

Abstract
Mitochondrial dysfunction involves defective insulin secretion by pancreatic beta-cells, and insulin resistance in insulin-sensitive tissues such as muscle and adipose tissue. Mitochondria are recognized as the most important cellular source of energy, and the major generator of intracellular reactive oxygen species (ROS). Intracellular antioxidative systems have been developed to cope with increased oxidative damage. In case of minor oxidative stress, the cells may increase the number of mitochondria to produce more energy. A mechanism called mitochondrial biogenesis, involving several transcription factors and regulators, controls the quantity of mitochondria. When oxidative damage is advanced beyond the repair capacity of antioxidative systems, then oxidative stress can lead to cell death. Therefore, this organelle is central to cell life or death. Available evidence increasingly shows genetic linkage between mitochondrial DNA (mtDNA) alterations and type 2 diabetes (T2D). Based on previous studies, the mtDNA 16189 variant is associated with metabolic syndrome, higher fasting insulin concentration, insulin resistance index and lacunar cerebral infarction. These data support the involvement of mitochondrial genetic variation in the pathogenesis of T2D. Importantly, phylogeographic studies of the human mtDNAs have revealed that the human mtDNA tree is rooted in Africa and radiates into different geographic regions and can be grouped as haplogroups. The Asian populations carry very different mtDNA haplogroups as compared to European populations. Therefore, it is critically important to determine the role of mtDNA polymorphisms in T2D.
Keywords: type 2 diabetes, mitochondrial DNA, reactive oxygen species, biosynthesis, phenotype
Abbreviations: ATP - adenosine-5'-triphosphate; AMPK - adenosine monophosphate-activated protein kinase; BMI - body mass index; Chaperon - protein between the mitochondrial membranes; Cu/ZnSOD - copper/zinc superoxide dismutase; D-loop - displacement loop; GPx - glutathione peroxidase; HVR - highly variable region; IGT - impaired glucose tolerance; MnSOD - manganese superoxide dismutase; mtDNA - mitochondrial DNA; mTFB - mitochondrial transcription factor B; NRF - nuclear respiratory factor; PGC-1α - PPARγ coactivator-1α; PPARγ - peroxisome proliferator-activated receptor γ; RCC - respiratory chain complex; rCRS - revised Cambridge reference sequence; ROS - reactive oxygen species; rRNA - ribosomal ribonucleic acid; SNP - single-nucleotide polymorphism; T2D - type 2 diabetes; Tfam - mitochondrial transcription factor A; TFB - transcription factor B; TIM - translocase of the mitochondrial inner membrane; TOM - translocase of the mitochondrial outer membrane; tRNA - transfer ribonucleic acid
Mitochondria and diabetes
There is evidence that mitochondria are involved in the development of diabetes. Epidemiological studies have revealed that the inherited transmission of type 2 diabetes (T2D) is maternally influenced [1-2]. Diabetes is also frequently associated with mitochondrial diseases [3-8]. Recent studies have added evidence that reactive oxygen species (ROS) generated from mitochondria play a major role in the pathogenesis of diabetic complications [9-11]. Inherited defects in mitochondrial oxidative phosphorylation were found in muscle cells of insulin-resistant offspring of diabetic parents [12]. These findings support the concept of mitochondrial involvement in T2D.
Mitochondria are the major source of cellular energy. Defects in mitochondrial DNA (mtDNA) may lead to dysfunction of enzymes involved in mitochondria respiratory chains. Also, increased ROS production, a consequence of ineffective electron transportation, may cause further cellular damage. Mitochondrial dysfunction may cause diabetes by the following mechanisms:
1. Defective mtDNA may impair ATP production caused by a dysfunction of encoded respiratory chain enzymes and impaired insulin secretion of beta-cells.
2. Defects in oxidative phosphorylation in insulin-sensitive tissue could impair insulin action.
Mitochondrial DNA
Mitochondria have their own DNA. However, mtDNA is dependent on nuclear genes for its replication and expression. Mitochondrial genome is highly compacted on the double-stranded circular mtDNA containing 16569 bp in length. The genes are arranged in two strands, the outer circle, a guanine-rich heavy (H) strand, and the inner circle, a cytosine-rich light (L) strand. They encode 13 polypeptides, 22 tRNA, and 2 rRNA which are required for oxidative phosphorylation [13]. The genes are tightly packed without introns. Mitochondrial DNA contains a displacement loop (D-loop), a non-coding control region of approximately 1.1 kb (between position 16024 and 576). Initiation of mtDNA replication in cells occurs within the D-loop [14, 15].
The D-loop is frequently associated with sequence variation. Greenberg et al. were the first to describe two separate regions with high nucleotide diversity i.e. the highly variable region 1 (HVR-1) between positions 16024 and 16365, and HVR-2 between positions 73 and 340 [16]. An additional HVR-3 region between positions 438 and 574 was later found by Lutz et al. [17]. Sequence data of the mtDNA D-loop have frequently been used in studies of population evolution, in anthropology applications, and in forensic practice studies [18-21].
Human mtDNA is more susceptible to oxidative damage and consequently acquires mutations at a higher rate than nuclear DNA. This is due to elevated exposure of mtDNA to high ROS levels generated during respiration, lack of protective histones, and limited capacity for repair of mtDNA damage [22, 23].
Mitochondrial biogenesis controls cell function and survival
Mitochondria are the intracellular organelles responsible for supplying most of the cellular energy needs. This is created by producing ATP through oxidative phosphorylation via the respiratory chain complex in its inner membrane. Each cell contains several hundreds to more than a thousand mitochondria. Each mitochondrion contains 2-10 copies of mtDNA. The abundance of mitochondria and mtDNA copies vary dramatically in different stages of energy demand and physiological conditions, and is tightly controlled by the mechanism called mitochondrial biogenesis. Mitochondrial biogenesis can vary in different organs reflecting the different requirements of each organ’s specific innate function [24-26].
