16), and we show that small scale changes in structure can result in significant changes in the fluorescence properties of this FP.
EXPERIMENTAL PROCEDURES
Plasmid and Reagents
A plasmid containing the gene encoding Venus was a kind gift from Prof. Atsushi Miyawaki at the Brain Science Institute, RIKEN, Japan. The gene was subcloned into a pET21 vector with a hexahistidine tag at the N terminus to aid purification. Ultra-pure GdmCl was purchased from MP Biomedicals, Cambridge, UK. All other chemicals were of analytical grade and purchased from Sigma, BDH, or Melford Laboratories. Millipore-filtered, double-deionized water was used throughout. YFP (Venus) also known as SEYFP-F46L variant contains the mutations F46L, F64L, S65G, V68L, S72A, M153T, V163A, S175G, and T203Y when compared with wild-type GFP.
Protein Expression and Purification
A single colony of transformed Escherichia coli cells (BL21(DE3)) harboring the Venus expression vector was picked from 2×TY ampicillin plates and used to inoculate 10 ml of 2×TY media containing 0.1 mg ml−1 of ampicillin. After growth at 37 °C overnight, this pre-culture was used to inoculate 1 liter of growth culture of 2×TY media containing 0.1 mg ml−1 of ampicillin. The medium was incubated at 37 °C on a shaker until the cell density at A600 nm reached 0.5–0.6. The cells were induced with 1 ml of 1 m isopropyl 1-thio-β-d-galactopyranoside and left shaking for 4 h. The yellow fluorescent cells were harvested by centrifugation (SLC 4000 Sorvall rotor, at 5000 rpm, 8 °C for 10 min), and the cell pellet was resuspended in 50 mm Tris, 0.3 m NaCl (pH 8.0) and lysed by sonication (Misonix Inc) on ice for 5-min power cycles (15 s pulse on and 45 s pulse off) at power level 8 with a 9-mm probe. The lysate was centrifuged at 18,000 rpm (SS34 Sorvall rotor) at 8 °C for 45 min. The supernatant was pooled and loaded onto a HisTrap column (GE Healthcare) at a flow rate of 2 ml min−1. After washing with 25 ml of 50 mm Tris, 0.3 m NaCl (pH 8.0), Venus was eluted using a linear gradient to 50 mm Tris (pH 8.0), 0.3 m NaCl, 0.3 m imidazole run over 5 column volumes. The yellow fractions were pooled and concentrated (Vivaspins 20; Vivascience). The concentrated protein (<10 ml) was gel-filtrated on a HiLoad 26/60 Superdex G-75 column (Amersham Biosciences), which was pre-equilibrated in 50 mm Tris (pH 7.4) using a flow rate of 2.5 ml min−1. The purity of the YFP eluted was confirmed by SDS-PAGE, and its concentration was determined spectrophotometrically using an extinction coefficient of 23,380 m−1 cm−1 at 280 nm.
Equilibrium Unfolding Experiments
The exact concentration of GdmCl solution was determined from its refractive index using an Atago 1T refractometer (Bellingham + Stanley Ltd., Kent, UK). GdmCl solution was then mixed with the appropriate buffer solution to give various concentrations of GdmCl (0–7 m) in 800-μl aliquots using a Hamilton Microlab apparatus (Taylor Scientific, St. Louis, MO). 100 μl of protein solution in the appropriate buffer was added to yield a final concentration of the protein of 10 μm. 50 mm MES buffer was used for pH 6.0 and pH 6.6 and 25 mm Tris buffer for pH 7.6 and pH 8.0. The protein/denaturant mixtures were incubated at 25 or 37 °C for at least 7 days before measurements were taken.
Far-UV Circular Dichroism Measurements
Far-UV CD spectra were recorded using a 0.1-cm path length cell on a Chirascan circular dichroism spectrometer (Applied Photophysics, Leatherhead, UK) with an emission bandpass of 2 nm. Scans were taken between 210 and 250 nm at a scan rate of 1 nm s−1. The largest difference in signal between the native and denatured states of YFP was observed at 220 nm. The integral of the CD signal between 210 and 250 nm was used to monitor unfolding. The concentration of protein was 10 μm with various concentrations of GdmCl (0–7 m).
Fluorescence Measurements
Fluorescence measurements were taken with a Cary Eclipse 200 spectrometer (Varian, Palo Alto, CA) using a 1-cm path length cuvette. For the yellow chromophore, the excitation wavelength was 488 nm with a bandpass of 5 nm for both excitation and emission. The largest difference in fluorescence between the native and denatured states was observed at 527 nm, and the emission at this wavelength was used in the subsequent analysis. For tyrosine fluorescence, the excitation wavelength was 275 nm. The largest difference in fluorescence between the native and denatured states was observed at 308 nm.
Determination of the pKa of the Yellow Chromophore
The pKa of the p-hydroxybenzylidene imidazolidinone chromophore of Venus was determined by monitoring the fluorescence (527 nm) or absorbance (516 nm) of the protein as a function of pH in a low ionic strength 10 mm MOPS-MES buffer. The pKa was measured in the absence of added salt in addition to the presence of 0.4 m NaCl or NaNO3. The data were fitted to Equation 1,
where F is the fluorescence; A and B are variables defining the base lines, and nH is the Hill coefficient. UV-visible measurements were taken with a Cary 400 Scan (Varian, Palo Alto, CA).
Two-state and Multistate Model Analysis
Equations for the fitting of unfolding data to a two-state and three-state model have been derived and described in detail elsewhere (22). Equation 2 is for a simple two-state model,
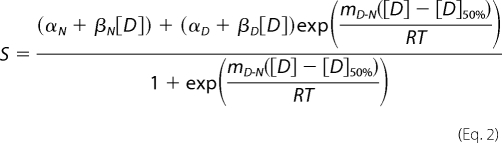 |
where S is the observed intensity of the optical property; βN and βD are the slopes of the native and denatured base lines, respectively, and αN and αD are the intensity of optical properties of the native state and denatured state, respectively, in the absence of denaturant; R is the gas constant; T is the absolute temperature; mD-N is a constant related to the average fractional change in the degree of exposure of residues on unfolding; and [D] is the concentration of denaturant. [D]50% is the midpoint of the unfolding transition and concentration of denaturant at which 50% of the sample is unfolded, and 50% is folded. ΔGD-NH2O, the difference in Gibbs' free energy of native and denatured states, can be calculated using Equation 3.
The data from yellow fluorescence, tyrosine/tryptophan fluorescence, and far-UV circular dichroism experiments were fitted globally to Equation 1 using a shared mD-N value and the software package from GraphPad Prism (Version 5).
