

Abstract
Responses of cells to mechanical properties of the adhesion substrate were examined by culturing normal rat kidney epithelial and 3T3 fibroblastic cells on a collagen-coated polyacrylamide substrate that allows the flexibility to be varied while maintaining a constant chemical environment. Compared with cells on rigid substrates, those on flexible substrates showed reduced spreading and increased rates of motility or lamellipodial activity. Microinjection of fluorescent vinculin indicated that focal adhesions on flexible substrates were irregularly shaped and highly dynamic whereas those on firm substrates had a normal morphology and were much more stable. Cells on flexible substrates also contained a reduced amount of phosphotyrosine at adhesion sites. Treatment of these cells with phenylarsine oxide, a tyrosine phosphatase inhibitor, induced the formation of normal, stable focal adhesions similar to those on firm substrates. Conversely, treatment of cells on firm substrates with myosin inhibitors 2,3-butanedione monoxime or KT5926 caused the reduction of both vinculin and phosphotyrosine at adhesion sites. These results demonstrate the ability of cells to survey the mechanical properties of their surrounding environment and suggest the possible involvement of both protein tyrosine phosphorylation and myosin-generated cortical forces in this process. Such response to physical parameters likely represents an important mechanism of cellular interaction with the surrounding environment within a complex organism.
Adhesions between cells and the extracellular matrix (ECM) are known to modulate numerous critical cellular events such as gene expression (1), embryonic development (1), and cell locomotion (2). This process involves interactions of ECM proteins, e.g., collagen, fibronectin, or vitronectin, with the integrin family of transmembrane receptors. Subsequent cascade of events include the phosphorylation of proteins at adhesion sites and the recruitment of various cytoskeletal proteins to form focal adhesions (3).
A number of observations suggest that cell adhesion reactions involve not only receptor binding but also physical interactions and the cytoskeleton (4–9). For example, it is well known that, to elicit a full response, ECM proteins must be immobilized or cross-linked (8, 9). In addition, previous studies have shown that cells can respond to forces exerted through surrounding fluid, adhered beads, or substrates (5, 6, 10–12). Thus, mechanical forces may play a role in adhesion responses, and, conversely, cells may actively probe and respond to mechanical cues in the surrounding environment. Consistent with this latter idea, a number of studies suggest that physical and chemical properties of the adhesion substrate can profoundly affect cell locomotion, growth, and differentiation (13–15).
Although previous observations are suggestive, there has been no direct demonstration that cells can probe and respond to the mechanical property of the substrate. To test this hypothesis directly, it is necessary to culture cells on substrates with variable physical properties while maintaining a constant chemical environment. In this study, we have developed a thin polyacrylamide-based, collagen-coated flexible substrate. By maintaining a constant total concentration of acrylamide while varying the concentration of bis-acrylamide, we were able to obtain a series of chemically identical substrates with a wide range of flexibility. By using imaging techniques, we show that cells can respond to differences in substrate flexibility by altering both their adhesion structures and motile behavior. Moreover, the response appears to involve both tyrosine phosphorylation and forces generated by the actin-myosin cytoskeleton.
MATERIALS AND METHODS
Preparation of Polyacrylamide Substrate and RIA.