Biosynthesis of mitochondrial proteins requires contribution from mitochondria and the nucleus, but most of them are encoded by nuclear genes and synthesized outside of mitochondria. The assembly and functioning of respiratory enzyme complexes in cells require coordinated expression and interaction between gene products of mitochondria and nuclear genomes [27-29]. Mitochondrial biogenesis is controlled by a complex cascade of events activated in response to environmental stress. The activation of the nuclear transcriptional coactivator PPARγ coactivator-1α (PGC-1α) gene, a major regulator of mitochondrial biogenesis induced by the environmental signals, triggers the process. After activation, PGC-1α regulates the expression of transcription factors involved in the coordinated expression of mitochondrial genes such as nuclear respiratory factors (NRF-1 and NRF-2). This event in turn triggers the expression of nuclear genes coding for polypeptides of the respiratory chain and proteins involved in transcription and replication of mtDNA. Both NRF-1 and NRF-2 can regulate the expression of mitochondrial transcription factor A (Tfam) and B (mTFB). These factors are then imported into the mitochondrial matrix, act on the promoters within the D-loop region of mtDNA, and regulate replication and transcription of the mitochondrial genome (Figure 1). This provides a unique mechanism for the cell to integrate the expression of nuclear DNA-encoded proteins with the transcription of genes encoded by mtDNA. Hence, it is possible that the effectiveness of mitochondrial biogenesis may be altered or impaired by polymorphisms in the nuclear genes of PGC-1α, NRF-1 and 2, Tfam, or by base changes in the D-loop of mtDNA. Such genetic variations may alter the activities of transcription factors and regulators.
Figure 1. Molecular pathway for mitochondrial biogenesis.
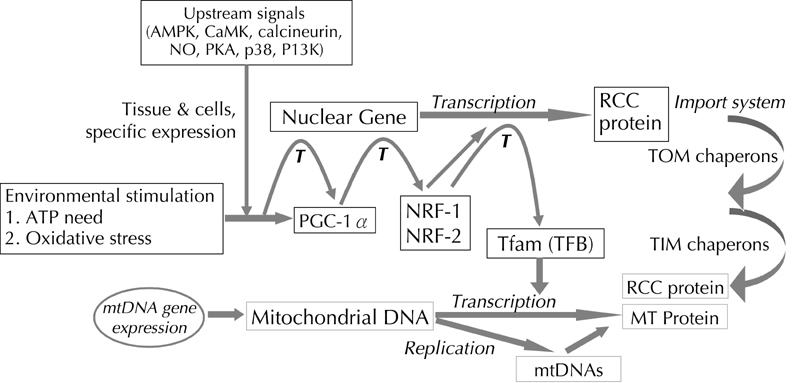
The figure shows the molecular pathway of mitochondrial biogenesis. The two major initial events are transcription and replication of mitochondrial DNA. These events are the pre-requisite for ATP production to fulfill the energy requirements of tissue cells. This is exactly controlled by sequential events in response to environmental or oxidative stress. The activation of the nuclear transcriptional coactivator-PGC-1α gene by these signals triggers the next step in the process. PGC-1α, acting as a coactivator, binds to the corresponding nuclear genes to help the translation of a series of nuclear DNA-encoded respiratory enzymes and mitochondrial transcription factors A (Tfam) and B (mTFB). The latter two proteins are also cooperatively transcribed by specific nuclear respiratory factors (NRF-1 and 2). These factors are then imported into the mitochondrial matrix to activate further processes.
Alterations in mitochondrial biogenesis could be the underlying pathologic factor for some important human diseases. This is particularly relevant in chronic diseases such as diabetes mellitus [9-11], renal insufficiency [30-33], liver disease [34-36], and neurodegenerative diseases [37-39]. Persistent cell damage by excessive exposure to free radicals resulting from the byproducts of mitochondrial biogenesis can contribute to the development and progressive deterioration of these diseases.
Mitochondia and reactive oxygen species (ROS)
Mitochondria are the main intracellular source and immediate target of ROS. Approximately 1-5% of the oxygen consumed by mitochondria in tissue cells is converted to ROS under normal physiological conditions. Defects in the respiratory chain in affected tissue of patients with mitochondrial disease or aged individuals contribute to increased production of superoxide anions by mitochondria [22, 40, 41]. Approximately 90% of oxygen in the cell is consumed by mitochondria and the mitochondrial respiratory chain is the source of continuing flux of oxygen radicals. Therefore, these organelles are susceptible to contribute to oxidative damage generated in situ [42].
To deal with the continuing ROS production by aerobic metabolism, cells have developed antioxidative enzymes, including mitochondrial manganese superoxide dismutase (MnSOD), copper/zinc superoxide dismutase (Cu/ZnSOD), glutathione peroxidase (GPx), and catalase [43]. Although these enzymes in combination with other antioxidants can dispose of most of the ROS and free radicals generated under normal condition, a fraction of ROS may escape the defense mechanism and cause damage to critical cellular macromolecules including nucleic acids, proteins, and lipids [44, 45].
When cells, with adequate antioxidant capacity and good quality of parental mitochondria, are activated in response to mild environmental oxidative stress, and if energy supply is decreased, then mitochondria can increase the abundance of structural proteins and mtDNA molecules [46]. This may result in an increase of energy supply by increased mitochondrial biogenesis. However, when the capacity of the antioxidant system is compromised, then the exposure of tissue cells to higher oxidative stress results in an increase of defective mitochondria and mutated mtDNA. mtDNA encodes essential polypeptides involved in oxidative phosphorylation. Respiratory enzymes containing defective protein subunits encoded by mutated mtDNA may cause mitochondrial dysfunction. On the other hand, restimulated mitochondrial ROS production and oxidative mtDNA damage eventually cause a decline in mitochondria function and cell death [11, 26, 40]. Therefore, another important function of mitochondria is to act as a regulator in the initiation and execution of programmed cell death, or apoptosis [47-49]. Recent evidence suggests that mitochondria play a crucial role in the determination of cell life or death [25].
Mitochondrial DNA variants and diabetes mellitus
There is compelling evidence for a genetic predisposition to diabetes [50-52]. Early in 1962, James Neel addressed the question of how diabetes, an apparently genetic disease with such an adverse effect on survival, could have become so common [53]. His observation of the high T2D frequency in previously undernourished communities raised the "thrifty genotype" hypothesis, which suggested that the predisposition to T2D carry some selective advantage in evolutionary history. The hypothesis is supported by the high prevalence and strong familial association of T2D in the population of Pima Indians [54, 55] and Polynesians [56]. The thrifty genotype may have contributed to their survival during centuries of poor nutrition, but increased the risk of diabetes following recent urbanization.