NMR HDX Experiments
600 μl of 15N-labeled Venus, at a concentration of 140 μm and buffered in 20 mm Tris-HCl (pH 8.0), was flash-frozen and lyophilized overnight prior to the HDX experiment. 600 μl of 99.9% D2O pre-warmed to 37 °C was added to the lyophilized NMR sample, and the resulting solution was immediately transferred to a 5-mm NMR tube. A series of two-dimensional {15N-1H} HSQC spectra were recorded over 48 h at 37 °C using a 700 MHz Bruker AVANCE NMR spectrometer equipped with a cryogenic probe head (Bruker BioSpin, Karlsruhe, Germany). One-dimensional proton spectra were recorded before and after the HDX series, and the peak intensity of the most upfield-shifted methyl resonance (δ1H = −1.0 ppm) was used as a reference for normalization. Over the next 13 months, the NMR sample was sealed with parafilm and incubated at 37 °C in between the NMR measurements, with intervals from days to months. For each time point, a one-dimensional proton spectrum was recorded for intensity normalization with respect to the initial time point, and a two-dimensional{15N-1H} HSQC spectrum was recorded to monitor the intensity changes of individual amide 15N-1H correlations, using the previously reported assignments (BMRB entry 15826) (28). The individual peak intensities were normalized and fit to a single-exponential decay, I(t) = I0e − kex·t, using the rate analysis module within Sparky (29). The observed HDX rate constants of amide protons, kex, were used to calculate the free energy of unfolding, ΔGHX, defined as shown in Equation 4,
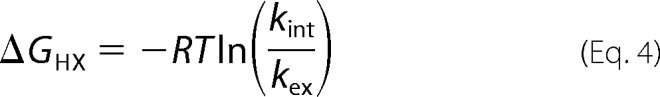 |
under EX2 conditions. The sequence-dependent intrinsic HDX rate constants, kint, and the corresponding free energy of unfolding were calculated using an Excel spreadsheet obtained from the Englander laboratory.
Chloride/Nitrate Ion Titration by NMR Spectroscopy
200 μl of 15N-labeled Venus, at a concentration of 140 μm, containing 10% D2O (v/v), 0.1 mm dithiothreitol and buffered in 10 mm MES (pH 6.0) (for chloride ion titration) or 10 mm MES/MOPS (pH 6.0) (for nitrate ion titration), was used as a starting sample for chloride or nitrate ion titration at 37 °C. A 5 or 2 m stock of either NaCl or NaNO3 solution was prepared by dissolving NaCl or NaNO3 into the appropriate buffer for titration. Aliquots of the stock solution were added progressively into the starting sample, resulting in final concentrations of NaCl or NaNO3 of 50, 100, 200, and 400 mm with a dilution factor at the end of the titration step being 25%. The titration was carried out using a 3-mm NMR tube to minimize the salt effect, which is particularly pronounced for a high field NMR spectrometer equipped with a cryoprobe; in our case, a 700 MHz Bruker AVANCE NMR spectrometer was used for the titration experiments.
RESULTS
Residue-specific Free Energy Analysis of Folding Equilibrium under Native Conditions by NMR HDX Experiments
To make a direct comparison between the stability and unfolding of Venus with that previously reported for GFPuv (22, 23), a detailed amide HDX study on Venus was undertaken. These experiments have been widely used to probe (un)folding intermediates and to extract the free energy of unfolding of proteins at a residue-specific level (see Ref. 30 and references therein). The latter requires that the system under investigation is in the so-called EX2 regime, and the exchange rate is proportional to the catalyst concentration, i.e. pH for a base-catalyzed exchange reaction, which has already been established for GFPuv under similar experimental conditions (22). Our previous studies on GFPuv revealed a super-stable core of amide groups that are highly resistant to exchange even in the presence of GdmCl over a period of 3 months (23). To obtain a similar quantitative description of the stability and super-stable core of Venus, we have extended the HDX experiments at 37 °C to over 13 months to measure, with confidence, the rates of HDX for even the slowest exchanging amide groups. These groups have exchange rates, which are in the range of 10−6 min−1, or once per year, e.g. Met-218 and Tyr-106 (Fig. 1). An initial qualitative comparison reveals that Venus exhibits a more extended degree of protection under native conditions (pH 8.0 and 37 °C) than GFPuv (where the amides of 13 residues Ile-14, Leu-15, Leu-60, Tyr-92, Val-93, Tyr-106, Val-112, Lys-113, Asn-120, Asn-121, Leu-201, Met-218, and Leu-220 are protected after 1 month of HDX in the presence of 0.5 m GdmCl; underlined residues remained protected after 12 weeks of HDX) (23). The additional highly protected residues in Venus are clustered between β-strands 10 and 11 (Ala-206, Val-219, and Leu-221) and at the ends of the β-barrel structure. Structural mapping of these highly protected amide groups reveal two well defined hydrogen bond networks, centered in β-strands 5 and 11 with Tyr-106 and Met-218 having the lowest exchange rates within the cluster. Structurally, these two hydrogen bond networks form an extended β-sheet (Fig. 1C), although topologically they cluster at the N and C termini of the primary sequence, particularly the latter where four out of the six highly protected hydrogen bonds involve four consecutive residues, Met-218 to Leu-221, in the last strand, β11 (Fig. 1D).
FIGURE 1.

NMR HDX experiments of Venus. A, 700 MHz {15N-1H} HSQC spectra of Venus recorded at 37 °C before (gray), after 3 months (blue), and after 13 months (red) of HDX. B, representative HDX profiles over a broad range of time scales. The region between the initial point and 3 months of HDX is highlighted in cyan. C, structural mapping of the highly protected amide groups in Venus. The backbone amide nitrogen atoms of the residues that retain significant cross-peak intensities after 3 months of HDX are shown in blue spheres and labeled with corresponding identities. The carbonyl carbon atoms that are involved in the stable hydrogen bonding with those highly protected amide groups are shown in yellow spheres; the corresponding hydrogen bonds are shown as sticks, bringing the hydrogen bond donors and acceptors. Additionally, those hydrogen bonds that correspond to the amide protons whose NMR intensities remained intense over 13 months of HDX are colored red. D, topological mapping of the highly stable hydrogen bond network of Venus. The labeling follows the same scheme as in C, except that the hydrogen bonds that are stable between 3 and 13 months are colored blue instead of yellow. The hydrogen bonds that are involved in the super-stable core in GFP, i.e. the amide proton that remained largely protected over 3 months of HDX under denaturing conditions, are highlighted in dashed green lines. The 11 β-strands are labeled accordingly and color ramped from blue to red. The three α-helices (α3–α5) connecting strands β3 and β4 are shown in light blue rectangular boxes. E, residue-specific free energy of unfolding, ΔGHX, derived from NMR HDX experiments under native conditions is shown as a function of residue numbers. The free energies of unfolding, ΔGHX, derived from GdmCl-induced unfolding experiments using the yellow fluorescence chromophore (Chr) and tyrosine fluorescence (Tyr) are indicated as horizontal bars with widths corresponding to the uncertainties of the fittings. Residues located in β-strands and α-helices are indicated in blue and gold, respectively. The position of the chromophore encompassing Gly-65, Tyr-66, and Gly-67 is indicated in red. F, structural mapping of the residue-specific free energy. Stereo representation of the backbone of Venus with the Cα atoms shown in spheres and ramp-colored according to the corresponding free energies of unfolding (left panel). The chromophore is shown as yellow sticks, and tyrosine and tryptophan residues are shown in semi-transparent white and green surfaces, respectively.
