

Abstract
Vitamin K antagonists such as warfarin inhibit the vitamin K-dependent γ-glutamyl carboxylation during protein processing and block the secretion of under-γ-carboxylated prothrombin (FII) in the rat but not in the human or bovine. Under-γ-carboxylated prothrombin is also secreted from warfarin-treated human (HepG2) cell cultures but is degraded in the endoplasmic reticulum in warfarin-treated rat (H-35) cell cultures. This differential response to warfarin has been shown to be determined by the structural difference in the proteins rather than by the origin of the cell line. When recombinant rat prothrombin (rFII) and human prothrombin (hFII) were expressed in a transformed human kidney cell line (HEK293), secretion of rFII but not hFII was drastically decreased in response to warfarin. To determine the structural signal required for this differential response, chimeric cDNAs with the propeptide/Gla domains, kringle domain, and serine protease domain exchanged between rFII and hFII were generated (FIIRHH and FIIHRR, FIIRRH and FIIHHR, FIIRHR and FIIHRH) and expressed in both warfarin-treated HEK293 cells and HepG2 cells. The presence of the hFII kringle domain changed the stability of rFII to that of hFII, and the rFII kringle domain changed the stability of hFII to that of rFII. The kringle domain therefore is critical in determining the metabolic fate of under-γ-carboxylated prothrombin precursors during processing. Prothrombin contains two kringle structures, and expression of additional rFII/hFII chimeras (FIIHrhH and FIIHhrH, FIIRrhR, and FIIRhrR) was used to determine that the first of the two kringles plays a more important role in the recognition process.
During biosynthesis, specific glutamyl residues of prothrombin (coagulation factor II) are carboxylated to γ-carboxyglutamyl (Gla) residues by a vitamin K-dependent hepatic microsomal enzyme (1). The coproduct of this reaction, vitamin K 2,3-epoxide is reduced to the enzymatically active hydronaphthoquinone form of the vitamin by a microsomal epoxide reductase (1). Warfarin, a 4-hydroxycoumarin-based anticoagulant, blocks γ-carboxylation by inhibiting the recycling of vitamin K from its epoxide form to the reduced form (2). In the acquired vitamin K deficiency produced in the presence of warfarin, under-γ-carboxylated forms of prothrombin appear in the plasma of some species (3, 4).
Secretion of under-γ-carboxylated prothrombin has been shown to be species-dependent. In the rat or in a rat hepatoma cell line (H-35), warfarin treatment results in a drastic decline of prothrombin secretion, and an intracellular accumulation of under-γ-carboxylated prothrombin that is degraded in a pre-Golgi compartment (5–7). In patients treated with oral anticoagulants (4) and in cultures of a human hepatoma (HepG2) cell line incubated with warfarin (8), under-γ-carboxylated prothrombin is secreted. Rat prothrombin (rFII) stably expressed in HepG2 cells treated with warfarin is not secreted but is degraded intracellularly (8), whereas secretion of endogenous human prothrombin (hFII) is not altered. These data suggest that a structural element within the prothrombin molecule determines the fate (secretion vs. retention and degradation) of the under-γ-carboxylated protein during its intracellular processing through the secretory pathway.
Mature plasma prothrombin consists of an amino-terminal Gla domain, a kringle domain containing two kringle structures, and a carboxyl-terminal serine protease catalytic domain (9). To define the structural signal present in prothrombin that is responsible for the differential processing of rat/human protein in response to warfarin, a number of chimeric rFII/hFII cDNAs were generated by using recombinant DNA techniques. These constructs were expressed in human embryonic kidney (HEK293) and hepatoma (HepG2) cell lines. The response of these chimeras to warfarin has been evaluated to determine the location of the structural difference between rat and human prothrombin that is responsible for their differential processing.
MATERIALS AND METHODS
Chimeric cDNA Constructs.
The cDNAs coding for rFII and hFII were cloned into pcDNA3 as EcoRI fragments as previously described (8). Chimeric cDNA construction was based on the PCR. Oligonucleotides PM1 to PM15 were designed with PcDNA3-rFII and hFII as templates, with necessary restriction sites at 5′ ends. BbsI class II restriction sites were created to ensure no disruption of the native sequence at the ligation junction sites (10). All fragments generated by PCR were sequenced to ensure no mutations were introduced. The constructs are shown in Fig. 4.
Figure 4.
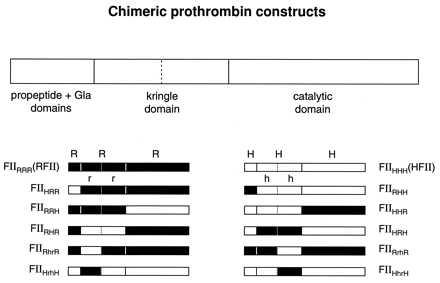
Chimeric prothrombin constructs. A terminal BbsI site was introduced via the PCR to prevent disruption of original sequences at the junction site (10). Mutations were introduced when necessary without altering the amino acid sequence. The kringle domain starts from amino acid 58(rFII) or 57(hFII) to the first factor X cleavage site of amino acid 270(rFII) or 271(hFII). There are two kringle structures in the kringle domain. The first kringle structure ends at the thrombin cleavage site of amino acid 154(rFII) or 155 (hFII). The catalytic domain contains the C-terminal sequence starting from the first factor X cleavage site (9, 11).