Equally convincing is the "thrifty phenotype" hypothesis [57]. A strong association between small size at birth and the risk of developing the metabolic syndrome in adult life has been reported since 1989 in studies of a number of cohorts followed up from birth on [58-65]. Barker and Hales proposed that maternal nutrition programs fetal metabolism and predisposes to T2D later in life [60]. Their hypothesis was supported by data from the Dutch famine study [66], which demonstrated that short periods of maternal malnutrition can permanently affect glucose homeostasis in the offspring. However, "thrifty genotype" and "thrifty phenotype" may not be exclusive to each other. Both concepts could be explained by genotype that promoted survival during earlier nutritional adversity but later add to the risk of T2D. Since mitochondria are responsible for supplying most of the cellular energy needs, it has been hypothesized that mitochondria might play a role in "thrifty genotype" [67].
Quite a number of case studies have found a link between the occurrence of diabetes and mitochondrial genetic variation such as point mutation [3-6], deletion [7] and duplication [8]. The commonest single mutation of mtDNA, which may lead to diabetes, is located at bp 3243 G:C relative to the reference sequence [68]. This point mutation causes maternally inherited diabetes and deafness (MIDD) [3], mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) syndrome or a progressive kidney failure [69]. Since the 3243A>G mutation coexists in cells with wild-type mtDNA, the mutation load in different tissue may affect the different phenotypes associated with this mutation. Even in the group of presumed normoglycemic 3243A>G-positive individuals, a substantial fraction has diabetes or impaired glucose tolerance in oral glucose tolerance test screening [70]. However, the commonest A3243G mutation has been found in only 1.5% among the idiopathic diabetes forms [71]. This is too rare to account for a major cause of diabetes.
In addition to qualitative changes caused by mtDNA variations, quantitative defects of mitochondria have also been proposed to be the underlying mechanism of insulin resistance in T2D [72-74]. Changes in mtDNA content in leukocytes of diabetes patients have been reported previously. However, controversy exists regarding the contribution of mtDNA content to the development of T2D, because changes of the mtDNA copy number may be secondary to hyperglycemia rather than being the major cause of insulin resistance [75-77]. In addition, discrepancies in the effect of glucose metabolism on mtDNA copy number may be attributed to the nature of cell types [78].
A common transitional variant at bp 16189 (T>C transition) in the first hypervariable segment of the mtDNA control region (D-loop) initially showed an unusually high incidence in the MELAS phenotype group as compared to controls (67.0% vs. 22.4%) [79]. It was suggested that the 16189 variant reflects a predisposition towards the formation or fixation of mtDNA mutation. This 16189 variant was later found to be associated with elevated fasting insulin levels in men born in Hertfordshire, UK, between 1920 and 1930, in whom the link between small birth size and impaired glucose tolerance (IGT) at age 64 was confirmed [80, 81]. Moreover, a population-based case-control study in Cambridgeshire, UK, demonstrated a significant association between the 16189 variant and T2D [82].
In Asia, a hospital-based case-control study found an association between the mtDNA 16189 variant and lacunar cerebral infarction in a Chinese population [83]. Another case-control study in Chinese aged 40 or older revealed that the mtDNA 16189 variant was associated with the metabolic syndrome. This association remained significant after correcting for age and body mass index (BMI). Furthermore, the 16189 variant occurred more frequently in individuals with an increasing number of metabolic syndrome traits [84]. A follow-up study of 1,054 Chinese adults revealed that the proportion of subjects with the 16189 variant increased with higher fasting insulin concentration and insulin resistance index. Increased BMI was an aggravating factor for the development of T2D in subjects carrying the 16189 variant. The data exemplify an additive effect on the pathogenesis of T2D caused by genetic and environmental factors [85].
Although some recent case-control studies in Europe could not replicate the association between the 16189 variant and T2D [86-89], a multinational study in Asians including 2,469 T2D patients and 1,205 controls from Korea, Japan, Taiwan, Hong Kong, and China confirmed the role of the mtDNA 16189 variant at least in Asian T2D patients [90]. The prevalence rate of the 16189 variant of mtDNA in Taiwan Chinese adults was 38.2% (34.6% in non-diabetic and 43.1% in diabetic subjects) [84]. This finding was similar to those reported for Koreans (28.8%) [91], Japanese (34.4%) [92], Mainland Chinese (30%; non-diabetes 20-26.6%, T2D 33-36.9 %) [93, 94], Indonesia (10-60%, major population 32-47%) [95, 96], and Kuna Amerinds of Panama (28.5%) [97]. However, it was higher than the prevalence reported for Anglo-Saxon Caucasians (8.2%; non-diabetes 6.4%, T2D 9.9%) [82] and Indians (12.2%) [98]. Following the trail of ancient human migrations from Africa to the Asian Pacific region [99], we can observe an increasing frequency of the mtDNA 16189 variant along this path (Figure 2). The magnitude of influence of a particular genetic variant on disease susceptibility may dependent on the prevalence of this variant in the population. We hypothesized that the rapid increase of T2D in Asians under the influence of Western lifestyle may partly be explained by the high prevalence of the mtDNA variant. This hypothesis still needs further clarification.
Figure 2. The prevalent rate of the mtDNA 16189 variant traced along the ancient path of human migration.
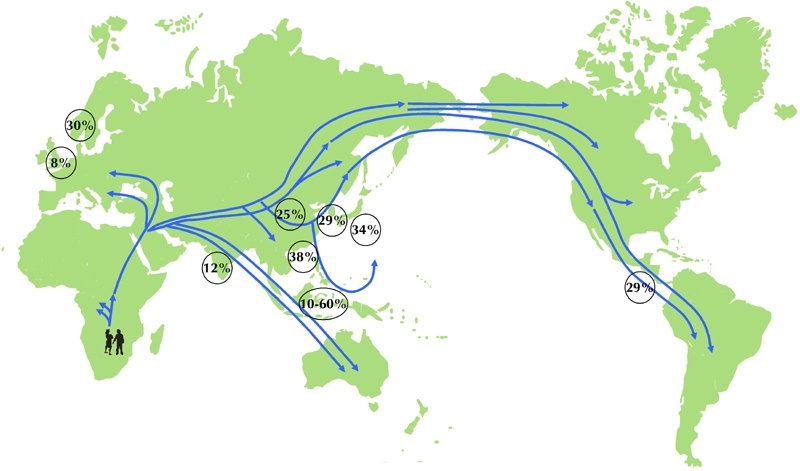
The encircled data represent the percentages of the population in a given country/region with the mtDNA 16189 variant according to available study data. The incidences are given for England (8%), Finland (30%), India (12%), China (25%), Korea (29%), Japan (34%), Taiwan (38%), Indonesian archipelago (10%-60%), and Panama (29%) [82, 85, 89, 90-98]. The world map shows possible migration routes of different people, as suggested by studies of mitochondrial DNA. The map is modified from [100].