The rates of exchange of individual amide protons, spanning more than 4 orders of magnitude, were used to derive the free energy of unfolding, ΔGHX, at the resolution of individual amide groups under native conditions. Plotting the results as a function of residue number reveals that the free energies of unfolding are higher for residues in β-strands with an average value of 12.5 ± 0.9 kcal mol−1 (n = 44) and lower for those in loop regions or α-helices with an average of 8.3 ± 0.8 kcal mol−1 (n = 13). The overall pattern of the observed free energies of unfolding are in line with the observed rates of exchange, with amides in β4-β6, β10, and β11 exhibiting markedly higher stabilities compared with those in other β-strands (Fig. 1).
Chemical Denaturation Studies and Stability of Venus as a Function of pH
A number of studies have used the fluorescence of the post-translationally formed chromophore as a sensitive probe of the state of the protein to monitor both the un/folding and stability of a number of FP variants, including Citrine, GFPuv, superfolder GFP, and DsRed, (19–23, 26, 27, 31–33). Previously, we have studied the chemical denaturation of both GFPuv and Citrine using the intrinsic green and yellow fluorescence, respectively (22, 23, 32). For GFPuv, large kinetic barriers to unfolding were observed, which result in the system taking a significant amount of time to reach equilibrium (∼3 months) (22). Here, we have measured the stability of Venus with the chemical denaturant GdmCl and, similar to the results described above, find that the chemical denaturation curves also shift as a function of incubation time (data not shown). Because of the very long time scale that the system requires to fully reach equilibrium, we have chosen to work under nonequilibrium conditions, and samples were incubated at 25 °C for 7 days prior to measurements (unless stated otherwise). Although the thermodynamic parameters that are obtained (Table 1) are not true equilibrium values, they are a good measure of the stability of the protein under different conditions, and a comparative analysis of the data is reliable.
TABLE 1.
Apparent thermodynamic data from the chemical denaturation of Venus
Values were obtained from the best fit of the yellow fluorescence (YF), tyrosine fluorescence (Tyr), and far-UV CD (CD) data to a two-state model (see Equation 2). For the YF data acquired at lower pH values, the initial drop in fluorescence, which we have shown is not due to an unfolding event, was not included in the fit.
| Conditions | mD-N | [D]50% | ΔGD-NH2O | [D]50%a | ΔGD-NH2Oa |
|---|---|---|---|---|---|
| kcal mol−1m−1 | m | kcal mol−1 | m | kcal mol−1 | |
| pH 8.0 37 °C | |||||
| YF | 2.22 ± 0.06 | 5.10 ± 0.01 | 11.3 ± 0.3 | 5.11 ± 0.02 | 8.74 ± 0.04 |
| Tyr | 1.51 ± 0.07 | 5.24 ± 0.03 | 7.9 ± 0.3 | 5.21 ± 0.02 | 8.93 ± 0.04 |
| CD | 2.08 ± 0.26 | 5.04 ± 0.05 | 10.5 ± 1.3 | 5.07 ± 0.06 | 8.7 ± 0.1 |
| pH 8.0 | |||||
| YF | 1.83 ± 0.09 | 5.92 ± 0.03 | 10.8 ± 0.5 | 5.81 ± 0.02 | 9.9 ± 0.2 |
| Tyr | 1.75 ± 0.11 | 5.80 ± 0.04 | 10.1 ± 0.6 | 5.89 ± 0.02 | 10.1 ± 0.2 |
| CD | 1.83 ± 0.23 | 6.19 ± 0.15 | 11.2 ± 1.3 | 6.12 ± 0.03 | 10.5 ± 0.2 |
| pH 7.6 | |||||
| YF | 1.91 ± 0.17 | 5.85 ± 0.06 | 11.2 ± 0.9 | 5.72 ± 0.03 | 9.8 ± 0.2 |
| Tyr | 1.52 ± 0.08 | 5.93 ± 0.06 | 9.0 ± 0.4 | 5.77 ± 0.02 | 9.9 ± 0.2 |
| CD | 1.65 ± 0.19 | 5.08 ± 0.15 | 8.4 ± 1.0 | 5.81 ± 0.03 | 9.9 ± 0.2 |
| pH 6.6 | |||||
| YF | 1.99 ± 0.07 | 5.22 ± 0.02 | 10.4 ± 0.3 | 5.16 ± 0.04 | 8.8 ± 0.2 |
| Tyr | 1.25 ± 0.05 | 5.23 ± 0.02 | 6.6 ± 0.3 | 5.23 ± 0.02 | 8.9 ± 0.2 |
| CD | 2.09 ± 0.55 | 4.78 ± 0.08 | 10.3 ± 2.5 | 5.19 ± 0.04 | 8.9 ± 0.2 |
| pH 6.0 | |||||
| YF | 2.20 ± 0.08 | 3.61 ± 0.02 | 8.0 ± 0.3 | 3.52 ± 0.17 | 6.0 ± 0.3 |
| Tyr | 2.10 ± 0.12 | 3.23 ± 0.02 | 6.8 ± 0.4 | 3.19 ± 0.03 | 5.5 ± 0.1 |
| CD | 2.48 ± 0.61 | 3.26 ± 0.10 | 8.8 ± 2.2 | 3.16 ± 0.03 | 6.2 ± 0.2 |
a Data were calculated using a global fit using a shared mD-N value of 1.71 kcal mol−1 m−1.
The chemical denaturation of Venus was measured over the pH range 6.0–8.0. At higher pH values (7.6 and 8.0), the protein was found to be very stable, with a single unfolding transition, which has a midpoint at ∼5.8 M GdmCl (Fig. 2A). Intriguingly, at pH 6.6 and below, an additional transition was observed at a much lower denaturant concentration (∼1.0 m GdmCl). This is very pronounced at pH 6.0 leading to a biphasic transition profile (Fig. 2A). One explanation for these results is that a partially structured intermediate state with decreased fluorescence is populated at low denaturant concentrations under these conditions.
FIGURE 2.