PcDNA3-FIIHRR and PcDNA3-FIIRHH.
A 0.27-kb fragment containing the HFII propeptide and Gla sequences was amplified from pcDNA3-HFII with PM1 and PM3, subcloned into pBluescript (+/−) to give pbl/sk-PGH. Primers PM4 and PM5 were used to amplify a 0.3-kb RFII fragment from pcDNA3-RFII, and the BbsI/ApaI digest of the PCR product was ligated with the PGH insert (BamHI/BbsI) of pbl/sk-PGH and the 3′ end ApaI/EcoRI fragment (1.46 kb) of RFII into pcDNA3 to give pcDNA3-FIIHRR. A 0.37-kb fragment containing the RFII propeptide and Gla sequences was amplified from pcDNA3-RFII with PM1 and PM2 and subcloned into pBluescript (+/−) to generate pbl/sk-PGR. PM4 and PM6 were used to amplify a 0.47-kb HFII fragment from pcDNA3-HFII, and the BbsI/BstEII digest of the PCR product was ligated with the PGR insert (BamHI/BbsI) of pbl/sk-PGR and the 3′ end BstEII/EcoRI fragment (1.3 kb) of HFII into pcDNA3 to give pcDNA3-FIIRHH.
PcDNA3-FIIHHR, PcDNA3-FIIRRH, PcDNA3-FIIHRH, PcDNA3-FIIRHR, PcDNA3-FIIHrhH, PcDNA3-FIIHhrH, PcDNA3-FIIRhrR, and PcDNA3-FIIRrhR.
The following sequences were amplified as the 0.7-kb human kringle (KH) with PM4 and PM10, the 0.7-kb rat kringle (KR) with PM4 and PM9, the 1.0-kb human protease domain (PH) with PM7 and PM11, the 1.0-kb rat protease domain (PR) with PM8 and PM11, the human kringle 1 domain (K1H) with PM15 and PM3, the human kringle 2 domain (K2H) with PM14 and PM10, the rat kringle 1 domain (K1R) with PM13 and PM3, and the rat kringle 2 domain (K2R) with PM12 and PM9. These PCR products were subcloned into pBluescript (+/−), respectively. PcDNA3-FIIHHR was generated by ligating the PGH insert (BamHI/BbsI) and the KH insert (BbsI) with the PR insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIRRH was generated by ligating the PGR insert (BamHI/BbsI) and the KR insert (BbsI) with the PH insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIHRH was generated by ligating the PGH insert (BamHI/BbsI) and the KR insert (BbsI) with the PH insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIRHR was generated by ligating the PGR insert (BamHI/BbsI) and the KH insert (BbsI) with the PR insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIHrhH was generated by ligating the PGH insert (BamHI/BbsI) and the K1R insert (BbsI) and K2H insert (BbsI) with the PH insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIHhrH was generated by ligating the PGH insert (BamHI/BbsI) and the K1H insert (BbsI) and K2R insert (BbsI) with the PH insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIRhrR was generated by ligating the PGR insert (BamHI/BbsI) and the K1H insert (BbsI) and K2R insert (BbsI) with the PR insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI). PcDNA3-FIIRrhR was generated by ligating the PGR insert (BamHI/BbsI) and the K1R insert (BbsI) and K2H insert (BbsI) with the PR Insert (BbsI/XbaI) into pcDNA3 (BamHI/XbaI).
Cell Culture and Transfections.
A human hepatoma cell line (HepG2) and a transformed human embryonic kidney cell line (HEK293) were cultured at 37°C in humidified air containing 5% CO2 in T-25 flasks (Corning) using DMEM supplemented with 10% FBS. Cells were transfected for 24 h by calcium phosphate coprecipitation (8) before fresh growth medium was added and incubated for 24 h. The transfected cells were then subjected to metabolic labeling and analysis of protein processing.
Metabolic Labeling, Immunoprecipitation, and SDS/PAGE.
Steady-state metabolic labeling was for 24 h as described previously (8). In pulse-labeling experiments, transfected cells were rinsed twice in methionine-depleted DMEM (DMEM-met) and incubated in the same medium for 30 min to deplete the endogenous methionine. DMEM-met supplemented with 10% dialyzed FBS and 250 μCi/ml [35S]methionine was then added and the cells were incubated for 5 min before the cell lysates were immunoprecipitated for prothrombin. Immunoprecipitation from cell lysates and media, and subsequent SDS/gel electrophoresis and fluorography were carried out as previously described (8). For detection of chimeric prothrombin in HEK293 cells, a mixture of 50% specific rabbit polyclonal anti-rFII and 50% specific rabbit polyclonal anti-hFII (8) was used for immunoprecipitation. For detection of chimeras (FIIHRR, FIIHHR, FIIRRH, FIIHHR, FIIHRH, FIIRHR, FIIHrhH, FIIHhrH, FIIRrhR, FIIRhrR) in HepG2 cells, the specific anti-rFII antibody was used. Citrate-washed BaSO4 was used to adsorb fully γ-carboxylated prothrombin, and under-γ-carboxylated prothrombin was defined as prothrombin remained in the supernatant after BaSO4 adsorption (8). Total protein secretion was measured by 10% trichloroacetic acid precipitation of cell media followed by quantitation of precipitated radioactivity with liquid scintillation spectrometry.