Polyacrylamide gels were attached to glass coverslips following a method described previously (16). In brief, a large piece of coverglass (No. 1, 45 mm × 50 mm; Fisher) was flamed in a Bunsen burner, soaked in 0.1N NaOH, and air dried. A small aliquot of 3-aminopropyltrimethoxysilane (Sigma) was spread evenly onto the glass surface. After 4–5 min, the coverslips were washed and soaked in distilled H2O. The coverslips were then immersed for 30 min in a solution of 0.5% glutaraldehyde (Polysciences) in PBS. Coverslips were then washed extensively in distilled H2O and air dried. Alternatively, coverslips were coated with nitrocellulose to increase the binding of the polyacrylamide (1% stock in amyl acetate; Ernest F. Fullam, Schenectady, NY). Twenty-five microliters of an acrylamide/bis-acrylamide mixture, containing 10% acrylamide and bis concentrations ranging from 0.26 to 0.03%, was then placed on the coverslip and covered with a small circular piece of coverglass (No. 1, 22-mm diameter; Fisher). After polymerization, the round coverglass was removed, and the gel was rinsed with 200 mM Hepes (Boehringer Mannheim, pH 8.5). The gel was then blot dried, and 200 μl of 50 mM sulfosuccinimidyl 6 (4′-azido-2′-nitrophenyl-amino) hexanoate (Sulfo-SANPAH; Pierce) in 200 mM Hepes, pH 8.5, was pipetted onto the surface. The coverslip chamber was exposed to the UV light of a sterile hood at a distance of 6 inches for 5 min. The Sulfo-SANPAH solution was then removed, and the photoactivation procedure was repeated. After photoactivation, the polyacrylamide sheet was washed several times in 200 mM Hepes (pH 8.5). A 0.2-mg/ml solution of type I collagen (United States Biochemical) was then layered onto the substrate and allowed to react overnight at 4°C. After washing with 200 mM Hepes, the gels were stored at 4°C. To form a cell culture chamber, coverslips were attached with vacuum grease (Dow Corning) to a 70 × 50 × 6-mm Plexiglass plate with a 35-mm diameter annulus bored through the center (17). Before plating cells, the gel was soaked for 30–45 min in culture medium at 37°C.
A modified RIA (18) using mAb against type I collagen (clone COL-1) and 125I-anti mouse IgG (Amersham) was used to determine the relative amount of collagen bound on the surface of polyacrylamide sheets.
Characterization of Polyacrylamide Gels.
The flexibility of polyacrylamide sheets with different acrylamide/bis-acrylamide ratios was determined both macroscopically and microscopically. Sheets of 69 mm × 30 mm × 1 mm were deformed with a force (F⊥) of 0.103 N (Fig. 1A), and Young’s modulus (19) was calculated according to the equation: Y = (F⊥/A)/(Δl/l), where l is the original length of the sheet, Δl is the change in length, and A is the cross-sectional area. Compliance or “stiffness” of thin gels used for cell culture was characterized microscopically by deforming with glass microneedles (20). The same needles were calibrated by measuring vertical movements of the tip after applying known submilligram weights. Gel compliance was calculated as N/μm deformation. Uncoated and collagen-coated substrates gave identical compliance and Young’s modulus values.
Figure 1.
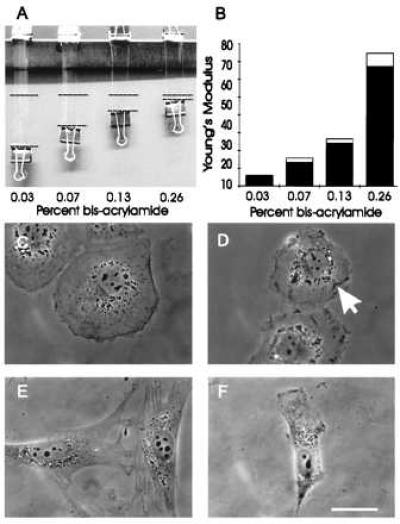
Mechanical characteristics of polyacrylamide substrates and effects on cell morphology. (A and B) identically sized strips of polyacrylamide with various acrylamide/bis-acrylamide ratios were fixed at one end and stretched at the other end with a downward force of 0.103 N. The dashed lines represent the amount of stretching caused by applied weight (A). The extent of stretching was then used for the calculation of Young’s modulus, expressed as N/m2 (B). (C–F) Phase morphology of NRK (C and D) or 3T3 (E and F) cells plated on substrates containing 0.26% bis- (C and E) or 0.03% bis-acrylamide (D and F). NRK cells on the more flexible substrate are less well spread and contain irregular ruffles on the ventral surface (D, arrow), as determined by optical sectioning at a high magnification. Similarly, 3T3 cells on the substrate of high flexibility are typically less well spread and with a polarized morphology (F). Bar = 10 μm.
Cell Culture and Drug Treatments.