Chemical denaturation of Venus monitored by the yellow fluorescence (A), tyrosine fluorescence (B), far-UV CD (C), and yellow fluorescence (D) of Venus as a function of GdmCl, urea, and NaCl concentration. A–C, 10 μm protein in 50 mm MES buffer at pH 6.0 (filled circles) and pH 6.6 (squares) or 25 mm Tris buffer for pH 7.6 (triangles) and pH 8.0 (diamonds). Measurements were taken after 7 days of incubation at 25 and 37 °C (open circles). The data are normalized relative to their individual maximum fluorescence intensity. D, normalized chromophore fluorescence intensity at 527 nm is plotted as a function of denaturant (closed symbols) or chloride ion concentration (open symbols). Aliquots of 10 μm Venus were incubated with various GdmCl or urea concentrations or NaCl in the absence of denaturants for 7 days prior to the measurements. The samples were in 50 mm MES buffer for pH 6.0 and pH 6.6 or 25 mm Tris buffer for pH 7.6.
To investigate this further, tyrosine and tryptophan fluorescences were used as independent structural probes of the GdmCl-induced unfolding (Fig. 2B). In good agreement with the results obtained using the yellow fluorescence, a progressive reduction in the stability of Venus was seen on decreasing the pH from 8.0 to 6.0. However, the initial decrease in signal observed at low concentrations of denaturant at pH 6.0 and 6.6 with yellow fluorescence was not detected. In addition, far-UV CD spectroscopy was also employed. In this case, a single unfolding transition was observed at all pH values (Fig. 2C) consistent with the tyrosine and tryptophan data. Together, these results suggest that the initial loss in yellow fluorescence observed at pH 6.0 and 6.6 at low concentrations of GdmCl is not due to an unfolding event but instead that quenching of the fluorescence occurs possibly as a direct result of using GdmCl and the interaction of chloride ions with Venus.
Urea and NaCl Titration Curves
One possible explanation for the results described above is that, contrary to what has been reported previously (16), the fluorescence of Venus is sensitive to chloride ions at pH values of 6.0 and below. To investigate the origin of the loss in fluorescence at pH 6.0 in the presence of low concentrations of GdmCl, experiments in urea were performed.
In contrast to GdmCl, urea is a nonionic chemical denaturant that has roughly half the denaturing strength of GdmCl. A titration of Venus with urea shows that the yellow fluorescence remains constant up to a denaturant concentration of 4 m (Fig. 2D), strengthening the hypothesis that the initial loss of yellow fluorescence observed in the GdmCl titration is the result of interactions between the protein and chloride ions, rather than an unfolding-induced solvent exposure of the chromophore. To confirm this, an additional titration of native Venus with sodium chloride was undertaken, and the results obtained are in good agreement with the GdmCl titration up to about 1 m in each case (Fig. 2D). Together, the results strongly suggest that under mildly acidic conditions chloride ions (from GdmCl or NaCl) have a profound effect on the yellow fluorescence of Venus.
Measurements of the pKa of the p-Hydroxybenzylidene Imidazolidinone Chromophore of Venus
One mechanism by which chloride ions may lead to a quenching of the yellow fluorescence of Venus is by inducing a shift in the pKa of the p-hydroxybenzylidene imidazolidinone chromophore, as it is well established that the anionic form of the phenolic chromophore has higher fluorescence at 527 nm compared with the neutral form (13). To investigate this possible mechanism, the absorbance and fluorescence of Venus was measured as a function of pH over the pH range 5.5 to 8.0 in a low ionic strength MOPS/MES buffer in the absence and presence of 0.4 m chloride ions, see Fig. 3 and supplemental Fig. S3. As a control, 0.4 m NaNO3 was also used to determine whether the effects were specific and due to halide ion binding or were nonspecific and due to changes in ionic strength. The data in Fig. 3A clearly show that both chloride and nitrate ions result in a significant loss in fluorescence, which is most pronounced at lower pH. Analysis of the data, however, shows that there is relatively little change in the pKa of the yellow chromophore on addition of either salt (Fig. 3B and Table 2). Similar results were obtained when absorbance was used (Fig. 3, C and D, and Table 2). The Hill coefficients varied from 0.7 to 1.2 but generally were close to 1, suggesting a single titratable group (Table 2). However, there was not a clear isosbestic point in the absorbance measurements (Fig. 3C) suggesting that the system shows complex behavior on protonation/deprotonation. In addition, a blue shift in the peak corresponding to the neutral form of the chromophore was observed on addition of chloride or nitrate ions (Fig. 3C). The results indicate that neither chloride nor nitrate ions shift the pKa value of the chromophore in Venus establishing that this cannot be the cause of the observed quenching. The results also suggest that the effects are nonspecific and are most likely attributable to changes in ionic strength. The addition of 0.4 m NaCl or NaNO3 to a solution of Venus in a high ionic strength citrate-phosphate buffer at pH 6.0 had no effect on the fluorescence (data not shown), confirming our hypothesis that the effects are largely due to changes in ionic strength.
FIGURE 3.

pKa measurements of the yellow chromophore in the presence of chloride and nitrate ions. The left panels show the fluorescence (A) and UV absorbance (C) spectra of Venus (black lines) in the presence of 400 mm chloride ions (dark gray lines) or nitrate ions (light gray lines) at the lowest and highest analyzed pH values (5.5 and 8.0); the right panels show the fluorescence (B) and UV absorbance (D) profiles of Venus in the presence of 400 mm chloride ions (dark gray circles) or nitrate ions (light gray triangles) at various pH values ranging from 5.5 to 8.3. Data were fitted to Equation 1, and the pKa values of the chromophore calculated are given in Table 2. Conditions are as follows: 10 mm MES/MOPS buffer mixture was used at 25 °C. AU, absorbance unit; mAU, milli-AU.