Drug Treatment of the Cells.
In steady-state metabolic labeling experiments, HepG2 and HEK293 cells were treated with vitamin K (10 μg phylloquinone/ml) or warfarin (1 μg/ml) in the presence of [35S]methionine for 24 h. In pulse-labeling experiments, cells first were treated with vitamin K (10 μg/ml) or warfarin (1 μg/ml) for 24 h before 5-min pulse labeling in the presence of vitamin K or warfarin. In some experiments, brefeldin A (10 μg/ml) alone or with nocodazole (20 μg/ml) was added to labeling cell medium, and the cells were incubated in the presence of vitamin K or warfarin for 24 h.
Enzymes and Reagents.
Oligonucleotides were synthesized by Ransom Hill Bioscience (Ramona, CA). Restriction enzymes, T4 DNA ligase, and Wizard DNA maxiprep kits were obtained from Promega. Calf intestine alkaline phosphatase was obtained from New England Biolabs. Mammalian expression vector pcDNA3 was purchased from Invitrogen.
The transformed human embryonic kidney cell line (HEK293) and the human hepatoma cell line (HepG2) were obtained from American Type Culture Collection. FBS was purchased from HyClone. [35S]methionine (43.5 TBq/mmol) was obtained from DuPont/NEN. Vitamin K was purchased from Abbott. Warfarin was provided by the Wisconsin Alumni Research Foundation (Madison, WI). Kodak x-ray film was purchased from Eastman Kodak. Fluoro-Hance solution was purchased from Research Products International. All other chemicals and reagents were purchased from Sigma.
RESULTS
Differential Processing of Rat (rFII) and Human (hFII) Prothrombin Through the Secretory Pathway of HEK293 Cells.
Plasmids of pcDNA3-RFII and pcDNA3-HFII were transfected into HEK293 cells, and prothrombin production was assessed. In the presence of vitamin K (Fig. 1), transiently expressed rFII and hFII were both efficiently secreted into the culture media and appeared to be fully γ-carboxylated based on their adsorption to BaSO4. The secretion of hFII was not altered by warfarin treatment (105% of vitamin K-sufficient secretion), whereas only 31% as much rFII was secreted in the presence of warfarin (see Fig. 6 for quantitation of secretion). To determine whether the effect of warfarin on rFII secretion in HEK293 cells was a result of alteration of general protein secretion, synthesis of total secreted protein was measured (Fig. 2A) and found to be uninfluenced by warfarin treatment. The rate of synthesis of rFII, as measured by a 5-min pulse of [35S]methionine, was not altered by the presence of warfarin (Fig. 2B), establishing that the warfarin-induced decrease in prothrombin secretion resulted from intracellular degradation rather than an altered synthesis rate.
Figure 1.
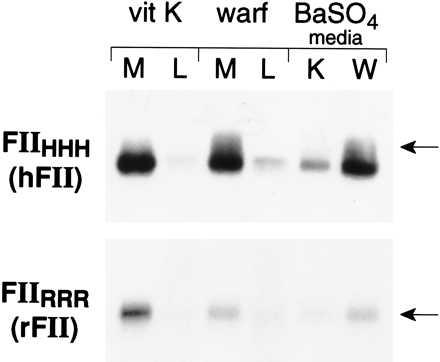
Expression of rFII and hFII in HEK293 cells. Cell cultures were transfected with pcDNA3-RFII or pcDNA3-HFII, and after 24 h cells were metabolically labeled with [35S]methionine (100 μCi/ml) in the presence of vitamin K (vit K) or warfarin (warf) for 24 h. Media (M) or lysates (L) were immunoprecipitated, electrophoresed, and detected as described in Materials and Methods. Under-γ-carboxylated prothrombin was defined as prothrombin remaining in solution after BaSO4 adsorption (lanes 5 and 6). The position of an 84-kDa molecular mass standard is indicated.
Figure 6.
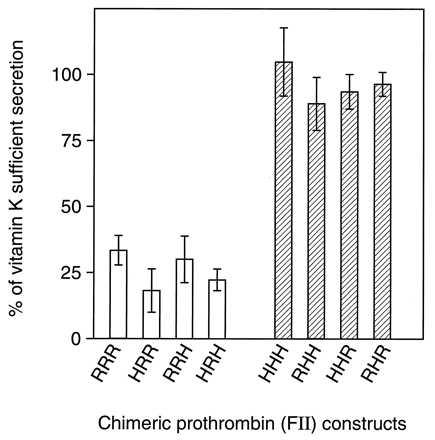
Quantitation of chimeric prothrombin construct expression in HEK293 cells. Gels from Figs. 1 and 5 were quantitated by densitometry. Ratios of lane 3 (medium from warfarin-treated cells) over lane 1 (medium from vitamin K-treated cells) were plotted as a percentage of vitamin K sufficient secretion. Data are mean ± SD (n = 3).