Normal rat kidney (NRK)-52E (American Type Culture Collection) were cultured in F-12K media (Sigma), supplemented with 10% fetal calf serum (JRH Biosciences, Lenexa, KS), 2 mM l-glutamine, 50 μg/ml streptomycin, 50 units/ml penicillin, and 250 ng/ml amphotericin B (GIBCO/BRL). Swiss 3T3 cells (ATCC) were cultured in DMEM (Sigma) supplemented with 10% donor calf serum (JRH Biosciences) and other additives as for NRK cells. For drug studies, phenylarsine oxide (Sigma) and KT5926 (Calbiochem) were each dissolved in dimethyl sulfoxide to generate stock solutions of 10 mM and 2 mM, respectively. Immediately before drug treatments, aliquots of inhibitor stocks were diluted in serum-containing media to obtain a final concentration of 5 μM for phenylarsine oxide and 20 μM for KT5926. 2,3-Butanedione monoxime (Sigma) was dissolved directly in culture medium to generate a working solution at the final concentration of 20 mM. Cells were incubated in KT5926 or 2,3-butanedione monoxime for 20 min and phenylarsine oxide for 10 min before fixation and staining.
Quantification of Cell Motility.
To measure the dynamics of the lamellipodia in NRK cells, time-lapse sequences of cells located at the periphery of colonies were recorded over a period of 10 min by using a Zeiss 40× F-achromat phase contrast lens. The leading edge was traced using coreldraw! 5.0 (Corel, Ottawa). The position of several randomly chosen points was determined as a function of time, and the SD was used as a measurement of the fluctuation of the leading edge. The motility of 3T3 cells was determined by plotting the migration of the center of nuclei in randomly chosen cells over a period of 60 min.
Fixation, Fluorescent Labeling, Immunoblotting, and Microscopy.
For fluorescence staining of vinculin, phosphotyrosine, and actin, cells were washed in 37°C PBS containing 1 mM sodium orthovanadate (Fisher), then fixed in 4% formaldehyde for 10 min (16% stock solution; Electron Microscopy Sciences, Fort Washington, PA) and extracted with 0.5% Triton X-100 (Boehringer Mannheim) in PBS for 5 min. Immunofluorescence staining was performed by using mAb against vinculin (clone VIN-11–5, Sigma) or phosphotyrosine (clone 4G10, Upstate Biotechnology, Lake Placid, NY), each at a dilution of 1:100. Rhodamine and fluorescein-conjugated secondary antibodies were obtained from Sigma. Staining of cells with fluorescein-phalloidin (Molecular Probes), microinjection of fluorescently labeled vinculin, and fluorescence microscopy were performed as described (21–23). A Nikon 60×, N.A. 1.2, PlanApo water immersion objective was used for the observation of fluorescent vinculin in living cells. Other fluorescence images were collected with either a Zeiss 63×, N.A. 1.25 Neofluar objective or a Zeiss 100×, N.A. 1.30 Neofluar objective lens. Both phase and fluorescence images were recorded with a cooled charged coupled device camera (TE/CCD-576EM; Princeton Instruments, Trenton, NJ, or CH250; Photometrics, Tucson, AZ). Immunoblotting was performed following published procedures (7).
RESULTS
We prepared a series of polyacrylamide substrates with 10% acrylamide and with bis-acrylamide ranging from 0.03 to 0.26%. Because cultured cells did not adhere to bare polyacrylamide surfaces, the substrate was activated chemically with a photoactivatible heterobifunctional reagent and reacted with type I collagen. RIAs indicated that the relative surface concentration of collagen varied by <3% among the substrates regardless of the flexibility. To facilitate observations with phase and fluorescence optics at high magnifications, we prepared sheets with a thickness of ≈40 μm, which were attached to coverslips through either nitrocellulose coating or a chemical reaction.