TABLE 2.
pKa and Hill coefficient nH values of the p-hydroxybenzylidene imidazolidinone chromophore of Venus in the presence and absence of additional salts
| pKa fluorescence | pKa absorbance | nH fluorescence | nH absorbance | |
|---|---|---|---|---|
| MES/MOPS buffer | 6.84 ± 0.04 | 7.01 ± 0.05 | 0.77 ± 0.07 | 0.68 ± 0.08 |
| +0.4 m NaCl | 6.89 ± 0.05 | 7.08 ± 0.07 | 0.95 ± 0.12 | 0.97 ± 0.16 |
| +0.4 m NaNO3 | 7.06 ± 0.04 | 7.15 ± 0.05 | 1.18 ± 0.11 | 1.19 ± 0.13 |
Structural Effects of Chloride and Nitrate Ions on Venus
To investigate the effects of both chloride and nitrate ions further, NMR experiments were undertaken in which 15N-labeled samples of Venus at pH 6.0 were titrated with sodium chloride or sodium nitrate. Perturbations to the chemical shifts were monitored as independent structural probes. In both cases, the results of the titrations show marginal changes in the overall appearance of the {15N-1H} correlations of Venus, indicating little significant conformational change upon the addition of either chloride or nitrate ions up to a maximum concentration of 0.4 m (supplemental Fig. S1). Additionally, the combined (15N and 1H) chemical shift perturbations (see “Experimental Procedures” for definition) of individual residues showed only a few significant changes for residues scattered in a number of β-stranded regions, particularly in β-strands 2 and 8–10 (Fig. 4A). Collectively, the {15N-1H} correlations of Venus exhibit progressive downfield shift changes on increasing chloride or nitrate ion concentrations (Fig. 4A and supplemental Fig. S1). Structural mapping of the residues, which show significantly large chemical shift perturbations, shows that most of these residues are located at the ends of the β-barrel structure, i.e. in the loops or helices in both lids or at the ends of individual β-strands (Fig. 4, B and C). For example, although the backbone amide group of Trp-57 (which is buried) shows limited chemical shift changes, those of the side-chain indole group (which is exposed) are significantly larger, suggesting that the effect may be associated with ion accessibility rather than specific coordination of an ion in the interior of the protein. The latter has been proposed based on previous crystallographic reports that have shown a well defined halide ion-binding site located in the proximity of the hydroxyl group of the tyrosyl ring of the chromophore and close to His-148, the residue that is associated with the pH sensitivity of GFP fluorescence (13). The maximum chemical shift perturbation of His-148 in this study is very small (0.076 ppm). In conclusion, our NMR data show little evidence to support a specific chloride or nitrate ion-binding site in Venus.
FIGURE 4.

Structural mapping of chemical shift perturbations in Venus upon addition of chloride and nitrate ions. A, chemical shift perturbations (Δδ) of Venus in the presence of 400 mm chloride (red) and nitrate ions (black) as a function of residue number. The weighted Δδ is defined as (Δδ(1H)2 + (0.65·Δδ(15N)2)1/2. The differences between chloride and nitrate ions (ΔΔδ) are shown in green with a shifted base line to illustrate the similar effects induced by the two different ions. The dashed horizontal line indicates the values of two standard deviations (2σ) of all Δδ. The residues that exhibit Δδ values larger than 2σ are indicated with corresponding residue identities. Residues that are located in the α-helical and β-stranded regions are shaded in pink an light blue, respectively, with the secondary structures shown on top of the panel. Structural mapping of the chemical shift perturbations induced by the addition of 400 mm chloride ions is shown in B and that induced by the addition of 400 mm nitrate ions is shown in C. The backbone nitrogen atoms are shown in spheres with various sizes proportional to the observed chemical shift perturbations (as indicated in the middle). The chromophore within the β-barrel is shown as yellow sticks.
DISCUSSION
GFP and its family members are central to modern biological and medical sciences because of their versatility and robustness in a multitude of imaging and spectroscopic applications (1, 4–6). Critical to the usefulness of FP variants is the ability to achieve efficient folding, rapid maturation of the chromophore, high quantum yields, i.e. brightness, and reduced sensitivity to environmental changes, such as pH and ion concentrations. For this reason, Venus has become one of the most commonly used FP variants for imaging and Förster resonance energy transfer studies both in vivo and in vitro, and it has been used in both ensemble measurements and single molecule experiments (1, 16, 34).
Recent studies on the stability and folding of GFP and some of its variants have established that the protein has a complex energy landscape with a number of different intermediate states, high energy barriers, and multiple pathways (18–27, 31, 32, 35). Fully characterizing this energy landscape, and establishing whether there is a general underlying folding mechanism for the complex β-barrel topology of FPs, represents a significant challenge. To gain a better understanding of the folding of FPs in general, we have carried out extensive biophysical studies on Venus using a variety of spectroscopic tools.
At pH values of 7.0 and higher, the native state of Venus is extremely stable with a midpoint of unfolding of over 5.8 m GdmCl (after 7 days of equilibration in denaturant at 25 °C). This is similar to previous studies that have reported high stabilities for GFP and other FPs (36). In our previous studies on the unfolding/folding behavior of a truncated version of GFPuv, we observe that the protein takes a long time to fully equilibrate in the presence of chemical denaturants due to the high energy barriers of unfolding (22). Venus shows similar behavior (data not shown); however, a full time-dependent analysis of the unfolding was not undertaken here. Instead, stability measurements at different temperatures and pH values were made after the same equilibration period (1 week) to enable us to perform a comparative analysis. The data clearly show that there is a rapid decrease in stability on lowering the pH from 7.6 to 6.0 (Fig. 2 and Table 1).
It is difficult to compare quantitatively the stability we have measured here for Venus with that previously measured for other FPs, as different experimental conditions and equilibration times have been used. However, measurements have made on GFPuv at pH 6.0 and 25 °C, after 5 and 13 days of incubation in GdmCl. In this case, the midpoints for unfolding are ∼2.75 and 2.50 m, respectively, which can be compared with the midpoint for Venus measured under similar conditions after 7 days of 3.5–3.6 m GdmCl (Table 1). Thus, Venus is more stable toward chemical denaturation than GFPuv.
To compare in further detail the unfolding/folding and stability of Venus with GFPuv, a comprehensive series of hydrogen-deuterium amide exchange measurements were made. GFPuv, we have established that many amide protons are very resistant to exchange, effectively undergoing no exchange at all even at high pH (8.6) and temperature (37 °C) and in the presence of 1 m GdmCl (22, 23). With the aim of obtaining accurate exchange rates for even the most slowly exchanging amide protons of Venus, HDX measurements were made at pH 8.0 and at 37 °C for over 13 months, and HDX rates spanning over 4 orders of magnitude were obtained (Fig. 1). Free energies of exchange were calculated and are shown in Fig. 1. For many of the amide groups in the β-strands of the barrel structure, high values for ΔGHX are obtained (∼12 kcal mol−1), indicating that exchange probably occurs only on global unfolding, which under the conditions used is a rare event. In comparison, amide groups in loop and helical regions have lower values that cluster around 8 kcal mol−1 suggesting that there are also local unfolding events that lead to exchange. A comparison between the free energies of unfolding calculated using the yellow fluorescence from the chemical-denaturation experiments and those calculated from the HDX measurements reveals that for some of the most protected amide groups in β-strands 4–6 and 11, values of ΔGHX are higher than for ΔGD-NH2O (Fig. 1E). Some caution has to be used when comparing the different datasets that are measured under different conditions (equilibrium versus nonequilibrium conditions), i.e. samples are equilibrated for 7 days in the chemical denaturation experiments and for 13 months in the NMR deuterium exchange experiments.