Figure 2.
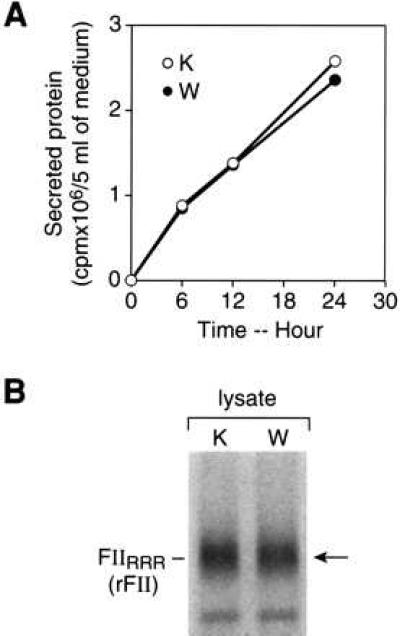
(A) Effect of vitamin K or warfarin on the synthesis of total secretory proteins in HEK293 cells. Confluent cells were incubated with vitamin K (K) or warfarin (W) in the presence of [35S]methionine. Media were withdrawn at the times indicated and centrifuged to remove cell debris, and protein was precipitated with 10% trichloroacetic acid. Precipitated radioactivity was determined by liquid scintillation spectrometry. (B) Effect of vitamin K or warfarin on the rate of biosynthesis of rFII in HEK293 cells. HEK293 cells were transfected with pcDNA3-RFII. After 24 h, the cells were incubated for 24 h followed by a 5-min pulse-labeling with [35S]methionine. Prothrombin in cellular lysates from three flasks was immunoprecipitated and detected by SDS/PAGE and fluorography. The position of an 84-kDa molecular mass standard is indicated. In two experiments, the amount of [35S]prothrombin in the warfarin-treated cells was 110% and 106% that of the vitamin K-treated cells.
Brefeldin A was used to block ER-to-Golgi transport in a 24-h metabolic labeling experiment (Fig. 3). The accumulation of rFII in the cellular lysate of warfarin-treated cells was only 35% of that observed in the presence of vitamin K, indicating that the degradation of rat prothrombin occurs in a pre-Golgi compartment. In contrast, the accumulation of hFII in cells treated with vitamin K or warfarin was not significantly different. Similar results were obtained in the presence of nocodazole, an ER–Golgi retrograde transport blocker, except that the general protein synthesis was decreased significantly in the presence of nocodazole (data not shown). These data suggest that structural differences in rFII and hFII must determine the fate of under-γ-carboxylated prothrombin in these cells. To assess these structural differences, chimeras between rFII and hFII cDNAs were constructed (Fig. 4).
Figure 3.
Effect of brefeldin A on the expression of rFII and hFII in HEK293 cells. HEK293 cells were transfected with pcDNA3-RFII or pcDNA3-HFII, and after 24 h the cells were metabolically labeled with [35S]methionine in the presence of brefeldin A and vitamin K (vit K) or warfarin (warf) for 24 h. Prothrombin in media (M) or lysates (L) was immunoprecipitated followed by SDS/PAGE and fluorography. The position of an 84-kDa molecular mass standard is indicated. Gels from three experiments were quantitated by densitometry, and the amount of prothrombin present in warfarin-treated cells was 95 ± 9.2% of that of the vitamin K-treated cells for hFII and 35 ± 3.6% for rFII.
Role of the Kringle, Catalytic, and Propeptide + Gla Domain of Prothrombin in Determining the Stability of Intracellular Under-γ-Carboxylated Prothrombin.
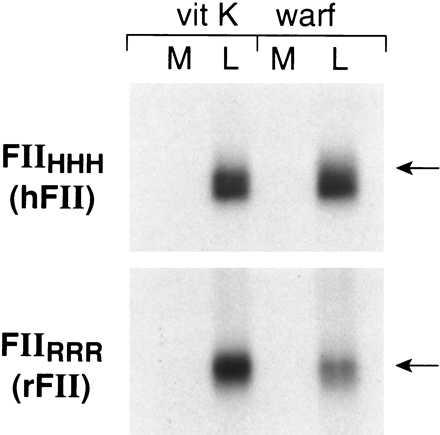
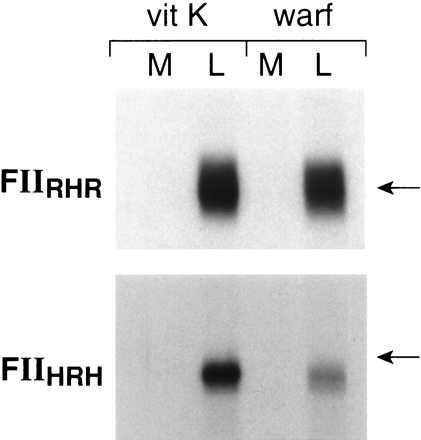
Plasmids containing the chimeric constructs shown in Fig. 4 were transfected into HEK293 cells, expressed, and metabolically labeled for 24 h in the presence of vitamin K or warfarin. The data in Figs. 5 and 6 indicate that in the presence of vitamin K, all chimeric proteins were secreted into the media and the secreted protein was largely adsorbed by BaSO4. In the presence of warfarin, constructs FIIRHH and FIIHHR were secreted to a similar extent as in the presence of vitamin K (Figs. 5 A and B), whereas the synthesis of constructs FIIHRR and RRH was drastically decreased in the presence of warfarin. These data suggested that neither the propeptide + Gla domain nor the catalytic domain was responsible for the instability of under-γ-carboxylated prothrombin. The decreased secretion of FIIHRH in the presence of warfarin compared with the lack of effect of warfarin on the secretion of FIIRHR (Fig. 5C) suggests that it is the kringle domain that is the determining structural feature in regulating degradation or secretion of under-γ-carboxylated prothrombin.