The elastic property of the substrate was tested first by applying known forces to large suspended sheets (Fig. 1A). The length of polyacrylamide sheets increased in proportion to the applied force and, upon the release of forces, recovered fully within a second. Thus, the substrate behaves as an ideal material for testing the effect of substrate elasticity on cell behavior. The Young’s modulus showed a 12-fold difference between sheets of 0.26 and 0.03% bis-acrylamide (Fig. 1 A and B). The substrates also were probed microscopically with a calibrated microneedle to determine substrate “stiffness” or compliance (20). The compliance displayed a 16-fold difference (≈7.3 × 10−7 N/μm vs. ≈4.6 × 10−8 N/μm) between substrates of 0.26 and 0.03% bis-acrylamide.
On more rigid substrates, both NRK epithelial cells and 3T3 fibroblasts were well spread and appeared indistinguishable from those cultured on glass or plastic surfaces (Fig. 1 C and E). When cells were cultured on increasingly flexible substrates, there was a corresponding change in morphology: NRK cells became less well spread and irregularly shaped (Fig. 1D). Phase-dense ruffles appeared not only along the periphery but on the ventral surface of the cell (Fig. 1D, arrow). 3T3 cells lost most of their stress fibers (not shown) and became increasingly spindle-shaped. Cells with an elongated body and a lamellipodium at one end increased from 47% on substrates of 0.26% bis- to 69% on substrates of 0.03% bis-acrylamide (Fig. 1F).
To rule out the possibility that the morphological changes observed were due to the differential loss of covalently bound collagen from the substrate, cells were cultured on either 0.26% or 0.03% substrate to a high density, then lysed with 0.5% Triton X-100. RIA with anti-type I collagen antibodies indicated that the collagen concentration did not decrease on either stiff or flexible substrates as a result of cell growth, when compared with control substrates not used for cell culture.
The altered cell morphology on flexible substrates suggests that there may be differences in the rate of motility. On rigid 0.26% bis-acrylamide substrates, 3T3 cells migrated at an average rate of 0.06 μm/min. This rate increased to 0.55 μm/min (Fig. 2C; n = 68) on soft substrates of 0.03% bis-acrylamide. On both rigid and soft substrates, NRK epithelial cells remained associated with colonies and underwent little net nuclear migration. However, lamellipodial ruffling along the periphery of the colony was much more active on soft substrates. As shown in Fig. 2A, the extent of fluctuation in lamellipodia boundary differed by ≈6-fold between rigid (0.26% bis-acrylamide) and flexible (0.03% bis-acrylamide) substrates (Fig. 2 A and B).
Figure 2.
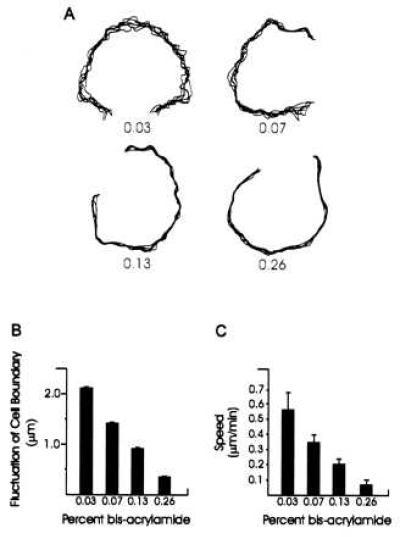
Measurements of lamellipodial activity and cell motility on substrates of various flexibilities. (A, B) Fluctuation of the lamellipodia of NRK cells cultured on substrates of varying flexibilities. Numbers in A indicate the percentage concentration of bis-acrylamide. Images of cells were recorded every 2 min over a period of 10 min and active lamellipoda were traced and overlaid to generate the plots. The degree of lamellipodial protrusion/retraction was then quantified based on the SD of the position of seven randomly chosen points along the active edge. Fifteen cells were analyzed under each bis-acrylamide concentration (B). Lamellipodia become less active with increasing rigidity of the substrate. (C) Rate of locomotion of 3T3 cells on substrates of varying flexibilities. Cells become less motile with increasing rigidity of the substrate.