For both GFPuv and Venus, there is evidence that β-strand 7 is the least stable β-strand based on the NMR HDX data (Fig. 1D). Consistent with this are previous simulation and NMR studies that have suggested that the formation of β-strand 7 is the last step in the folding pathway, given the observed internal dynamics that are most abundant in this region in the native state (37, 38). In addition, a recent in vitro biomolecular fluorescence complementation (BiFC) study has provided further evidence that the folding of β-strand 7 is involved in the last step of the β-barrel closure of a circularly permuted and truncated GFPuv variant (39).
A comparison of the results reported here for Venus with those previously published for GFPuv (22, 23) shows that the super-stable core of highly protected amide groups in Venus is similar to GFPuv and includes residues in β-strands 1–2 and 4–6. These residues form a mini β-sheet, which for the GFPuv we have postulated forms a critical folding nucleus (22). However, the results on Venus show that this region is more extended and also includes further residues located in β-strands 2, 4, and 6 but also many in β-strands 10 and 11. This leads to two hydrogen-bonded networks that center around residues in β-strands 5 and 11 and that together cluster and link N- and C-terminal regions of the protein (Fig. 1D). The exact reasons why Venus displays a larger super-stable core compared with GFPuv are not known; however, it is noted that Venus has a higher global stability than GFPuv.
For GFPuv, the highly protected super-stable core of amide groups is proposed to form a potential folding nucleus as it constitutes the most stable region of structure within the protein. For Venus, the folding nucleus is more extensive, and may well be more stable (Fig. 1F). Notably, it incorporates regions of the protein at both the N and C termini of the structure as described above. It is therefore interesting to speculate that the formation of such a stable nucleus leads to faster folding kinetics and an accelerated maturation of the chromophore, as has been observed (16). This is approaching the time scale that is required for translation of the complete primary sequence of the protein. Therefore, the chromophore formation of Venus may be coupled to protein synthesis in a co-translational manner, for which we have devised an experimental strategy to probe by solution state NMR spectroscopy (40, 41).
Robust co-translational folding of FPs is crucial in the context of studying protein stabilities through the use of FPs as fluorescent fusion tags (42–44). It has been shown that fusion of GFPuv at the N or C termini of the target proteins can lead to an increased aggregation propensity (17). Although a number of “superfolder” FPs have been developed to achieve very robust refolding efficiencies (45), there is still room for improvement through protein engineering, e.g. by supercharging to prevent aggregation (46).
Not only is it crucial to obtain robust folding efficiencies and stabilities for full-length FPs but also for enhanced structural integrities of FP fragments. In addition, spectroscopic properties that are insensitive to the local environment are critical for many applications, including BiFC. In this technique, binary complex formation is monitored through the emergence of fluorescence upon FP fragment complementation (47). A recent in vitro BiFC study that used the superfolder YFP as a model system showed that BiFC can be biased by the nature of the binding events, which are salt-dependent; the two examples presented therein exhibited opposite salt dependence when the yellow fluorescence was used as a readout (48). Interestingly, high pH (7.6–8.8) facilitated BiFC, whereas a complete loss of BiFC was observed at neutral pH (7.2) in the presence of 150 mm NaCl.
Through the use of GdmCl in our unfolding experiments, in addition to other experiments reported here, we have established that Venus is sensitive to chloride ions under very mildly acidic conditions (Figs. 2 and 3). This important result is in contrast to previous studies that suggested that Venus is less sensitive to proton and halide ion concentrations than other FPs (16).
To understand the mechanism by which chloride ions result in a decrease in fluorescence at pH 6.0, the pKa of the p-hydroxybenzylidene imidazolidinone chromophore of Venus was measured under different conditions (Fig. 3 and Table 2). Although a significant loss in fluorescence was observed at pH 6.0 in the presence of 0.4 m chloride or nitrate ions (Fig. 3A), it could not be attributed to changes in pKa, which varied little (Table 2). The Hill coefficients associated with the pKa fits were found to be near unity suggesting a single titratable site (Table 2). However, the absorbance measurements did not show a clear isosbestic point (Fig. 3C) suggesting more complex behavior. In addition, a blue shift in the absorbance of the neutral form of the chromophore was observed on addition of chloride or nitrate ions (Fig. 3C).
A number of groups have studied the pH and anion dependence of different fluorescent proteins and have shown that the pKa value of the chromophore can be modified both by mutation of residues close to the chromophore or by halide ion binding. For example, Remington and co-workers (49) showed that mutation of His-148 to glutamine or glycine increases the pKa value of the chromophore from 7 to 8 in a YFP variant. However, these mutations also caused the loss of the isosbestic point in pH titrations (50), similar to our observations on Venus, indicating that multiple processes occur simultaneously. In addition, they observed a blue shift in the absorption maximum of the neutral form of the chromophore upon the addition of chloride (50). Venus also demonstrates this behavior, and there is a blue shift in the neutral form from 421 to 400 nm upon addition of 0.4 m chloride or nitrate ions (Fig. 3C), which suggests that destabilization of the transition dipole of the neutral form results from a decrease in the polarity of the environment near the chromophore, providing evidence that this is a ground-state effect. CD analysis in the visible region showed two negative peaks at 395 and 516 nm (data not shown) corresponding to the anionic and neutral forms of the chromophore, respectively. The addition of either chloride or nitrate ions to Venus did not alter the peak positions or intensities in contrast to YFP-H148Q suggesting that Venus does not have a specific anion-binding site that alters the symmetry of the chromophore environment.
Halide ion binding has been shown to quench the green fluorescence of E2GFP, a GFP variant that contains F64L/S65T/T203Y/H231L mutations (where E2 corresponds to the underlined residues) (13). The origin of the fluorescence quenching has been attributed to a localized binding of the halide ion to a region close to residues Val-42, Val-68, Gln-69, Tyr-203, and Val-224 and mediated by a number of water molecules inside the β-barrel. In contrast to the crystallographic study of Venus, which associated the insensitivity of the protein toward chloride ions to the substitution of F64L that is thought to prevent chloride ion binding through a series of conformational rearrangements (51), the crystal structure of E2GFP showed that the F64L replacement is responsible for creating a specific halide-binding site, and the sensitivity is further increased by the S65T and T203Y substitutions (13).
YFP-H148Q was reported to contain a specific halide-binding site (near the chromophore) altering its pKa from 7.14 in the absence of anions to 7.86 in 150 mm chloride (50, 52). A crystal structure of this variant identified this binding site (53).
A definitive-binding site for either chloride or nitrate ions, however, was not apparent from the NMR titrations performed on Venus (Fig. 4). Changes in absorbance and fluorescence with increasing chloride or nitrate ion concentrations (Fig. 3) occur concomitantly and show these anions affect molar absorbance rather than quantum yield. In agreement with our findings, fluorescence lifetimes of YFP-H148Q were independent of both pH (6.0–8.0) and chloride ion concentration (0–400 mm) suggesting a ground state effect (50).