Figure 5.
Expression of chimeric prothrombins in HEK293 cells. Transfection, expression, metabolic labeling, immunoprecipitation, fluorography, and assay of metabolically labeled prothrombin as described in Fig. 1. The position of an 84-kDa molecular mass standard is indicated.
The decreased secretion of FIIHRH in the presence of warfarin (Fig. 5C) was not related to an effect on synthesis rate, because the amount of FIIHRH synthesized in warfarin-treated cells during a 5-min pulse-labeling period was found to be 103% of that seen in vitamin K-treated cells. When brefeldin A was included in the 24-h labeling medium to block the transport of proteins from ER to Golgi, much less FIIHRH accumulated in the warfarin-treated cells than in the vitamin K-treated cells (Fig. 7). These data suggest that the warfarin-induced intracellular degradation of FIIHRH within a pre-Golgi compartment was as extensive as it was for native rFII.
Figure 7.
Effect of brefeldin A on the expression of FIIHRH in HEK293 cells. Transfection with pcDNA3-FIIHRH and pcDNA3-FIIRHR, metabolic labeling, and analysis was performed as described in Fig. 3. Gels from three experiments were quantitated by densitometry, and the amount of prothrombin present in warfarin-treated cells was 90 ± 1% of that of the vitamin K-treated cells for FHRHR and 30 ± 6% for FIIHRH.
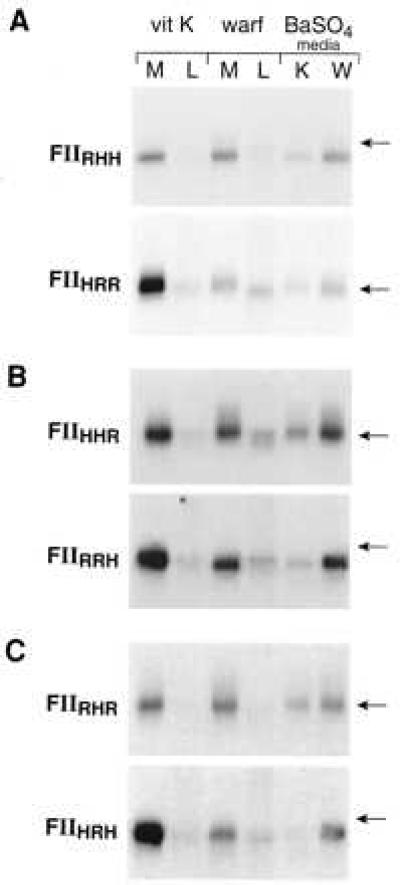
Expression, Processing, and Secretion of Chimeras in HepG2 Cells.
Because HEK293 cells do not synthesize endogenous vitamin K-dependent coagulation proteins, the protein processing machinery in this cell line may not be identical to that present in hepatocytes. The chimeric cDNAs, with the exception of pcDNA3-FIIRHH, were transfected into HepG2 cells, and their expression and secretion were analyzed (Fig. 8). Warfarin treatment drastically decreased the secretion of rat prothrombin carrying a human propeptide and Gla domain (FIIHRR) and increased the retention of this protein in the cell. Expression of chimeras with the catalytic domain switched (FIIRRH and FIIHHR) in vitamin K- or warfarin-treated HepG2 indicated that warfarin treatment almost completely abolished the secretion of FIIRRH whereas FIIHHR was efficiently secreted. In the presence of vitamin K, both FIIRHR and FIIHRH (kringle domain switched) were efficiently secreted. However, in the presence of warfarin, the secretion of FIIHRH was eliminated whereas a large amount of FIIRHR was secreted with only limited intracellular retention. This difference in the response of FIIHRH and FIIRHR to warfarin is identical to that of rFII and hFII and establishes that the kringle domain plays an important role in the intracellular processing of prothrombin in HepG2 cells as well as in HEK293 cells.
Figure 8.
Effect of vitamin K or warfarin on synthesis of prothrombin chimeras in HepG2 cells. HepG2 cells were transfected with pcDNA3-FIIHRR, FIIHHR, FIIRRH, FIIHRH, and FIIRHR, and prothrombin expression was analyzed as described in Fig. 1. A specific rabbit polyclonal antibody to rFII that has no detectable cross-reactivity to hFII(P) was used. The position of an 84-kDa molecular mass standard is indicated.