Thus, it is possible that cells can respond to differences in substrate adhesion structures on substrates of different flexibility. We speculate that the responses of cells to substrate flexibility most likely originate at cell substrate adhesion sites, where mechanical input might be translated into intracellular signals through the associated cytoskeleton or enzyme complexes. To test this hypothesis, we first microinjected NRK cells cultured on polyacrylamide sheets with fluorescently labeled vinculin. Cells on more rigid substrates (0.26% bis-acrylamide) formed arrays of stable, elongated focal adhesions, which showed only minor changes over a period of 10 min (Fig. 3 A and C, arrows). On flexible substrates (0.03% bis-acrylamide) the adhesion sites appeared as irregular punctate structures; many of which appeared and disappeared within 10 min (Fig. 3 B and D, arrows). The behavior of adhesion structures on flexible substrates appears similar to that seen in transformed cells (21).
Figure 3.
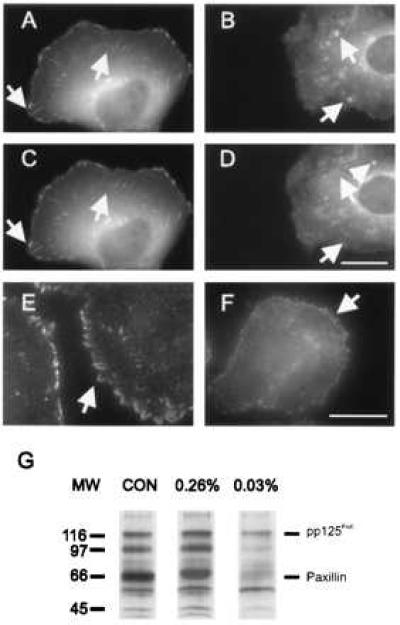
Distribution of vinculin and phosphotyrosine in NRK cells cultured on substrates with 0.26% bis-acrylamide (A, C, E) or 0.03% bis-acrylamide (B, D, F). (A–D) Cells were injected with rhodamine-labeled vinculin and imaged over a period of 10 min. On more rigid substrates (A, C), vinculin is incorporated into elongated focal adhesions, which show only minor changes during the period of observation. On highly flexible gels (B, D), vinculin is localized at punctate structures of irregular sizes and shapes, many of which appear and disappear over a period of 10 min (arrows). (E, F), Immunofluorescence of phosphotyrosine. Phosphotyrosine is localized at elongated focal adhesions in cells cultured on more rigid gels (E), and at punctate structures in cells cultured on highly flexible gels (F). (G) Anti-phosphotyrosine immunoblotting of whole cell lysates from NRK cells cultured on different substrates. On plastic dishes (CON) and rigid 0.26% bis-acrylamide substrates (0.26%), pp125FAK, paxillin and a 97-kDa protein are heavily phosphorylated after 48 hr of culture. Cells cultured on soft 0.03% bis-acrylamide substrates (0.03%) show a significantly lower extent of phosphorylation at these bands. Bar = 10 μm.
Because signaling at focal adhesions is believed to involve protein tyrosine kinases/phosphatases (1, 3, 7, 24–27), we asked whether there is a change in phosphotyrosine distribution in response to substrate flexibility. Immunofluorescence staining revealed that phosphotyrosine organization paralleled that of vinculin: When cells were plated on increasingly flexible substrates, the site of staining changed from wedge-shaped focal adhesions into irregular punctate structures (Fig. 3 E and F, arrows). Parallel immunoblot studies with anti-phosphotyrosine Py20 antibody indicated that, on flexible substrates, there was a large reduction in the overall extent of phosphorylation (Fig. 3G) compared with cells plated on plastic or rigid surfaces. Two of the major tyrosine-phosphorylated bands were identified as pp125FAK and paxillin, based on immunoreactivity with FAK-specific antibodies and electrophoretic mobilities, respectively.