Galietta et al. (52) screened a mutant library of 1536 clones of the YFP-H148Q and selected variants with increased halide ion sensitivity. They measured halide ion binding affinities and associated shifts in pKa for a subset of these. Similar to the results reported here, the fluorescence of the mutants was found to be both pH-sensitive and also influenced by the presence of anions (52).
To further understand the effects of chloride ions on Venus fluorescence, and to establish whether there is a halide ion-binding site in the protein, NMR was used to obtain residue-specific information on the structural changes induced in Venus on the addition of high concentrations (up to 0.4 m) of sodium chloride. In particular, we wanted to see if chloride ions induced changes in the chemical shifts of the residues shown in E2GFP and YFP-H148Q to be located near the halide ion-binding site. In our NMR backbone assignments, we could not identify the resonances of Leu-64 (adjacent to the chromophore in the central α-helix), Val-224 (located in β-strand 11), and Tyr-203 (adjacent to a segment of β-strand 10 that is at the center of a patch of unassigned residues, the surface of which overlaps with the dimer interface of Venus in the crystal structure (28)). These residues are likely to undergo breathing motions as a result of the ill-defined hydrogen bonding network centered around β-strand 7. However, we were able to observe the chemical shift perturbations of the amide groups of Gln-69 and Val-24 upon titration with 0.4 m of chloride ions at pH 6.0 and found that they are moderate with corresponding Δδ values of 0.16 and 0.20 ppm, respectively, compared with other loop residues that exhibit significantly larger changes; the mean chemical shift change 〈Δδ〉 is 0.25 ppm with a standard deviation of 0.21 ppm (Fig. 4). Many of these have charged groups, and hence we attribute the dominant effect of chloride ions to nonspecific electrostatic interactions or electrostatic screening (Fig. 4). This is consistent with our results using sodium nitrate instead of sodium chloride.
The chemical shift perturbation profiles of the two titration series are essentially the same (Fig. 4 and supplemental Fig. S1) with a mean pairwise chemical shift difference, ΔΔδ, of only 0.04 ppm and a standard deviation of 0.10 ppm (Fig. 4). In fact, nitrate ions induced slightly larger chemical shift perturbations compared with chloride ions, 〈Δδ〉 = 0.29 ppm. We have found a good spatial correlation between the observed chemical shift changes in response to changes in ionic strength with those observed in response to changes in pH values from 6.0 to 9.6 (28) that are localized in the top and bottom lids of the barrel structure (supplemental Fig. S2). In addition, we identified a number of cavities within the barrel structure of Venus that are located in close proximities to the residues that underwent significant chemical shift changes upon pH changes. Water molecules can in principle fit in these cavities and modulate the yellow fluorescence of the chromophore. It is therefore plausible that the addition of ions as well as changes in pH can induce a series of local conformational rearrangements leading to water-mediated fluorescence quenching.
Recent studies using intrinsic fluorescence (19–22, 27), exogenous fluorophores (32), and NMR (22, 23, 28, 31, 38, 54) in addition to the work we present here have begun to map out the complex energy landscape for both the in vitro and in vivo folding of this class of protein. Although many unusual features, such as parallel pathways and kinetic traps due to chromophore formation have been clearly established, much more work is needed to fully characterize and understand how large β-barrel FPs fold. In particular, it is becoming clear that FPs engineered to have different spectral and biophysical properties can show quite marked differences in their behavior toward environmental factors and intrinsic differences in stability and folding.
Supplementary Material
Acknowledgments
We thank Prof. Atsushi Miyawaki for providing the DNA plasmid of Venus and Dr. John Christodoulou for helpful discussions. We also thank the staff and the use of the Biomolecular NMR Facility, Department of Chemistry, University of Cambridge.
This work was supported in part by Access to Research Infrastructures Activity in the 6th Framework Program of the EC Contract RII3-026145, EU-NMR.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- GFP
- green fluorescent protein
- YFP
- yellow fluorescent protein
- FP
- fluorescent protein
- MES
- 4-morpholineethanesulfonic acid
- MOPS
- 4-morpholinepropanesulfonic acid
- HDX
- hydrogen-deuterium exchange
- GdmCl
- guanidinium chloride
- BiFC
- biomolecular fluorescence complementation.
REFERENCES
- 1.Shaner N. C., Patterson G. H., Davidson M. W. (2007) J. Cell Sci. 120, 4247–4260 [DOI] [PubMed] [Google Scholar]
- 2.Tsien R. Y. (1998) Annu. Rev. Biochem. 67, 509–544 [DOI] [PubMed] [Google Scholar]
- 3.Cubitt A. B., Heim R., Adams S. R., Boyd A. E., Gross L. A., Tsien R. Y. (1995) Trends Biochem. Sci. 20, 448–455 [DOI] [PubMed] [Google Scholar]
- 4.Giepmans B. N., Adams S. R., Ellisman M. H., Tsien R. Y. (2006) Science 312, 217–224 [DOI] [PubMed] [Google Scholar]
- 5.VanEngelenburg S. B., Palmer A. E. (2008) Curr. Opin. Chem. Biol. 12, 60–65 [DOI] [PubMed] [Google Scholar]
- 6.Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002) Science 296, 913–916 [DOI] [PubMed] [Google Scholar]
- 7.Ormo M., Cubitt A. B., Kallio K., Gross L. A., Tsien R. Y., Remington S. J. (1996) Science 273, 1392–1395 [DOI] [PubMed] [Google Scholar]
- 8.Reid B. G., Flynn G. C. (1997) Biochemistry 36, 6786–6791 [DOI] [PubMed] [Google Scholar]
- 9.Shyu Y. J., Suarez C. D., Hu C. D. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karasawa A., Tsuboi Y., Inoue H., Kinoshita R., Nakamura N., Kanazawa H. (2005) J. Biol. Chem. 280, 41900–41911 [DOI] [PubMed] [Google Scholar]