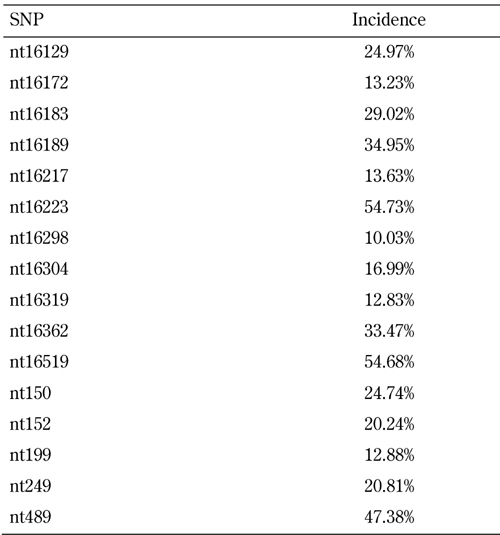
We have studied the polymorphism of mitochondrial D-loop DNA in 1,754 samples of peripheral leucocytes from a Chinese population. DNA sequences were analyzed by the DNASTAR Sequencing Analysis Software and compared with the revised Cambridge reference sequence (rCRS, last updated 05/16/2007). We found 324 variable positions within the 1100 bp mtDNA segment (nt16000 ~ nt502) in comparison to the rCRS. Table 1 shows the common genetic variants (>10%) in our series. The positions with the highest frequency of polymorphism (>30%) included nt16189, nt16223, nt16362, nt16519, and nt489. In this study, the T16217C mutation was more significantly associated with T2D than the T16189C mutation. Interestingly, we found that the T16217C mutation occurred simultaneously with the T16189C mutation, suggesting the existence of a linkage disequilibrium between T16217C and T16189C [101].
Table 1. Common polymorphisms (>10%) in a Chinese population compared with the revised Cambridge reference sequence (rCRS).
The possible mechanisms involved in the association of the 16189 variant with T2D are as follows:
1. Since the 16189C variant is located in the control region of mtDNA replication, it was suggested that the T>C transition results in a polycytosine tract that in turn may predispose the mtDNA to defects in replication [102, 103]. This hypothesis was not supported by two recent studies [88, 89].
2. T2D patients carrying the 16189C variant had impaired ability to respond properly to oxidative stress [104].
3. The mtDNA 16189 variant has a lower binding affinity to mitochondrial single-stranded DNA-binding protein, which is involved in mtDNA replication [90], and may impair mitochondrial biogenesis.
4. Possibly there is a linkage of the 16189 variant with other potential SNPs associated with diabetes. It is reasonable that haplotype analysis for determination of racial-specific characteristics will be more informative.
In our unpublished data, the haplotype B4 in the Chinese population is associated with T2D and the T16217C mutation. The latter occurred simultaneously with the T16189C mutation. This is a critical single-nucleotide polymorphism (SNP) for haplotype B4 classification. Tanaka et al. studied genotypes for 25 polymorphisms in the coding region of the mitochondrial genome in 1,337 unrelated Japanese individuals. Among the 10 haplogroups identified (F, B, A, N9a, M7a, M7b, G1, G2, D5, and D4), group N9a was significantly associated with resistance to the metabolic syndrome in women [105]. A second study involving 2,906 unrelated Japanese individuals and 1,365 unrelated Korean individuals further confirmed that haplogroup N9a is a resistant genotype to T2D [106].
Conclusions
Diabetes is accompanied by a group of risk factors of metabolic origin and an increased risk for cardiovascular disease. Identifying diabetes-susceptible genetic variants in humans has been challenging. Genome-wide association studies have detected at least 10 T2D-associated variants in nuclear DNA and emphasized the contribution of multiple variants of modest effect [107-110]. Although mtDNA is different from nuclear DNA, there is coordinated expression and interaction between the gene products of mitochondria and nuclear genomes. The role of defective oxidative metabolism related to mitochondrial dysfunction is not fully known and needs further clarification.
Disclosures: The authors report no conflict of interests.
Acknowledgments
This work was supported by research grants NSC 91-2314-B-182A-040, NSC 92-2314-B-182A-119, and NSC 91-2314-B-182A-132 from the National Science Council (Republic of China), and by grants CMRPG840551, CMRPG 850261, and CMRPG 850263 from Chang Gung University College of Medicine.