Thus, it is possible that stiff substrates stimulate an increase in tyrosine phosphorylation, which then leads to the formation of stable focal adhesions. To test this possibility, cells on highly flexible substrates were treated with phenylarsine oxide (PAO), a tyrosine phosphatase inhibitor (27), to cause an artificial increase in tyrosine phosphorylation. The treatment inhibited ruffling activities and caused the appearance of elongated focal adhesions at the cell boundary (Fig. 4 A–D, arrows), indistinguishable from those on rigid substrates without PAO (Fig. 3 A, C, E, arrows). Time-lapse recording of cells injected with fluorescent vinculin indicated that these focal adhesions were as stable as those on stiff substrates (not shown).
Figure 4.
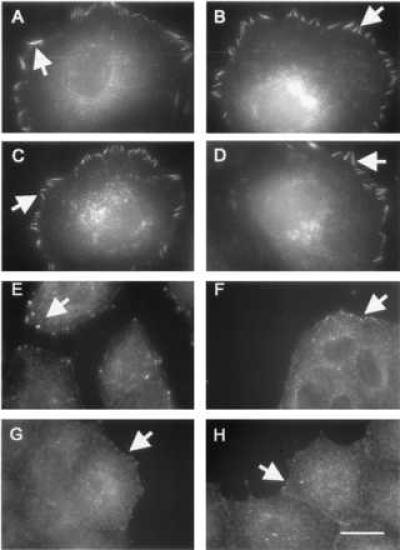
Role of tyrosine phosphorylation and myosin in the modulation of adhesion structures on flexible substrates. Cells were treated with PAO (A–D) or 2,3-butanedione monoxime (E–H) and processed for vinculin (A, B, E, F) or phosphotyrosine (C, D, G, H) immunofluorescence. Treatment of cells with PAO resulted in the formation of large focal adhesions in cells on both soft (0.03% bis-acrylamide; B, D) and rigid (0.26% bis-acrylamide; A, C) substrates. Treatment with 2,3-butanedione monoxime disrupted adhesion structures of cells cultured on substrates with either 0.26% bis-acrylamide (E, G), or 0.03% bis-acrylamide (F, H), and caused vinculin (E, F) and phosphotyrosine (G, H) to localize at small punctate structures regardless of the substrate flexibility. Bar = 10 μm.
To test the role of the cortical cytoskeleton in cellular responses to substrate flexibility, we treated cells on polyacrylamide substrates with KT5926, a potent inhibitor of the myosin light chain kinase (28), or 2,3-butanedione monoxime, an inhibitor of myosin motors (7, 29). Both compounds caused the disappearance of normal focal adhesions on firm substrates and the formation of irregular punctate vinculin-containing structures similar to those seen on highly flexible substrates. Parallel changes were observed with the staining of phosphotyrosine (Fig. 4 E–H, arrows). However, unlike cells on flexible substrates, myosin-inhibited cells showed neither enhanced motility nor rapid changes in adhesion structures, which likely reflect the involvement of myosin-generated forces in these processes.
DISCUSSION
Although it may be possible to address this issue with the silicone rubber substrates developed by Harris and improved in a recent study (20, 30–31), the ECM-coated polyacrylamide substrate has several important features. (i) It allows systematic and reproducible control of the flexibility of the substrate by changing the relative concentration of acrylamide and bis-acrylamide. (ii) Its superb optical quality and minimal thickness permits the observation of both immunofluorescence and microinjected fluorescent analogs at a high magnification. (iii) The substrate uses specific ECM molecules as the ligand for cell adhesion whereas polyacrylamide itself shows no detectable interaction with the cell surface. (iv) The porous nature of the polyacrylamide gel provides a more physiological environment for cell culture (32), particularly for epithelial cells. (v) Mechanical properties of the substrate can be characterized on both macro- and microscopic scales, with a nearly ideal elastic behavior in both cases.
Our results with 3T3 and NRK cells demonstrate the ability of cultured cells to detect the flexibility of the surrounding environment, and to regulate their adhesion structures and motility accordingly. On flexible substrates, fibroblasts migrate at a faster rate, and epithelial cells show elevated lamellipodial protrusion/retraction activities. These responses may be a result of destabilized adhesion, as indicated by microinjected fluorescent vinculin, and/or responses to signals that originate at the adhesion sites.