- 11.Millington M., Grindlay G. J., Altenbach K., Neely R. K., Kolch W., Bencina M., Read N. D., Jones A. C., Dryden D. T., Magennis S. W. (2007) Biophys. Chem. 127, 155–164 [DOI] [PubMed] [Google Scholar]
- 12.Kneen M., Farinas J., Li Y., Verkman A. S. (1998) Biophys. J. 74, 1591–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arosio D., Garau G., Ricci F., Marchetti L., Bizzarri R., Nifosì R., Beltram F. (2007) Biophys. J. 93, 232–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C. J., Heim R., Lu P., Pu Y., Tsien R. Y., Chang D. C. (1999) J. Cell Sci. 112, 1567–1577 [DOI] [PubMed] [Google Scholar]
- 15.Nagai T., Yamada S., Tominaga T., Ichikawa M., Miyawaki A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10554–10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai T., Ibata K., Park E. S., Kubota M., Mikoshiba K., Miyawaki A. (2002) Nat. Biotechnol. 20, 87–90 [DOI] [PubMed] [Google Scholar]
- 17.Chang H. C., Kaiser C. M., Hartl F. U., Barral J. M. (2005) J. Mol. Biol. 353, 397–409 [DOI] [PubMed] [Google Scholar]
- 18.Chirico G., Cannone F., Diaspro A. (2006) Eur. Biophys. J. 35, 663–674 [DOI] [PubMed] [Google Scholar]
- 19.Enoki S., Maki K., Inobe T., Takahashi K., Kamagata K., Oroguchi T., Nakatani H., Tomoyori K., Kuwajima K. (2006) J. Mol. Biol. 361, 969–982 [DOI] [PubMed] [Google Scholar]
- 20.Enoki S., Saeki K., Maki K., Kuwajima K. (2004) Biochemistry 43, 14238–14248 [DOI] [PubMed] [Google Scholar]
- 21.Fukuda H., Arai M., Kuwajima K. (2000) Biochemistry 39, 12025–12032 [DOI] [PubMed] [Google Scholar]
- 22.Huang J. R., Craggs T. D., Christodoulou J., Jackson S. E. (2007) J. Mol. Biol. 370, 356–371 [DOI] [PubMed] [Google Scholar]
- 23.Huang J. R., Hsu S. T., Christodoulou J., Jackson S. E. (2008) HFSP J. 2, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson S. E., Craggs T. D., Huang J. R. (2006) Expert Rev. Proteomics 3, 545–559 [DOI] [PubMed] [Google Scholar]
- 25.Mickler M., Dima R. I., Dietz H., Hyeon C., Thirumalai D., Rief M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 20268–20273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews B. T., Gosavi S., Finke J. M., Onuchic J. N., Jennings P. A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 12283–12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrews B. T., Schoenfish A. R., Roy M., Waldo G., Jennings P. A. (2007) J. Mol. Biol. 373, 476–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu S. T., Behrens C., Cabrita L. D., Dobson C. M. (2009) Biomol. NMR Assign. 3, 67–72 [DOI] [PubMed] [Google Scholar]
- 29.Goddard T. D., Kneller D. G. (2008) Sparky, NMR Assignment and Integration Software Version 3.114, University of California, San Francisco [Google Scholar]
- 30.Englander S. W., Mayne L., Krishna M. M. G. (2007) Q. Rev. Biophys. 40, 287–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan F., Kuprov I., Craggs T. D., Hore P. J., Jackson S. E. (2006) J. Am. Chem. Soc. 128, 10729–10737 [DOI] [PubMed] [Google Scholar]
- 32.Orte A., Craggs T. D., White S. S., Jackson S. E., Klenerman D. (2008) J. Am. Chem. Soc. 130, 7898–7907 [DOI] [PubMed] [Google Scholar]
- 33.Zapata-Hommer O., Griesbeck O. (2003) BMC Biotechnol. 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai L., Friedman N., Xie X. S. (2006) Nature 440, 358–362 [DOI] [PubMed] [Google Scholar]
- 35.Xie J. B., Zhou J. M. (2008) Biochemistry 47, 348–357 [DOI] [PubMed] [Google Scholar]
- 36.Stepanenko O. V., Verkhusha V. V., Kazakov V. I., Shavlovsky M. M., Kuznetsova I. M., Uversky V. N., Turoverov K. K. (2004) Biochemistry 43, 14913–14923 [DOI] [PubMed] [Google Scholar]
- 37.Helms V., Straatsma T. P., McCammon J. A. (1999) J. Phys. Chem. B 103, 3263–3269 [Google Scholar]
- 38.Seifert M. H., Georgescu J., Ksiazek D., Smialowski P., Rehm T., Steipe B., Holak T. A. (2003) Biochemistry 42, 2500–2512 [DOI] [PubMed] [Google Scholar]
- 39.Huang Y. M., Bystroff C. (2009) Biochemistry 48, 929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu S. T., Cabrita L. D., Fucini P., Christodoulou J., Dobson C. M. (2009) J. Am. Chem. Soc. 131, 8366–8367 [DOI] [PubMed] [Google Scholar]
- 41.Hsu S. T., Fucini P., Cabrita L. D., Launay H., Dobson C. M., Christodoulou J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 16516–16521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rücker E., Schneider G., Steinhäuser K., Löwer R., Hauber J., Stauber R. H. (2001) Protein Expr. Purif. 21, 220–223 [DOI] [PubMed] [Google Scholar]
- 43.Wang H., Chong S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 478–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yen H. C., Xu Q., Chou D. M., Zhao Z., Elledge S. J. (2008) Science 322, 918–923 [DOI] [PubMed] [Google Scholar]
- 45.Pédelacq J. D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S. (2006) Nat. Biotechnol. 24, 79–88 [DOI] [PubMed] [Google Scholar]
- 46.Lawrence M. S., Phillips K. J., Liu D. R. (2007) J. Am. Chem. Soc. 129, 10110–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerppola T. K. (2006) Nat. Rev. Mol. Cell Biol. 7, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ottmann C., Weyand M., Wolf A., Kuhlmann J., Ottmann C. (2009) Biol. Chem. 390, 81–90 [DOI] [PubMed] [Google Scholar]
- 49.Elsliger M. A., Wachter R. M., Hanson G. T., Kallio K., Remington S. J. (1999) Biochemistry 38, 5296–5301 [DOI] [PubMed] [Google Scholar]
- 50.Jayaraman S., Haggie P., Wachter R. M., Remington S. J., Verkman A. S. (2000) J. Biol. Chem. 275, 6047–6050 [DOI] [PubMed] [Google Scholar]
- 51.Rekas A., Alattia J. R., Nagai T., Miyawaki A., Ikura M. (2002) J. Biol. Chem. 277, 50573–50578 [DOI] [PubMed] [Google Scholar]
- 52.Galietta L. J., Haggie P. M., Verkman A. S. (2001) FEBS Lett. 499, 220–224 [DOI] [PubMed] [Google Scholar]
- 53.Wachter R. M., Yarbrough D., Kallio K., Remington S. J. (2000) J. Mol. Biol. 301, 157–171 [DOI] [PubMed] [Google Scholar]
- 54.Seifert M. H., Ksiazek D., Azim M. K., Smialowski P., Budisa N., Holak T. A. (2002) J. Am. Chem. Soc. 124, 7932–7942 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.