The Kringle 1 Structure Is the More Important Determinant.
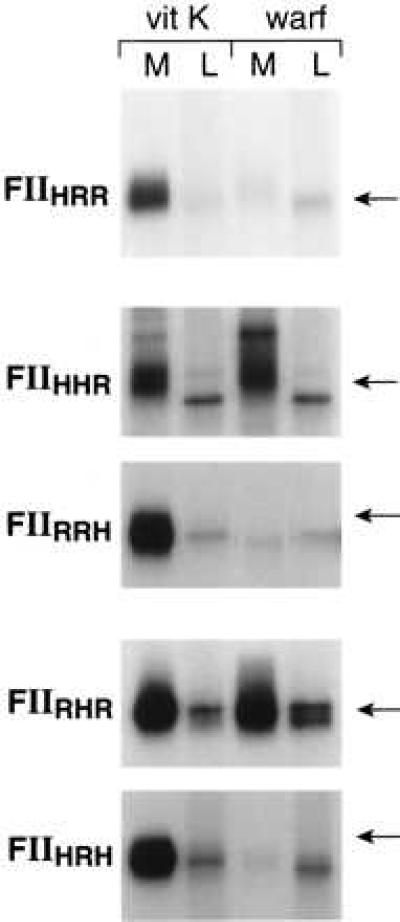
There are two separate kringle structures in the kringle domain of prothrombin. To determine which of the kringles is more important in influencing intracellular stability, rFII and hFII chimeras with individual kringle structures exchanged were transiently expressed in both 293 cells and HepG2 cells. The data in Fig. 9 demonstrate that in HEK293 cells both FIIHhrH (kringle 2 of hFII replaced by that of rFII) and FIIHrhH (kringle 1 of hFII replaced by that of rFII) were expressed normally in the presence of vitamin K as shown by their efficient secretion and adsorption to BaSO4. In the presence of warfarin, both chimeras were also efficiently secreted but under-γ-carboxylated because they were not adsorbed to BaSO4. However, the secretion of FIIHhrH was not altered in response to warfarin in HEK293 cells, whereas the secretion of FIIHrhH was decreased significantly. These data suggest that kringle 1, not kringle 2, of rFII is most important in destabilizing the under-γ-carboxylated chimera protein precursors in the HEK293 cells. Data in Fig. 10 demonstrate a similar trend in HepG2 cells. In the presence of vitamin K, both of these two chimeras are efficiently secreted. The secretion of FIIHhrH was not altered by warfarin treatment and was identical to the endogenous hFII, whereas the secretion of FIIHrhH was decreased significantly (about 50%) in response to warfarin. Because the secretion of FIIHRH was completely abolished by warfarin treatment in HepG2 cells (Fig. 8), it appears that the kringle 1 structure of rFII destablizes under-γ-carboxylated chimera protein precursors in HepG2 cells but to a lesser extent than the whole kringle domain. Expression of two other chimeras, FIIRhrR and FIIRrhR, in HEK293 cells and HepG2 cells also suggested that both kringle structures were involved in determining the stability of under-γ-carboxylated prothrombin, but the kringle 1 structure again appeared to be more important in this regard (data not shown).
Figure 9.
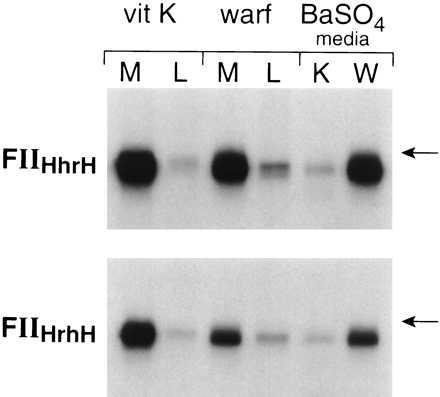
Expression of FIIHrhH and FIIHhrH in HEK293 cells. Transfection, expression, metabolic labeling, immunoprecipitation, and autoradiography were as described in Fig. 1. The position of an 84-kDa molecular mass standard is indicated.
Figure 10.
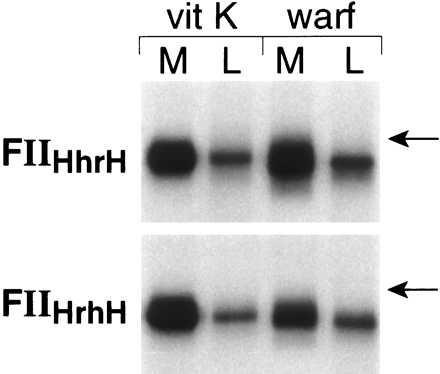
Expression of FIIHrhH and FIIHhrH in HepG2 cells. Transfection, expression, metabolic labeling, immunoprecipitation, and fluorography were as described in Fig. 8. The position of an 84-kDa molecular mass standard is indicated.