References
- 1.Alcolado JC, Alcolade R. Importance of maternal history of non-insulin dependent diabetic patients. BMJ. 1991;302 (6786):1178–1180. doi: 10.1136/bmj.302.6786.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin RS, Lee WC, Lee YT, Chou P, Fu CC. Maternal role in type 2 diabetes mellitus: indirect evidence for a mitochondrial inheritance. Int J Epidemiol. 1994;23(5):886–890. doi: 10.1093/ije/23.5.886. [DOI] [PubMed] [Google Scholar]
- 3.Van den Quweland JM, Lemkes HH, Ruitenbeek W, Sandkuijl LA, deVijlder MF, Struyvenberg PA, van de Kamp JJ, Maassen JA. Mutation in mitochondrial tRNA (leu) (UUR) gene in a large pedigree with maternally transmitted type 2 diabetes mellitus and deafness. Nat Genet. 1992;1(5):368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- 4.Tawata M, Hayashi JI, Isobe K, Ohkuno E, Ohtaka M, Chen J, Aida K, Onaya T. A new mitochondrial DNA mutation at 14577 T/C is probably a major pathogenic mutation for maternally inherited type 2 diabetes. Diabetes. 2000;49(7):1269–1272. doi: 10.2337/diabetes.49.7.1269. [DOI] [PubMed] [Google Scholar]
- 5.Reardon W, Ross RJ, Sweeney MG, Luxon LM, Pembrey ME, Harding AE, Trembath RC. Diabetes mellitus associated with a pathogenic point mutation in mitochondrial DNA. Lancet. 1992;340 (8832):1376–1379. doi: 10.1016/0140-6736(92)92560-3. [DOI] [PubMed] [Google Scholar]
- 6.Liou CW, Huang CC, Wei YH. Molecular analysis of diabetes mellitus-associated A3243G mitochondrial DNA mutation in Taiwanese cases. Diab Res Clin Pract. 2001;54(Suppl 2):S39. doi: 10.1016/s0168-8227(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 7.Ballinger SW. Maternally transmitted diabetes and deafness associated with a 10.4 Kb mitochondrial DNA deletion. Nat Genet. 1992;1(1):11–15. doi: 10.1038/ng0492-11. [DOI] [PubMed] [Google Scholar]
- 8.Rotig A. Maternally inherited duplication of the mitochondrial genome in a syndrome of proximal tubulopathy, diabetes mellitus, and cerebellar ataxia. Am J Hum Genet. 1992;50(2):364–370. [PMC free article] [PubMed] [Google Scholar]
- 9.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 10.Rosen P, Nawroth PP, King G, Moller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCOMCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17(3):189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- 11.Ceriello A, Ihnat MA, Thorpe JE. The "metabolic memory": is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab. 2009;94(2):410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- 12.Peterson KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grossman LI, Watson R, Vinograd J. The presence of ribonucleotides in mature closed-circular mitochondrial DNA. Proc Natl Acad Sci USA. 1973;70(12):3369–3343. doi: 10.1073/pnas.70.12.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnberg A, van Bruggen EF, Borst P. The presence of DNA molecules with a displacement loop in standard mitochondrial DNA preparations. Biochim Biophys Acta. 1971;246(2):353–357. doi: 10.1016/0005-2787(71)90147-x. [DOI] [PubMed] [Google Scholar]
- 15.Yasukawa T, Yang MY, Jacobs H, Holt IJ. A bidirectional origin of replication maps to the major noncoding region of human mitochondrial DNA. Mol Cell. 2005;18(6):651–662. doi: 10.1016/j.molcel.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg BD, Newbold IE, Sugino A. Intraspecific nucleotide sequence variability surrounding the origin of replication in human mitochondrial DNA. Gene. 1983;21(1-2):33–49. doi: 10.1016/0378-1119(83)90145-2. [DOI] [PubMed] [Google Scholar]
- 17.Lutz S, Weisser HJ, Heizmann J, Pollak S. A third hypervariable region in the human mitochondrial D-loop. Hum Gene. 1997;101(3):384. [PubMed] [Google Scholar]
- 18.Wallace D, Brown M, Lott M. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238(1):211–230. doi: 10.1016/s0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- 19.Castro JA, Picornell A, Ramon M. Mitochondrial DNA: a tool for populational genetics studies. Int J Legal Med. 1998;1(4):327–332. [PubMed] [Google Scholar]
- 20.Ballinger SW, Schurr TG, Torroni A, Gan YY, Hodge JA, Hassan K, Chen KH, Wallace DC. Southern Asian mitochondrial DNA analysis reveals genetic continuity of ancient mongoloid migrations. Genetics. 1992;130(1):139–152. doi: 10.1093/genetics/130.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coskun PE, Ruiz-Pesini E, Wallace DC. Control region mtDNA variants: Longevity, climatic adaptation, and a forensic conundrum. Proc Natl Acad Sci USA. 2003;100(5):2174–2176. doi: 10.1073/pnas.0630589100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yakes FM, van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94(2):514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croteau DL, Stierum RH, Bohr VA. Mitochondrial DNA repair pathways. Mutat Res. 1999;434(3):137–148. doi: 10.1016/s0921-8777(99)00025-7. [DOI] [PubMed] [Google Scholar]
- 24.Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- 25.Lee HC, Wei YH. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000;7(1):2–15. doi: 10.1007/BF02255913. [DOI] [PubMed] [Google Scholar]
- 26.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell. 2005;37(4):822–834. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Silva P, Enriquez A, Montoya J. Replication and transcription of mammalian mitochondrial DNA. Exp Physiol. 2003;88(1):41–56. doi: 10.1113/eph8802514. [DOI] [PubMed] [Google Scholar]
- 28.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576(1-2):1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- 29.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol. 2005;25(4):1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doleris LM, Hill GS, Chedin P, Nochy D, Bellanne-Chantelot C, Hanslik T, Bedrossian J, Caillat-Zucman S, Cahen-Varsaux J, Bariety J. Focal segmental glomerulosclerosis associated with mitochondrial cytopathy. Kidney Int. 2000;58(5):1851–1858. doi: 10.1111/j.1523-1755.2000.00356.x. [DOI] [PubMed] [Google Scholar]