Immunofluorescene and immunoblots of phosphotyrosine suggest that signals elicited by a stiff substrate involve either the stimulation of a tyrosine kinase or inhibition of a tyrosine phosphatase. In addition, by artificially maintaining a high level of tyrosine phosphorylation through PAO-inhibition of tyrosine phosphatases, cells can bypass the stimulus elicited by stiff substrates and always assume a morphology characteristic of that on rigid surfaces. Although it is difficult to rule out indirect effects, these results are consistent with the idea that increases in tyrosine phosphorylation, in response to substrate stiffness, cause the formation of mature, stable focal adhesions and possibly also reduced ruffling activities.
How could cells sense the difference among substrates of identical chemical properties but varying flexibilities? The most plausible mechanism involves active pushing/pulling of its integrin receptors through the associated cytoskeleton; the response then leads to changes in tyrosine phosphorylation. Our observations with myosin inhibitors are consistent with the idea that a myosin motor is involved in probing substrate mechanical properties. Presumably, upon the inhibition of myosin, cells lose their ability to detect the resistance to adhesion forces and interpret all substrates as being flexible. Although alternative explanations are possible, this interpretation is supported by a recent observation that myosin is required for the increase in tyrosine phosphosphorylation induced by lysophosphatidic acid (7). In addition, treatment of endothelial cells with cytochalasin D causes a decrease in the extent of FAK tyrosine phosphorylation (33).
An attractive hypothesis is that, when receptors become anchored to a rigid substrate or cross-linked, the resistance to cytoskeleton-generated forces causes an increase in tension at adhesion structures and activates downstream signals through a force-sensitive enzyme complex. However, to detect flexibility, it is necessary for the cells to modulate and measure the probing force in response to different substrate resistance (otherwise, cells will simply deform soft substrates to an increasing extent until they experience a similar resistance as on stiff substrates). Alternatively, cells may be able to measure the amount of substrate deformation as they apply a defined probing force. These possibilities are speculative at present, and the precise mechanism for cells to probe and respond to substrate flexibility remains to be elucidated.
The present results, together with a growing list of observations suggesting that cells can respond to both the magnitude and distribution of adhesion forces (34), strongly indicate that communications through physical signals are as important as communications through chemical messengers. Physiologically, mechanical properties of cells’ surrounding environment could be modulated by synthesis/degradation of ECM proteins (1), the movement of surrounding cells, or pressure/fluid shear of the blood flow (10, 11). Such events are likely to occur frequently during embryonic development and wound healing (1, 35) and may play an important role in guiding cell movement and regulating cell functions.
Acknowledgments
We thank Drs. S. P. Wheatley and E. Luna for critical comments on the manuscript, Drs. E. Luna and A. Hitt for assistance with the RIA, and T. Pelham for technical assistance. This study was funded by National Institutes of Health Grant GM-32476
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: ECM, extracellular matrix; PAO, phenylarsine oxide; NRK, normal rat kidney.