DISCUSSION
The different response to warfarin of prothrombin secretion in the rat and human is well recognized (3–5, 7, 8). Because the same differential response to warfarin was observed between the two prothrombins in HepG2 cells stably expressing rFII (8), the response must be based on structural differences. The data reported here demonstrate the same differential response to warfarin in HEK293 cells transiently expressing rFII and hFII despite the lack of the warfarin-induced retention of under-γ-carboxylated proteins observed in hepatocytes (Fig. 1). In this cell line, the secretion of rFII but not hFII was greatly decreased by warfarin. This decline in secretion was caused by selective degradation of under-γ-carboxylated rFII as evidenced by equal rates of rFII biosynthesis in the presence of vitamin K or warfarin (Fig. 2B) and by the decreased accumulation of rFII in warfarin-treated cells (Fig. 3). These data demonstrate that HEK293 cells are a suitable model to study this well recognized differential response of rFII and hFII to warfarin and to define the corresponding structural determinant.
The C-terminal catalytic domain constitutes more than 50% of the prothrombin molecule, and prothrombin from rat and human shares about 86% amino acid sequence identity in this region (11). It is clear (Fig. 6) that this region of the protein does not contain the necessary signal for the preferential degradation of the under-γ-carboxylated form of rFII rather than hFII. Similarly, efficient secretion of FIIRHH indicates that differences in the propeptide and Gla domains of rFII and hFII do not determine their different metabolic fate of the under-γ-carboxylated proteins. The sequence identity in this region is also high (86%) between the two proteins (11). Although the available data demonstrate that structural differences in the propeptide and Gla domains are not responsible for the different fate of the under-γ-carboxylated rFII and hFII, these structures are important in the overall process. The Gla domain is the region containing 10 Glu residues that are specifically γ-carboxylated into Gla residues, and the propeptide domain, which is cleaved in the Golgi compartment, is the targeting sequence of the protein substrates for this vitamin K-dependent γ-glutamyl carboxylation.
The kringle domain of prothrombin contains the sequence of the highest diversity (11) between rat and human species (69% amino acid sequence identity). The data in Figs. 5 and 6 clearly demonstrate that structural differences in the kringle domain are sufficient to cause the observed differential secretion/degradation of under-γ-carboxylated rFII or hFII in HEK293 cells. The significance of the kringle domain in the intracellular processing/degradation of prothrombin was strongly supported by data from a human hepatoma HepG2 cell line (Fig. 8), where endogenous vitamin K-dependent proteins are constitutively produced and secreted. The data in Figs. 9 and 10 also suggest that although the kringle 1 structure apparently plays a more important role in determining the stability of under-γ-carboxylated prothrombin precursors in the ER, structural features of both kringles are important. Kringle domains have been identified in prothrombin and other coagulation and fibrinolytic proteins such as plasminogen, tissue-type plasminogen activator (t-PA), and urokinase, as well as apolipoprotein(a) and hepatic growth factor (12, 13). The main function of kringle domains has been thought to involve interactions with substrate cofactors or receptors (14). In prothrombin, the kringle 2 structure is implicated in binding to its cofactor, factor Va (15). These studies have defined a new role of the kringle domain—that of regulating prothrombin processing through the secretory pathway.
X-ray structural analysis of fragment 1 (containing the Gla and kringle 1 structures) of bovine prothrombin (16, 17) and NMR analysis of the Gla and epidermal growth factor domains of factor X (18) have shown that the presence or absence of Ca2+ results in drastic structural alterations in the Gla domain of prothrombin and factor X. These conformational changes in the Gla domain might cause global conformation alterations, especially in the adjacent kringle structures. The selective degradation of under-γ-carboxylated prothrombin is a γ-carboxylation-related phenomenon, and the propeptide and Gla domains might interact in some fashion with the adjacent kringle domain during protein processing in the ER. When γ-carboxylation fails, the interactions between the kringle domain and the propetide/Gla domain may be disrupted in a manner that signals the selective degradation of under-γ-carboxylated rFII precursors. However, under-γ-carboxylated hFII apparently is protected from the degradation process and is secretion-competent in HepG2 cells. It is possible that certain cellular factors are involved in the degradation process and that the mode of interaction between these putative protein factors and under-γ-carboxylated precursors is dictated by the structural differences of the kringle domain between the two prothrombins. These quality-control mechanisms may be responsible for the endoplasmic reticulum degradation of unassembled and misfolded proteins, such as the unassembled subunits of T cell receptor and the mutant cystic fibrosis transmembrane conductance regulator (ΔCFTR) (19–22). A similar quality-control mechanism might be present for the processing of vitamin K-dependent proteins.
Although the most recent evidence suggests that the ER degradation of mutant integral membrane proteins is initiated by export of proteins to the cytoplasmic side followed by a proteasome-dependent pathway (see recent review, ref. 23), the mechanisms of degradation of mutant-soluble proteins in the ER are less clear. Recently, the carboxyl-terminal region of factor IX was found to be essential for its secretion, and mutant factor IX proteins carrying mutations in this region are subjected to a proteasome-dependent degradation (24). Tokunaga et al. (25) have reported a rapid ER degradation of protein C in warfarin-treated HEK293 cells and have implicated a cysteine protease in this degradation process. During warfarin therapy and chronic/acute liver diseases, conditions in which vitamin K utilization is impaired, the plasma levels of vitamin K-dependent proteins are depressed to varying degrees. The data presented here suggest that ER degradation confers a general quality-control mechanism for this group of proteins in the protein-trafficking pathway. This study is one of the first efforts to identify the molecular determinants for this process and explains the apparent differences in this quality-control mechanism between the prothrombin of two species.