- 31.Hagiwara M, Yamagata K, Capaldi RA, Koyama A. Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int. 2006;69(7):1146–1152. doi: 10.1038/sj.ki.5000207. [DOI] [PubMed] [Google Scholar]
- 32.Ortiz A, Justo P, Catalan MP, Sanz AB, Lorz C, Egido J. Apoptotic cell death in renal injury: the rationale for intervention. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2(2):181–192. [PubMed] [Google Scholar]
- 33.Takebayashi S, Kaneda K. Mitochondrial derangement: possible initiator of microalbuminuria in NIDDM. J Diabt Complications. 1991;5(2-3):104–106. doi: 10.1016/0891-6632(91)90034-m. [DOI] [PubMed] [Google Scholar]
- 34.Medina J, Fernandez-Salazar LI, Garcia-Buey L, Moreno-Otero R. Approach to the pathogenesis and treatment of nonalcoholic steatohepatitis. Diabetes Care. 2004;27(8):2057–2066. doi: 10.2337/diacare.27.8.2057. [DOI] [PubMed] [Google Scholar]
- 35.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37(5):1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 36.Mehta K, Van Thiel DH, Shah N, Mobarhan S. Nonalcoholic fatty liver disease: pathogenesis and the role of antioxidants. Nutr Rev. 2002;60(9):289–293. doi: 10.1301/002966402320387224. [DOI] [PubMed] [Google Scholar]
- 37.de la Torre JC. Pathophysiology of neuronal energy crisis in Alzheimer's disease. Neurodegener Dis. 2008;5(3-4):126–132. doi: 10.1159/000113681. [DOI] [PubMed] [Google Scholar]
- 38.Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G. Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Alzheimer disease. Neurotox Res. 2003;5(7):491–504. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 39.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17(3):331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Wei YH, Lu CY, Lee HC Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- 41.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA. 1988;85(17):6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Limon-Pacheco J, Gonsebatt ME. The role of antioxidants and antioxidant-related enzymes in protective responses to environmentally induced oxidative stress. Mutat Res. 2009;674(1-2):137–147. doi: 10.1016/j.mrgentox.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 44.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 45.Ballinger SW. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med. 2005;38(10):1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 46.Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochemical Journal. 2000;348(Pt2):425–432. [PMC free article] [PubMed] [Google Scholar]
- 47.Gogvadze V, Orrenius S. Mitochondrial regulation of apoptotic cell death. Chem Biol Interact. 2006;163(1-2):4–14. doi: 10.1016/j.cbi.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 48.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 49.Gronski MA, Weinem M. Death pathways in T cell homeostasis and their role in autoimmune diabetes. Rev Diabet Stud. 2006;3(2):88–95. doi: 10.1900/RDS.2006.3.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newman B, Selby JV, King MC, Slemenda C, Fabsitz R, Friedman GD. Concordance for type 2 (non-insulin-dependent) diabetes mellitus in male twins. Diabetologia. 1987;30(10):763–768. doi: 10.1007/BF00275741. [DOI] [PubMed] [Google Scholar]
- 51.Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Stengard J, Kesaniemi YA. Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia. 1992;35(11):1060–1067. doi: 10.1007/BF02221682. [DOI] [PubMed] [Google Scholar]
- 52.Medici F, Hawa M, Ianari A, Pyke DA, Leslie RD. Concordance rate for type II diabetes mellitus in monozygotic twins: actuarial analysis. Diabetologia. 1999;42(2):146–150. doi: 10.1007/s001250051132. [DOI] [PubMed] [Google Scholar]
- 53.Neel JV. Diabetes Mellitus: a "thrifty" genotype rendered detrimental by "Progress"? Am J Hum Genet. 1962;14:353–362. [PMC free article] [PubMed] [Google Scholar]
- 54.Knowler WC, Bennett PH, Hamman RF, Miller M. Diabetes incidence and prevalence in Pima Indians: a 19-fold greater incidence than in Rochester, Minnesota. Am J Epidemiol. 1978;108(6):497–505. doi: 10.1093/oxfordjournals.aje.a112648. [DOI] [PubMed] [Google Scholar]
- 55.Hanson RL, Elston RC, Pettitt DJ, Bennett PH, Knowler WC. Segregation analysis of non-insulin dependent diabetes mellitus in Pima Indians: evidence for a major-gene effect. Am J Hum Genet. 1995;57(1):160–170. [PMC free article] [PubMed] [Google Scholar]
- 56.King H, Rewers N. Diabetes in adults is now a Third World problem. The WHO Ad Hoc Diabetes Reporting Group. Bull World Health Organ. 1991;69(6):643–648. [PMC free article] [PubMed] [Google Scholar]
- 57.Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35(7):595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- 58.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2(8663):577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 59.Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303(6809):1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36(1):62–67. doi: 10.1007/BF00399095. [DOI] [PubMed] [Google Scholar]
- 61.Rich Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, Willett WC, Hennekens CH. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ. 1997;315(7105):396–400. doi: 10.1136/bmj.315.7105.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lithell HO, McKeigue PM, Berglund L, Mohsen R, Lithell UB, Leon DA. Relation of size at birth to non-insulin dependent diabetes and insulin concentrations in men aged 50-60 years. BMJ. 1996;312(7028):406–410. doi: 10.1136/bmj.312.7028.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geremia C, Cianfarani S. Insulin sensitivity in children born small for gestational age (SGA) Rev Diabet Stud. 2004;1(2):58–65. doi: 10.1900/RDS.2004.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leon DA, Lithell HO, Vagero D, Koupilova I, Mohsen R, Berglund L, Lithell UB, McKeigue PM. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15000 Swedish men and women born 1915-1929. BMJ. 1998;317(7153):241–245. doi: 10.1136/bmj.317.7153.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ. 1999;318(7181):427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, Bleker OP. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351(9097):173–177. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- 67.Poulton J. Does a common mitochondrial DNA polymorphism underlie susceptibility to diabetes and the thrifty genotype? Trend Genet. 1998;14(10):387–389. doi: 10.1016/s0168-9525(98)01529-7. [DOI] [PubMed] [Google Scholar]