References
- 1.Juliano R L, Haskill S J. J Cell Biol. 1993;120:577–585. doi: 10.1083/jcb.120.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein L R, Liotta L A. Curr Opin Oncol. 1994;6:106–113. doi: 10.1097/00001622-199401000-00015. [DOI] [PubMed] [Google Scholar]
- 3.Craig S W, Johnson R P. Curr Opin Cell Biol. 1996;8:74–85. doi: 10.1016/s0955-0674(96)80051-2. [DOI] [PubMed] [Google Scholar]
- 4.Palecek S P, Loftus J C, Ginsberg M H, Lauffenburger D A, Horowitz A F. Nature (London) 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 5.Wang N, Butler J P, Ingber D E. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- 6.Ingber D E. J Cell Sci. 1993;104:613–627. doi: 10.1242/jcs.104.3.613. [DOI] [PubMed] [Google Scholar]
- 7.Chrzanowska-Wodnicka M, Burridge K. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyamoto S, Akiyama S K, Yamada K M. Science. 1995;267:883–885. doi: 10.1126/science.7846531. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto S, Teramoto H, Coso O A, Gutkind J S, Burbelo P D, Akiuama S K, Yamada K M. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girad P R, Nerem R M. J Cell Phys. 1995;163:179–193. doi: 10.1002/jcp.1041630121. [DOI] [PubMed] [Google Scholar]
- 11.Davies P F. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choquet D, Felsenfeld D P, Sheetz M P. Cell. 1997;88:39–48. doi: 10.1016/s0092-8674(00)81856-5. [DOI] [PubMed] [Google Scholar]
- 13.Harris A K. Exp Cell Res. 1973;77:285–297. doi: 10.1016/0014-4827(73)90579-x. [DOI] [PubMed] [Google Scholar]
- 14.Folkman J, Moscona A. Nature (London) 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 15.Ingber D E, Folkman J. J Cell Biol. 1989;109:317–330. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alpin J D, Hughes C. Anal Bichem. 1981;113:144–148. doi: 10.1016/0003-2697(81)90057-9. [DOI] [PubMed] [Google Scholar]
- 17.McKenna N M, Wang Y-l. Methods Cell Biol. 1989;29:105–205. doi: 10.1016/s0091-679x(08)60195-8. [DOI] [PubMed] [Google Scholar]
- 18.Goding J W. Monoclonal Antibodies: Practice and Principals. New York: Academic; 1983. pp. 75–77. [Google Scholar]
- 19.Heuvelen A V. Physics: A General Introduction. Brown, New York: Little; 1982. p. 299. [Google Scholar]
- 20.Lee J, Leonard M, Oliver T, Ishihara A, Jacobson K. J Cell Biol. 1994;127:1957–1964. doi: 10.1083/jcb.127.6.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stickel S K, Wang Y-l. J Cell Biol. 1987;104:1521–1526. doi: 10.1083/jcb.104.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meigs J B, Wang Y-l. J Cell Biol. 1986;102:1430–1438. doi: 10.1083/jcb.102.4.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelham R J, Jr, Lin J J-C, Wang Y-l. J Cell Sci. 1996;109:981–989. doi: 10.1242/jcs.109.5.981. [DOI] [PubMed] [Google Scholar]
- 24.Parsons J T. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- 25.Burridge K, Turner C E, Romer L H. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maher P A, Pasquale E B, Wang J Y J, Singer S J. Proc Natl Acad Sci USA. 1985;82:6570–6580. doi: 10.1073/pnas.82.19.6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maher P A. Proc Natl Acad Sci USA. 1993;90:11177–11181. doi: 10.1073/pnas.90.23.11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakanishi S, Yamada K, Iwanashi K, Kuroda K, Kase K. Mol Pharmacol. 1990;37:482–488. [PubMed] [Google Scholar]
- 29.Higuchi H, Takemori S. J Biochem (Tokyo) 1989;105:638–643. doi: 10.1093/oxfordjournals.jbchem.a122717. [DOI] [PubMed] [Google Scholar]
- 30.Harris A K, Wild P, Stopak D. Science. 1980;208:177–179. doi: 10.1126/science.6987736. [DOI] [PubMed] [Google Scholar]
- 31.Burton K, Taylor D L. Nature (London) 1997;385:450–454. doi: 10.1038/385450a0. [DOI] [PubMed] [Google Scholar]
- 32.Simons K, Fuller S D. Annu Rev Cell Biol. 1985;1:243–288. doi: 10.1146/annurev.cb.01.110185.001331. [DOI] [PubMed] [Google Scholar]
- 33.Defilippi P, Retta S F, Olivo C, Palmieri M, Venturino M, Silengo L, Tarone G. Exp Cell Res. 1995;221:141–152. doi: 10.1006/excr.1995.1361. [DOI] [PubMed] [Google Scholar]
- 34.Chen C S, Mrksich M, Huang S, Whitesides G M, Ingber D E. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 35.Schaffer C J, Nanney L B. Int Rev Cytol. 1996;169:151–181. doi: 10.1016/s0074-7696(08)61986-5. [DOI] [PubMed] [Google Scholar]