Acknowledgments
We gratefully acknowledge the support of the College of Agricultural and Life Sciences and The Medical School of the University of Wisconsin–Madison, and Grant DK14881 from the National Institutes of Health, Bethesda, MD.
References
- 1.Suttie J W. FASEB J. 1993;7:445–452. doi: 10.1096/fasebj.7.5.8462786. [DOI] [PubMed] [Google Scholar]
- 2.Suttie J W. In: The New Dimensions of Warfarin Prophylaxis. Wessler S, Becker C G, Nemerson Y, editors. New York: Plenum; 1987. pp. 3–16. [Google Scholar]
- 3.Stenflo J. Acta Chem Scand. 1970;24:3762–3763. doi: 10.3891/acta.chem.scand.24-3762. [DOI] [PubMed] [Google Scholar]
- 4.Ganrot P O, Nilehn J E. Scand J Clin Lab Invest. 1968;22:23–28. [PubMed] [Google Scholar]
- 5.Shah D V, Swanson J C, Suttie J W. Thromb Res. 1984;35:451–458. doi: 10.1016/0049-3848(84)90236-6. [DOI] [PubMed] [Google Scholar]
- 6.Yamanaka Y, Yamano M, Yasunaga K, Shike T, Uchida K. Thrombosis Res. 1990;57:205–214. doi: 10.1016/0049-3848(90)90320-c. [DOI] [PubMed] [Google Scholar]
- 7.Zhang P, Suttie J W. Blood. 1994;84:169–175. [PubMed] [Google Scholar]
- 8.Wu W, Bancroft J D, Suttie J W. Thromb Haemostasis. 1996;76:46–52. [PubMed] [Google Scholar]
- 9.Mann K G. In: Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 3rd Ed. Colman R W, Hirsh J, Marder V J, Salzman E W, editors. Philadelphia: Lippincott; 1994. pp. 184–199. [Google Scholar]
- 10.Stemmer W P C, Morris S K. Biotechniques. 1992;13:215–220. [PubMed] [Google Scholar]
- 11.Degen S J F. Semin Thromb Hemostasis. 1992;18:230–242. doi: 10.1055/s-2007-1002429. [DOI] [PubMed] [Google Scholar]
- 12.McLean J W, Tomlinson J E, Kuang W-J, Eaton D L, Chen E Y, Fless G M, Scanu A M, Lawn R M. Nature (London) 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura T, Nishizawa T, Hagiya M, Saki T, Shimonishi M, Sugimura A, Tashero T, Shimuzu S. Nature (London) 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 14.Patthy L, Trexler M, Vali Z, Banyai L, Varadi A. FEBS Lett. 1984;171:131–136. doi: 10.1016/0014-5793(84)80473-1. [DOI] [PubMed] [Google Scholar]
- 15.Esmon C T, Jackson C M. J Biol Chem. 1974;249:7791–7797. [PubMed] [Google Scholar]
- 16.Tulinsky A, Park C H, Skrzypczak-Jankun E. J Mol Biol. 1988;202:885–901. doi: 10.1016/0022-2836(88)90565-7. [DOI] [PubMed] [Google Scholar]
- 17.Soriano-Garcia M, Park C H, Tulinsky A, Ravichandran K G, Skrzypczak-Jankun E. Biochemistry. 1989;28:6805–6810. doi: 10.1021/bi00443a004. [DOI] [PubMed] [Google Scholar]
- 18.Sunnerhagen M, Forsen S, Hoffren A M, Drakenberg O T, Stenflo J. Nat Struct Biol. 1995;2:504–509. doi: 10.1038/nsb0695-504. [DOI] [PubMed] [Google Scholar]
- 19.Rajagopalan S, Xu Y H, Brenner M B. Science. 1993;263:387–390. doi: 10.1126/science.8278814. [DOI] [PubMed] [Google Scholar]
- 20.Ward C L, Kopito R R. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 21.Rose J K, Doms R W. Annu Rev Cell Biol. 1988;4:257–288. doi: 10.1146/annurev.cb.04.110188.001353. [DOI] [PubMed] [Google Scholar]
- 22.Hurtley S M, Helenius A. Annu Rev Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- 23.Brodsky J L, McCracken A A. Trends Cell Biol. 1997;7:151–156. doi: 10.1016/S0962-8924(97)01020-9. [DOI] [PubMed] [Google Scholar]
- 24.Kurachi S, Pantazatos D P, Kurachi K. Biochemistry. 1997;36:4337–4344. doi: 10.1021/bi962002v. [DOI] [PubMed] [Google Scholar]
- 25.Tokunaga F, Wakabayashi S, Koide T. Biochemistry. 1995;34:1163–1170. doi: 10.1021/bi00004a009. [DOI] [PubMed] [Google Scholar]