- 68.Anderson S, Bankier AT, Barrell BG de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290(5806):457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 69.Massen JA, van Essen E, van den Quweland JM, Lemkes HH. Molecular and clinical aspects of mitochondrial diabetes mellitus. Exp Clin Endocrinol Diabetes. 2001;109(3):127–134. doi: 10.1055/s-2001-14834. [DOI] [PubMed] [Google Scholar]
- 70.Frederiksen AL, Jeppesen TD, Vissing J, Schwartz M, Kyvik KO, Schmitz O, Poulsen PL, Anderson PH. High prevalence of impaired glucose homeostasis and myopathy in asymptomatic and oligosymptomatic 3243A>G mitochondrial DNA mutation-positive subjects. J Clin Endocrinol Metab. 2009;94(8):2872–2879. doi: 10.1210/jc.2009-0235. [DOI] [PubMed] [Google Scholar]
- 71.Gerbitz KD, van den Quweland MW, Maassen JA, Jaksch M. Mitochondrial diabetes mellitus: a review. Biochim Biophys Acta. 1995;1271(1):253–260. doi: 10.1016/0925-4439(95)00036-4. [DOI] [PubMed] [Google Scholar]
- 72.Song J, Yound J, Sung YA, Pak YK, Park KS, Lee HK. Peripheral blood mitochondrial DNA content is related to insulin sensitivity in offspring of type 2 diabetic patients. Diabetes Care. 2001;24(5):865–869. doi: 10.2337/diacare.24.5.865. [DOI] [PubMed] [Google Scholar]
- 73.Lee HK, Song JH, Shin CS, Park DJ, Park KS, Lee KU, Koh CS. Peripheral blood mitochondrial DNA content can predict the development of diabetes. Diabetes. 1997;46(Suppl 1):175A. [Google Scholar]
- 74.Park KS, Lee KU, Song JH, Choi CS, Shin CS, Park DJ, Kim SK, Koh JJ, Lee HK. Peripheral blood mitochondrial DKA content is inversely correlated with insulin secretion during hyperglycemic clamp studies in healthy young men. Diabetes Res Clin Pr. 2001;52(2):97–102. doi: 10.1016/s0168-8227(00)00237-0. [DOI] [PubMed] [Google Scholar]
- 75.Weng SW, Lin TK, Liou CW, Chen SD, Wei YH, Lee HC, Chen IY, Hsieh CJ, Wang PW. Peripheral blood mitochondrial DNA content and dysregulation of glucose metabolism. Diabetes Res Clin Pr. 2009;83(1):94–99. doi: 10.1016/j.diabres.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 76.Singh R, Hattersley AT, Harries LW. Reduced peripheral blood mitochondrial DNA content is not a risk factor for type 2 diabetes. Diabetic Med. 2007;24(7):784–787. doi: 10.1111/j.1464-5491.2007.02164.x. [DOI] [PubMed] [Google Scholar]
- 77.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM Jr, Klein JB, Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287(5):E896–E905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 78.Wang PW. Tissue-specific difference of mitochondrial DNA content in type 2 diabetes. The 10th symposium on molecular diabetology in Asia and The 1st Sun Yat-Sen diabetes forum; Guangzhou, China. Nov 28-30, 2008. [Google Scholar]
- 79.Morten KJ, Poulton J, Sykes B. Multiple independent occurrence of the 3243 mutations in mitochondrial tRNA leuUUR in patients with the MELAS phenotype. Hum Mol Genet. 1995;4(9):1689–1691. doi: 10.1093/hmg/4.9.1689. [DOI] [PubMed] [Google Scholar]
- 80.Casteels K, Ong K, Phillips D, Bendall H, Pembrey M. Mitochondrial 16189 variant, thinness at birth, and type 2 diabetes. ALSPAC study team. Avon Longitudinal Study of Pregnancy and Childhood. Lancet. 1999;353(9163):1499–1500. doi: 10.1016/s0140-6736(98)05817-6. [DOI] [PubMed] [Google Scholar]
- 81.Poulton J, Brown MS, Cooper A, Marchington DR, Philips DI. A common mitochondrial DNA variant is associated with insulin resistance in adult life. Diabetologia. 1998;41(1):54–58. doi: 10.1007/s001250050866. [DOI] [PubMed] [Google Scholar]
- 82.Poulton J, Luan J, Macaulay V, Hennings S, Mitchell J, Wareham NJ. Type 2 diabetes is associated with a common mitochondrial variant: evidence from a population-based case-control study. Hum Mol Genet. 2002;11(13):1581–1583. doi: 10.1093/hmg/11.13.1581. [DOI] [PubMed] [Google Scholar]
- 83.Liou CW, Lin TK, Huang FM, Chen TL, Lee CF, Chuang YC, Tan TY, Chang KC, Wei YH. Association of the mitochondrial DNA 16189 T to C variant with lacunar cerebral infarction: evidence from a hospital-based case-control study. Ann N Y Acad Sci. 2004;1011:317–324. doi: 10.1007/978-3-662-41088-2_31. [DOI] [PubMed] [Google Scholar]
- 84.Weng SW, Liou CW, Lin TK, Wei YH, Lee CF, Eng HL, Chen SD, Liu RT, Chen JF, Chen IY, Chen MH, Wang PW. Association of mitochondrial DNA 16189 variant (T->C transition) with metabolic syndrome in Chinese adults. J Clin Endocr Metab. 2005;90(9):5037–5040. doi: 10.1210/jc.2005-0227. [DOI] [PubMed] [Google Scholar]
- 85.Liou CW, Lin TK, Weng HH, Lee CF, Chen TL, Wei YH, Chen SD, Chuang YC, Weng SW, Wang PW. A common mtDNA variant and increased body mass index as associated factors for development of type 2 diabetes: Additive effects of genetic and environmental factors. J Clin Endocrinol Metab. 2007;92(1):235–239. doi: 10.1210/jc.2006-0653. [DOI] [PubMed] [Google Scholar]
- 86.Saxena R, Bakker PIW, Singer K, Mootha V, Burtt N, Hirschhorn JN, Gaudet D, Isomaa B, Daly MJ, Groop L, Ardlie KG, Altshuler D. Comprehensive association testing of common mitochondrial DNA variation in metabolic disease. Am J Hum Genet. 2006;79(1):54–61. doi: 10.1086/504926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohlke KL, Jackson AU, Scott LJ, Peck EC, Suh YD, Chines PS, Watanabe RM, Buchanan TA, Conneely KN, Erdos MR. et al. Mitochondrial polymorphisms and susceptibility to type 2 diabetes-related traits in Finns. Hum Genet. 2005;118(2):245–254. doi: 10.1007/s00439-005-0046-4. [DOI] [PubMed] [Google Scholar]
- 88.Chinnery PF, Elliott HR, Patel S, Lambert C, Keers SM, Durham SE, McCarthy MI, Hitman GA, Hattersley AT, Walker M. Role of the mitochondrial DNA 16184-16193 poly-C tract in type 2 diabetes. Lancet. 2005;366(9497):1650–1651. doi: 10.1016/S0140-6736(05)67492-2. [DOI] [PubMed] [Google Scholar]
- 89.Das S, Bennett AJ, Sovio U, Ruokonen A, Martikainen H, Pouta A, Hartikainen AL, Franks S, Elliott P, Poulton J, Järvelin MR, McCarthy MI. Detailed analysis of variation at and around mitochondrial position 16189 in a large Finnish cohort reveals no significant association with early growth or metabolic phenotypes at age 31 years. J Clin Endocrinol Metab. 2007;92(8):3219–3223. doi: 10.1210/jc.2007-0702. [DOI] [PubMed] [Google Scholar]
- 90.Park KS, Chan JC, Chuang LM, Sukui S, Araki E, Nanjo K, Ji L, Ng M, Nishi M, Furuta H. et al. A mitochondrial DNA variant at position 16189 is associated with type 2 diabetes mellitus in Asians. Diabetologia. 2008;51:602–608. doi: 10.1007/s00125-008-0933-z. [DOI] [PubMed] [Google Scholar]
- 91.Kim JH, Park KS, Cho YM, Kang BS, Kim SK, Jeon HJ, Kim SY, Lee HK. The prevalence of the mitochondrial DNA 16189 variant in non-diabetic Korean adults and its association with higher fasting glucose and body mass index. Diabet Med. 2002;19(8):681–684. doi: 10.1046/j.1464-5491.2002.00747.x. [DOI] [PubMed] [Google Scholar]
- 92.Horai S, Hayasaka K. Intraspecific nucleotide sequence differences in the major noncoding region of human mitochondrial DNA. Am J Hum Genet. 1990;46(4):828–842. [PMC free article] [PubMed] [Google Scholar]
- 93.Ji L, Gao L, Han X. Association of 16189 variant (T>C transition) of mitochondrial DNA with genetic predisposition to type 2 diabetes in Chinese populations. Zhonghua Yi Xue Za Zhi. 2001;81(12):711–714. [PubMed] [Google Scholar]
- 94.Tang DL, Zhou X, Li X, Zhao L, Liu F. Variation of mitochondrial gene and the association with type 2 diabetes mellitus in a Chinese population. Diabetes Res Clin Pract. 2006;73(1):77–82. doi: 10.1016/j.diabres.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 95.Malik S, Sudoyo H, Pramoonjago P, Sukarna T, Darwis D, Marzuki S. Evidence for the de novo regeneration of the pattern of the length heteroplasmy associated with the T16189C variant in the control (D-loop) region of mitochondrial DNA. J Human Genet. 2002;47(3):122–130. doi: 10.1007/s100380200013. [DOI] [PubMed] [Google Scholar]
- 96.Malik S, Sudoyo H, Pramoonjago P, Suryadi H, Sukarna T, Nyunting M, Sahiratmadja E, Martuki S. Nuclear mitochondrial interplay in the modulation of the homopolymeric tract length heteroplasmy in the control (D-loop) region of the mitochondrial DNA. Hum Genet. 2002;110(5):402–411. doi: 10.1007/s00439-002-0717-3. [DOI] [PubMed] [Google Scholar]
- 97.Batista O, Kolman CJ, Bermingham E. Mitochondrial DNA diversity in the Kuna Amerinds of Panama. Hum Mol Genet. 1995;4(5):921–929. doi: 10.1093/hmg/4.5.921. [DOI] [PubMed] [Google Scholar]
- 98.Mountain JL, Hebert JM, Bhattacharyya S, Underhill PA, Ottolenghi C, Gadgil M, Cavalli-Sforza LL. Demographic history of India and mtDNA-sequence diversity. Am J Hum Genet. 1995;56(4):979–992. [PMC free article] [PubMed] [Google Scholar]
- 99.Templeton A. Out of Africa again and again. Nature. 2002;416(6876):45–51. doi: 10.1038/416045a. [DOI] [PubMed] [Google Scholar]
- 100.Cavalli-Sforza LL. Genes, peoples, and languages. University of California Press; Berkeley and Los Angeles: 2000. Technological revolutions and gene geography; pp. 92–132. [Google Scholar]
- 101.Hung YT, Wang PW, Hsieh CJ, Weng SW, Lin TK, Tiao MM, Chen JB, Liou CW. Sequence polymorphism of mitochondrial D-loop DNA in the southern Taiwanese. 2008 Conference of Taiwan Society for Mitochondrial Research and Medicine; Keelung, Taiwan. Feb 16-17, 2008. [Google Scholar]
- 102.Marchington DR, Poulton J, Sellar A, Holt IJ. Do sequence variations is in the major non-coding region of mitochondrial genome influence mitochondrial mutations associated with disease? Hum Mol Genet. 1996;5(4):473–479. doi: 10.1093/hmg/5.4.473. [DOI] [PubMed] [Google Scholar]
- 103.Gill-Randall R, Sherratt EJ, Thomas AW, Gagg JW, Lee A, Alcolado JC. Analysis of a polycytosine tract and heteroplasmic length variation in the mitochondrial DNA D-loop of patients with diabetes, MELAS syndrome and race-matched controls. Diabet Med. 2001;18(5):413–416. doi: 10.1046/j.1464-5491.2001.00477.x. [DOI] [PubMed] [Google Scholar]
- 104.Lin TK, Chen SD, Wang PW, Wei YH, Lee CF, Chen TL, Chuang YC, Tan TY, Chang KC, Liou CW. Increased oxidative damage with altered antioxidative status in type 2 diabetic patients harboring the 16189 T to C variant of mitochondrial DNA. Ann N Y Acad Sci. 2005;1042:64–69. doi: 10.1196/annals.1338.007. [DOI] [PubMed] [Google Scholar]
- 105.Tanaka M, Fuku N, Nishigaki Y, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Ito M, Nozawa Y, Yamada Y. Women with mitochondrial haplogroup N9a are protected against metabolic syndrome. Diabetes. 2007;56(2):518–521. doi: 10.2337/db06-1105. [DOI] [PubMed] [Google Scholar]
- 106.Fuku N, Park KS, Yamada Y, Nishigaki Y, Cho YM, Matsuo H, Segawa T, Watanabe S, Kato K, Yokoi K, Nozawa Y, Lee HK, Tanaka M. Mitochondrial haplogroup N9a confers resistance against type 2 diabetes in Asians. Am J Hum Genet. 2007;80(3):407–415. doi: 10.1086/512202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes for BioMedical Research. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316(5829):1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 108.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM. et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU. et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D'Agostino RB Sr, Cupples LA. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359(21):2208–2219. doi: 10.1056/NEJMoa0804742. [DOI] [PMC free article] [PubMed] [Google Scholar]